

Abstract
Antiretroviral therapy (ART) has rendered HIV-1 infection a manageable illness for those with access to treatment. However, ART does not lead to viral eradication due to the persistence of replication-competent, unexpressed proviruses in long-lived cellular reservoirs. The potential for long-term drug toxicities and the lack of access to ART for most people living with HIV-1 infection have fueled scientific interest in understanding the nature of this latent reservoir. Exploration of HIV-1 persistence at the cellular and molecular level in resting memory CD4+ T cells, the predominant viral reservoir in patients on ART, has uncovered potential strategies to reverse latency. Here, we review recent advances in pharmacologically-based ‘shock and kill’ HIV-1 eradication strategies, including comparative analysis of early clinical trials.
The successes and limitations of ART
The introduction of combination antiretroviral therapy (ART) represents a groundbreaking achievement in the effort to combat HIV-1 infection[1, 2]. Durable blockade of viral replication by combinations of antiretroviral drugs has transformed HIV-1 infection from a lethal condition characterized by progressive immune deficiency into a manageable medical problem[3–7]. However, long-term ART does not result in HIV-1 eradication due the presence of long-lived viral reservoirs[8–10]. ART cessation results in viral rebound within a matter of weeks that arises from resting memory CD4+ T cells harboring HIV-1 proviral DNA integrated into the cellular genome (Figure 1). This reservoir does not decay significantly during the lifespan of an HIV-1 infected patient[11, 12]. These latently infected cells are thought to sporadically re-activate leading to derepression of silenced HIV-1[13]. The process likely gives rise to the low-level viremia observed in patients on ART and is thought to be the source of productive infection and viral rebound in those who stop taking antiretrovirals[14, 15]. Multiple ART intensification trials have resulted in no change in residual viremia[16–19], which underscores the need for strategies that directly target or suppress the latent reservoir[20].
Figure 1. Sources and kinetics of plasma viremia on antiretroviral therapy (ART).
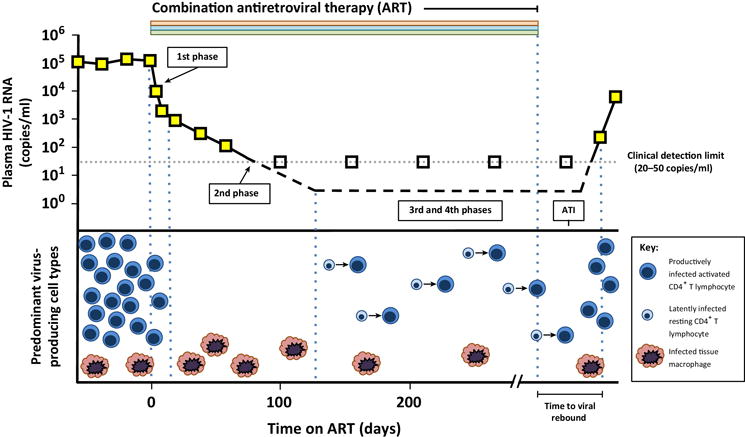
The initiation of ART results in a biphasic decay in plasma viremia. The first phase reflects the death of productively infected CD4+ T lymphocytes. Infected cells with a longer half-life, such as tissue macrophages, are thought to produce the second phase of viral decay, during which the viral load falls below the detection limit of commercial assays (20-50 copies/mL). Patients maintain low-level viremia during ART that likely arises from spontaneous reactivation of latently infected resting CD4+ T cells. The contribution of non-T cell reservoirs including chronically infected tissue macrophages to residual viremia remains incompletely understood. Analytical treatment interruption (ATI) consists of study participants stopping ART with close monitoring for adverse effects of unchecked viral replication and quantifiable viral rebound. The time to viral rebound during ATI is thought to provide an estimate of the efficacy of the intervention in reducing reservoir size.
Figure adapted from Durand C.M. et al. Trends in Immunology 2012 [ref 22]
The description of the first (and only) durable cure of HIV-1 infection[21] has invigorated HIV-1 “cure” research and has given rise to unique eradication strategies[22–24]. The mechanism(s) of reservoir eradication in the ‘Berlin patient,’ who underwent allogeneic stem cell transplant to treat acute myelogenous leukemia with donor cells homozygous for the C-C chemokine receptor 5 (CCR5) delta32 mutation [21], is still a matter of debate. However the lack of CCR5 expression, the major co-receptor required for HIV-1 cellular entry, on engrafted donor immune cells is likely to have played a significant role. Evidence from non-human primate models suggest that CCR5-deficient cells can suppress replication of CCR5-tropic virus[25]. Indeed, gene therapy approaches have been developed that disrupt the CCR5 coding sequence in patient T cells ex vivo. Autologous, CCR5-deficient T cells can then be expanded and re-infused into patients in an attempt to reduce the frequency of target cells for viral replication. A recent clinical trial evaluated the safety of this approach in vivo and demonstrated engraftment and persistence of these cells in the circulation and in tissues months after infusion[26].
From another standpoint, recognition of key cytokines that govern T cell activation status, trafficking and homeostasis in vivo has led to a number of immune-based strategies to target the HIV-1 latent reservoir. Three clinical trials studying the role of administering exogenous interferon alpha (IFNα) on latent reservoir dynamics are ongoing (NCT01935089, NCT01295515, NCT02227277). One trial, adding recombinant interleukin 7 (IL-7) to intensified ART regimens has been completed, but the results are not yet published (NCT01019551). The safety and efficacy of recombinant IL-15 in reducing reservoir size will be evaluated in an approved clinical trial that is not yet open for enrollment (NCT02191098).
The focus of this review is a pharmacologic approach to reservoir elimination, also known as the ‘shock and kill’ strategy[27], in which ART could be supplemented for a discrete time period with drugs that selectively re-awaken dormant viruses in the latent reservoir (induced proviral transcription) and render infected cells susceptible to virus-induced apoptosis or immune-mediated clearance. After depletion of the latent reservoir, ART could then be stopped without subsequent return of viremia, resulting in a functional cure, depending on the complete or partial elimination of viral sequences (see Glossary). Several classes of latency reversing agents, or LRAs, have been intensively studied and thorough reviews have recently been published describing the characteristics of mechanistically distinct LRA classes[23, 28, 29].
Despite considerable scientific and therapeutic advances in the three decades since the discovery of HIV-1, persistent inequities in global resource allocation and modest gains in terms of disease prevention underscore the urgent necessity of adjunctive strategies to augment ART. A pharmacologic approach to eliminate the latent reservoir with latency reversing agents may represent a scalable strategy with the potential to turn the tide of the AIDS epidemic[30, 31]. A small number of LRAs have now reached pilot clinical trials. The rationale, design, execution and results of these studies are reviewed here, with particular attention to the different means by which outcomes are measured, unique aspects of the trials themselves, and where the field is heading in light of the results of these pioneering studies.
Measuring HIV-1 Persistence
Pilot eradication trials have employed differing means of measuring latency reversal and reservoir perturbation in vivo[32–34]. These outcome measures constitute a spectrum connecting the molecular mechanisms of viral latency to its clinical phenotype (Table 1). Based on the consensus definition of HIV-1 latency as a state of transcriptionally silent but potentially inducible genomic integration, the most proximal measure of proviral reactivation is quantification of intracellular unspliced HIV-1 RNA via rtPCR. Unlike single or multiple spliced viral RNA species that may be present at low frequency without bona fide viral reactivation[35, 36], unspliced viral RNA transcripts are most likely to represent genomic RNA to be packaged into a nascent virion and are therefore a necessary precursor for virion production.
Table 1.
Measuring Latency Reversal in vivo
| Outcome measurement | Utility | Drawbacks | References |
|---|---|---|---|
| cell-associated unspliced viral RNA (caRNA) | Detection of intracellular unspliced viral RNA species correlate with active viral transcription | LRAs can activate cellular genes upstream from latent proviruses, inducing ‘read-through’ transcripts | [35] |
| cell-free plasma viremia (“single copy assay” or SCA) | Highly sensitive rtPCR can quantify small changes in low level viremia down to one HIV-1 RNA copy / mL | Threshold between stochastic fluctuations and true viral reactivation from reservoir is not defined. SCA is not suitable for high-throughput use | [39] |
| cell-associated proviral DNA qPCR | Allows for direct quantification of HIV-1 DNA species from patient cells that can be used as a measure of reservoir size | Total proviral DNA identifies unintegrated species unlikely to contribute to persistence; integrated DNA PCR (alu-PCR) identifies defective integrants. Both over-estimate reservoir size | [34, 104, 105] |
| Viral outgrowth from resting CD4+ T cells (quantitative viral outgrowth assay or QVOA) | The gold standard for determining reservoir size quantifies replication-competent proviruses via limiting dilution co-cultures | QVOA underestimates reservoir size. It isaA time-consuming assay not suitable for high-throughput use. A new assay (‘TILDA’)was recently described though yet to be validated against existing assays | [37] |
| Time to viral rebound (analytical treatment interruption or ATI) | Directly quantifies time of aviremic remission after LRA treatment and provides unambiguous measurement of primary objective | ATI is potentially risky to participants. Risk must be balanced against likelihood of positive outcome. No current LRAs appear capable of perturbing reservoir | [57, 58] |
An increase in unspliced HIV-1 RNA transcripts detected by rtPCR, often reported as a fold change over pre-intervention baseline, is a frequently employed measure for viral reactivation from latency in vivo. This measurement is not without pitfalls, however. Many latency reversing agents, including the HDAC inhibitors, act upon host promoters upstream from transcriptionally silent proviruses, inducing transcription of cellular genes and in turn producing ‘read-through’ viral transcripts (Figure 2) [35, 37]. rtPCR assays for unspliced intracellular viral RNA may be unable to distinguish these read-through transcripts, representing LRA-induced activation of cellular genes, from LRA-induced reactivation of viral transcription initiated at the authentic viral Cap site. Detection of read-through transcripts decreases the specificity of this assay. For this reason, fold-change increases in unspliced intracellular HIV-1 RNA detected in vivo in the absence of other evidence for reactivation fail to rule out the possibility of an off-target LRA effect and may not represent true latency reversal.
Figure 2. Induction of proviral transcription in latently infected resting CD4+ T cells.
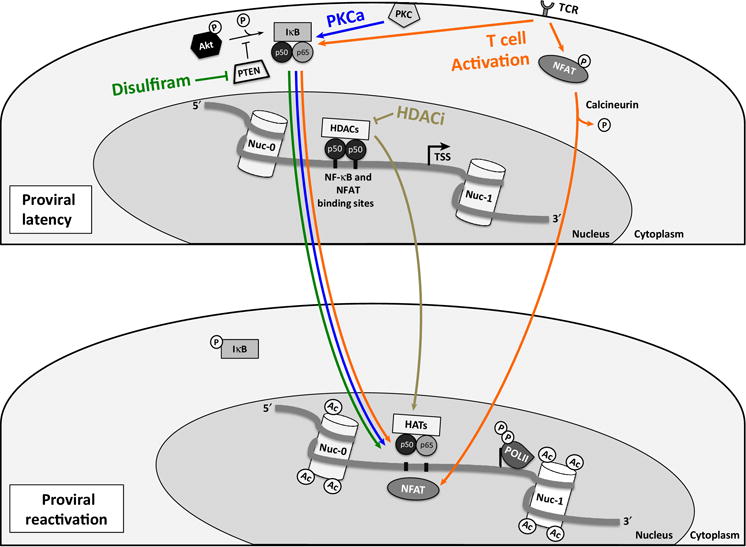
In the latent state, the 5′LTR NF-κB and NFAT binding sites are occupied by inactive p50 homodimers, while the active p50/p65 NF-κB isoform is bound to IκB and sequestered in the cytoplasm. NFAT is phosphorylated and also remains in the cytoplasm. Histone deacetylases are recruited to the 5′LTR and repress transcriptional activity. Additional mechanisms of latency that are not pharmacologic targets in pilot clinical trials have been omitted for clarity. Disulfiram (green arrows) blocks the activity of the Akt-inhibitory protein PTEN. Akt signaling pathway activation results in release of the active positive transcription elongation factor b (p-TEFb, not shown) as well as phosphorylation of IKK and subsequent degradation of IκB, which allows the p50/p65 NF-κB isoform to enter the nucleus, bind to the 5′LTR and induce transcription. Cytoplasmic PKC isoforms are activated by PKC agonists and also target IκB for degradation (blue arrows), which leads to active NF-κB entry into the nucleus. Stimulation of the T cell receptor (TCR) leads to cellular activation (orange arrows), which results in translocation of both transcription factors NF-κB and NFAT into the nucleus. Histone deacetylase inhibitors (HDACi, brown arrows) block the activity of histone deacetylases and lead to acetylation of Nuc-0 and Nuc-1. Active NF-κB bound to the 5′LTR leads to the recruitment of histone acetyltransferases (HATs).
Figure adapted from Xing S. et al Drug Discov. Today 2013 [ref 29] and Van Lint C. et al Retrovirology 2013 [ref 24]
All patients who achieve viral suppression on ART remain viremic at levels that are below the limit of currently available commercial viral load assays (20-50 copies of HIV-1 RNA/mL). The source of this low-level viremia, which typically fluctuates around a median frequency of one copy/mL[38, 39], is thought to be spontaneous reactivation of viral production from the latent reservoir. An increase in low-level viremia temporally related to the administration of a latency reversing agent is a reasonable surrogate for viral reactivation. A highly sensitive rtPCR assay, known as the single copy assay, has been used frequently in ART intensification and eradication studies[39].
Several unanswered questions are raised by the use of the single copy assay. Low level viremia fluctuates in a stochastic fashion over time in patients on ART[38, 40], and highly sensitive PCR assays to detect minute changes in viremia have the capacity to amplify these fluctuations. Similar to measurements of cell-associated viral RNA discussed above, pre-study power calculations prior to intervention and careful interpretation of results afterwards are required to reliably segregate stochastic changes in baseline low level viremia from de novo virion release from the reactivated latent reservoir. Many studies employ both a commercial rtPCR assay as well as the single copy assay as a means of balancing sensitivity and specificity when assaying for increases in viremia.
A detectable burst of viremia in the setting of LRA administration represents a reasonable surrogate outcome for anti-latency activity. However, it remains possible that modest transcriptional activation may occur without leading to detectable changes in viremia. Latently infected resting T cells induced to initiate proviral transcription in vivo without undergoing cellular activation may not be able to efficiently produce and release complete, infectious virions. It is likely that abundant defective proviral species will be able to reactivate and drive expression of a subset of viral genes (those not affected by mutations) without being able to produce infectious particles[41, 42]. Importantly, release of infectious virions may not be a necessary event for these cells to be eliminated through viral cytopathic effects or immune mechanisms. An LRA that could induce viral transcription such that viral peptides are presented to effector cells could conceivably result in targeting and depletion of infected cells without virion production or a detectable change in viremia.
Measuring change in reservoir size is a short-term, proximate measure for HIV-1 eradication. While the time between ART cessation and viral rebound is variable between patients because activation of latently infected T cells is a stochastic process[43, 44], reservoir size has been shown to correlate inversely with time to viral rebound[40, 45–50]. A recent comparative analysis of in vivo reservoir measurement modalities identified discordance between PCR-based and viral outgrowth detection methods[34]. PCR-based methods are likely to provide an over-estimation of reservoir size as most integrated proviruses are defective due to mutations or genetic deletions[41, 42]. These proviruses, though likely to be identified by sensitive PCR techniques, are unlikely to contribute to viral rebound and may not need to be a target of eradication strategies. Alternatively, virus outgrowth techniques, of which the quantitative viral outgrowth assay (QVOA) represents the gold standard, have recently been shown to underestimate the frequency of replication competent proviruses[41]. Therefore, the measurements of viral reservoir size are intimately linked to the methods used for its assessment, and this represents a critical area for method development. To that end, transcriptome and proviral analysis on single cells, performed at high-throughput, may represent the next generation of technologies propelling progress in the field[51].
The consequences of the sensitivity limits of reservoir size estimation became evident in three well-publicized cases of what initially appeared to be functional cures. Two HIV-1 positive patients who underwent allogeneic bone marrow transplant to treat lymphoma and subsequently experienced graft-versus-host disease[52], and an infant who was infected at delivery and started immediately on ART[53] experienced prolonged periods without rebound viremia after ART cessation (range: 2.8 – 27.6 months). During these aviremic intervals, virus outgrowth from the latent reservoir could not be detected in any of these patients, raising the possibility of reservoir eradication (or lack of reservoir formation in the case of the early-ART-treated infant). However rebound viremia ultimately developed in all three cases, indicating that HIV-1 persisted at a level sufficient to re-kindle productive infection but below the limit of detection of gold standard reservoir assays[54, 55]. These cases were a sobering reminder of the inherent challenges of reservoir eradication.
The ultimate goal of pharmacologic HIV-1 eradication strategies is to induce a prolonged remission from viral replication in the absence of antiretroviral treatment. Therefore controlled cessation of ART paired with close monitoring for development of viremia represents a clinical outcome measure closely tied to the primary clinical objective with highly desirable performance characteristics: development of viremia after treatment interruption within the time frame delineated by multiple, well-powered treatment interruption studies, is indicative of an unsuccessful anti-latency therapy. In contrast, a prolonged aviremic period off ART and after administration of an LRA beyond the expected time to viral rebound is highly predictive of reservoir perturbation.
The concerns raised by including analytical treatment interruption (ATI) in eradication trials arise not from interpretation of the results, but rather from the potential to cause harm to participants who stop and have to re-start ART. The Strategies for Management of Antiretroviral Therapy (SMART) study, a clinical trial in which participants were randomized to continuous or intermittent ART[56], identified a significantly higher risk of opportunistic infections or death for those in the intermittent therapy arm, and has served as the basis for current Department of Health and Human Services guideline recommendations to maintain continuous and indefinite ART for all HIV-1 infected individuals. The increase in mortality with intermittent ART is likely due to highly pro-inflammatory effects of ongoing viral replication[56]. The decision to include ATI in an eradication trial therefore must take into account the likelihood of successful reservoir depletion by an LRA and balance this with a trial framework that is able to identify viral rebound as early as possible, and swiftly re-initiate ART in order to minimize participant exposure to the pro-inflammatory effects of ongoing viral replication[57, 58].
First, Do No Harm: clinical trial outcomes
T cell activation
Activated CD4+ T cells are responsible for the vast majority of HIV-1 detectable in the plasma of untreated, viremic patients and are thought to have a life-span of one to two days in vivo [59, 60], while latently infected resting memory CD4+ T cells persist for years[11, 12] and harbor proviruses that are transcriptionally silent until these cells become activated. The first clinical trials to directly target the latent reservoir were based on the recognition that the activation state of an HIV-1 infected T cell correlates directly with both production of infectious virions and inversely with cellular life span[60, 61]. These trials hypothesized that activating resting T cells in vivo would lead to virion production by latently infected cells, which in turn would lead to rapid cell turnover and reservoir depletion. Participants were maintained on ART during the intervention in order to prevent de novo infection from occurring in bystander T cells. Four trials administering cytokines (IL-2 with or without IFNγ) to patients on stable ART demonstrated no change in viral reservoirs or time to viral rebound when therapy was stopped[14, 62–64]. Two trials directly and more aggressively targeted T cell activation by employing a combination of murine antibodies against human CD3 (OKT3) followed by IL-2[65, 66].
The first OKT3 trial, by Prins et al.[66], employed multiple outcome measures including quantitation of plasma HIV-1 RNA and proviral DNA in PBMCs, viral outgrowth in CD4+ T cell cultures ex vivo and direct HIV-1 RNA in situ hybridization on lymph node biopsies, obtained before and after the intervention. 5mg of OKT3 was infused daily on days one through five, and 4.5 million IU of recombinant human IL-2 was infused twice daily on days two through six. The trial was designed to have two sequential infusion courses two weeks apart. However, two of the three participants opted out of the second course due to adverse effects. Participants experienced unremitting fever, headaches, nausea, vomiting and diarrhea throughout the six day infusion period. They also developed anemia and lymphopenia, which resolved after the infusions ceased. One participant suffered hemodialysis-dependent acute kidney injury due to acute tubular necrosis as well as seizures requiring administration of anti-epileptic agents. Magnetic resonance imaging of the brain demonstrated white matter changes consistent with published reports of IL-2-induced neurotoxicity[67].
While all participants demonstrated evidence of T cell activation and development of antibodies against murine OKT3, perturbation of the latent reservoir was not observed. One participant experienced a transient increase in plasma viremia to 1,500 RNA copies/mL on day 5 of infusion [67]. Of note, this participant had a low-level detectable viral load (110 RNA copies/mL) just prior to the start of the intervention. The other two participants had undetectable viral loads at enrollment and remained undetectable with respect to plasma HIV-1 RNA throughout the study. In two subjects, a mild increase in HIV-1 RNA (two- to three-fold) was observed in lymph node specimens. Treatment-induced lymphopenia obscured the detection of proviral DNA levels in PMBC and virus outgrowth in CD4+ T cell cultures during the treatment phase, and no significant changes were observed afterward compared to baseline levels (Table 2).
Table 2.
Pilot HIV-1 Eradication Trials
| LRA class | LRA tested | n (# male) | Plasma viremia | Viral outgrowth | Viral RNA /DNA in tissue | ATI | Adverse Effects | Results | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Direct TCR stimulation | OKT3 + rhIL2 | 3 (NR) | rtPCR | rCD4 culture | LN bx caDNA from PBMCs | not performed | fever, HA, N/V/D, anemia, Dialysis-dependent AKI, Sz, hypothyroidism, lymphocytopenia | 1/3 pts: transient viremia to 1500 copies/mL (day 5) 0/3 pts: no change in DNA copies/106 rCD4 2 pts: viral RNA in LN increased (in situ hybridization) |
[66] |
| Direct TCR stimulation | OKT3 + rhIL2 | 3 (3) | rtPCR | rCD4 co-culture | Tonsil bx 2LTR circles in PBMC rtPCR seminal fluid | all subjects (n=3) | fever, chills, myalgias, stiff neck, headache (?aseptic meningitis), lymphopenia, hyponatremia (1 pt), neutropenia (1 pt) | 0/3 pts show viral load above 50 copies/mL 0/1 pt: 2LTR circles show no change 0/3 pts: tonsil bx completely negative for HIV-1 RNA 3/3 pts experience viral rebound w/in 6wks of STI |
[65] |
| HDAC inhibitor | valproic acid | 4 (NR) | Commercial Roche rtPCR and SCA | QVOA | rCD4 integrated gag DNA rtPCR seminal fluid (2/4 pts) | not performed | T20 injection site rxns (4/4 pts) Anemia (attributed to AZT + valproate; 1/4 pts) |
0/4 pts: SCA at or near 1 copy/mL (9 copies = max; 1 pt) 0/2 pts: no change in seminal RNA 0/4 pts: no decline in integrated DNA 3/4 pts: decline in IUBP >50% after 18 wks of rx |
[71] |
| HDAC inhibitor | valproic acid | 9 (6) | not performed | QVOA | not performed | not performed | None reported (cross sectional and longitudinal study) |
No change in IUPM noted in patients taking valproate + ART over six month period or compared to patients taking ART alone (historical cohort from previously published studies) | [74] |
| HDAC inhibitor | valproic acid | 11 (7) | not performed | limiting dilution rCD4 culture | PBMC DNA limiting-dilution Alu-PCR CD4 | 3/11 pts | None reported (observational cohort study) |
No difference in viral DNA in PBMCs between groups No difference in integrated viral DNA in CD4 cells No difference in viral outgrowth from rCD4 cells Viral rebound w/in 8 weeks of STI in three patients |
[73] |
| HDAC inhibitor | valproic acid | 11 (11) | SCA | QVOA | Seminal RNA | not performed | Minor, self-limited in 4 pts (not further specified) | 4/11 pts: >50% decrease in IUPB post-treatment No changes in SCA correlating with treatment 0/5 pts: seminal fluid RNA <400 copies/mL |
[76] |
| HDAC inhibitor | valproic acid | 11 (11) | SCA | QVOA | not performed | not performed | None reported | No changes in IUPB No changes in SCA |
[75] |
| HDAC inhibitor | valproic acid | 56 (47) | not performed | limiting dilution rCD4 culture | not performed | not performed | 5 pts: mood changes, GI side effects 1 pt: pulmonary embolism |
No changes in IUPB at any time point in either arm No viral load blips via commercial assay |
[77] |
| HDAC inhibitor | vorinostat (SAHA) | 8 (NR) | SCA | not performed | gag caRNA rtPCR in rCD4 | not performed | No adverse events > grade I; none attributable to SAHA | 0/8 pts: No changes in SCA (results not shown) 8/8 pts: increase in HIV-1 gag caRNA compared to baseline; mean 4.8 fold (1.5-10 fold) |
[83] |
| PTEN inhibitor | disulfiram | 15 (14) | SCA | QVOA | not performed | not performed | No adverse events > grade II; none attributable to disulfiram | 0/15 pts: No change in reservoir size by QVOA 0/15 pts: No change in SCA during dosing interval 4/15 pts: VL increase 2 hours post-first dose |
[81] |
| HDAC inhibitor | vorinostat (SAHA) | 5 (5) | SCA | QVOA | caRNA and DNA in CD4 at baseline, doses 11 and 22 | not performed | GI symptoms and headache <grade I 15-35% decline in platelet counts in all pts Close CNS monitoring showed no adverse events |
gag caRNA shows no increase after doses 11 or 22 for any pt RNA and DNA from PBMCs shows no changes No IUPB changes or changes in SCA |
[82] |
| HDAC inhibitor | vorinostat (SAHA) | 20 (19) | SCA and commercial assay | TILDA | unspliced caRNA in circulating and rectal CD4 proviral DNA in CD4 and rectal tissue | not performed | 40% of pts: diarrhea, lethargy, thrombocytopenia 20% of pts: dysgeusia, nausea / vomiting, headache, impaired concentration 10% of pts: abnormal LFTs |
caRNA in CD4 cells increases over 84 days during and after treatment (peak = 7.4 fold; IQR 3.4-9.1) No changes in plasma RNA, caDNA or inducible virus |
[84] |
| HDAC inhibitor | panobinostat | 15 (15) | TMA (qualitative) | QVOA | caRNA rtPCR in total CD4 2LTR, integrated and total proviral DNA in total CD4 | 9/15 pts | 7/15 pts: fatigue 2/15 pts: diarrhea 1/15 pts: rash, palpitations, stomachache, nausea, vomiting, sleeplessness |
unspliced caRNA increased 3.5 fold (2.1-14.4 fold) during pano (data analysed for whole cohort) No changes to integrated proviral DNA or IUPM 9/9 pts: viral rebound post-ATI at 17 days (14-56) |
[40] |
| PTEN inhibitor | disulfiram | 30 (NR) | SCA | not performed | unspliced caRNA in CD4 HIV DNA | not performed | 500mg arm: 3/10 pts hypophosphatemia 1000mg arm: 2/10 headache, lethargy, hypophosphatemia; 3/10 dysgeusia, nausea 2000mg arm: 4/10 dysgeusia, 3/10 headache, lightheadedness; 2/10 lethargy, nausea, hypophosphatemia |
caRNA in CD4 cells increases 1.5-2 fold in each arm SCA shows 2 fold increase in 2000mg arm after 3rd dose |
CROI 2015 abstract 428LB |
| HDAC inhibitor | romidepsin | 6 (5) | SCA and commercial assay | TILDA | unspliced caRNA in CD4 | not performed | 35 grade I events | caRNA induced in all pts 5/6 pts: viremia from undetectable to detectable No decrease in proviral DNA |
[89] |
These modest results paired with the severe adverse effects of the intervention helped inform the design of a second pilot trial attempting to validate the anti-CD3 / IL-2 strategy[65]. Kulkosky et al. administered OKT3 and IL2 to three subjects with notable protocol differences from Prins et al.[66] Initially, participants underwent ‘intensification’ of their ART regimens with didanosine and hydroxyurea prior to the study intervention to ensure maximal viral suppression. The dosing and frequency of OKT3 were significantly reduced: rather than 5mg daily for five days, Kulkosky et al. administered a 0.4mg intravenous dose once. This was followed by administration of IL-2 (1.2 million IU/m2/day) for a fifteen day period. Outcome measures were also different: for the first time, an analytical treatment interruption was included for participants in whom ex vivo viral outgrowth was below detection limits after the intervention. Plasma and seminal fluid were obtained for HIV-1 RNA quantification by rtPCR, CD8+-depleted PBMC were obtained for co-cultures assaying viral outgrowth, and tonsil biopsies were obtained for HIV-1 RNA in situ hybridization.
Despite the lower dose and single exposure to OKT3, all three participants developed symptoms compatible with aseptic meningitis (fevers, chills, myalgias, headache, stiff neck). Headaches persisted for up to a week after a single infusion. Participants also developed transient lymphopenia after OKT3 (7-14 days in duration). Plasma HIV-1 RNA remained below 50 copies/mL in all subjects during the intervention. Tonsil biopsies demonstrated no evidence of viral RNA in any specimen. All three subjects agreed to participate in treatment interruption, and all developed detectable viremia over a three to six week period and were re-started on ART with subsequent viral suppression.
The high toxicity and lack of efficacy observed in the OKT3 trials have informed all subsequent efforts at HIV-1 eradication using latency reversing agents. While it is widely acknowledged today that T cell activation via TCR engagement is an untenable eradication strategy based largely on the results of these trials, it is worth noting that these early studies built upon evidence compiled from contemporaneous pilot trials in which OKT3 and IL-2 were used for solid organ transplant and renal cell carcinoma[67, 68]. At the time of the design of these early HIV-1 eradication trials in the late 1990s, combination ART was itself a relatively novel approach to HIV-1 management that carried significant adverse effects and whose long-term efficacy was not yet assured. The availability of newer-generation ART combinations and a deeper understanding of the durability of viral suppression they provide have decreased the tolerance for potential adverse events in pilot HIV-1 eradication trials.
Valproic Acid
Studies of the molecular mechanism of viral latency have revealed the crucial role of epigenetic modifications by cellular enzymes[69]. Chromatin remodeling, and in particular, histone de-acetylation by cellular histone deacetylases (HDAC), contributes to silencing of proviral gene expression[70], while inhibition of HDAC enzymes leads to increases in cell-associated viral RNA in both latency models in vitro and aviremic patient cells ex vivo[23, 28]. Preclinical evidence demonstrating that HDAC inhibitors can induce reactivation of latent proviruses without global T cell activation made these compounds attractive alternatives to OKT3 and IL-2. This drug class represents the most widely tested latency reversing agent in vivo, with four unique HDAC inhibitors having been in pilot trials to date.
The first HDAC inhibitor to reach clinical trials was valproic acid[71]. Valproate (US brand name: Depakote) is an FDA-approved drug used to treat epilepsy and bipolar disorder. Lehrman et al. enrolled four ART-treated subjects whose regimens were intensified with the fusion inhibitor enfuvirtide (also known as T20) for four to six weeks prior to twice-daily oral administration of 500-750mg valproate [71]. Participants were treated with valproate for three months, and drug dosing was individualized based on a goal plasma valproate concentration of 50-100mg/L. Outcome measures included both commercial and single copy rtPCR to quantify viremia, rtPCR on seminal fluid, quantitative viral outgrowth and integrated proviral gag DNA from circulating resting CD4+ T cells.
The trial interventions were well-tolerated by participants. Enfuvirtide, administered subcutaneously, caused injection site reactions, and in one participant the combination of zidovudine (part of the baseline ART regimen) and valproate led to transient anemia. No changes in T cell activation profiles were observed. No significant changes occurred in low-level viremia or seminal fluid viral RNA. The frequency of resting CD4+ T cells harboring proviruses measured by viral outgrowth decreased after the 18 week intervention compared to baseline in all participants (range: 29% to 84% decrease). The investigators concluded that HDAC inhibition appeared to lead to partial reservoir depletion in these participants. The contribution of ART intensification to this outcome was unclear and became the subject of a subsequent study by the same group (discussed below).
The Lehrman et al. study was the first published report of a targeted reduction in the HIV-1 reservoir in vivo, and generated both excitement and passionate debate in the nascent field of HIV-1 persistence research[72]. Two independent groups conducted observational studies to determine whether viral reservoirs were diminished or absent in patients who were prescribed ART as well as long-term valproate for clinical indications[73, 74]. The first of these studies, by Siliciano et al., employed a cross-sectional design in recruiting nine ART-treated individuals who had been taking valproate for a minimum of six months (range 6-38 months). These subjects underwent phlebotomy at two time points (zero and six months) for quantification of viral outgrowth, which allowed investigators to model reservoir decay kinetics on valproate. The change in the frequency of latently infected cells in valproate-treated patients was also compared to the reservoir decay rate observed in a previously studied cohort of aviremic subjects on ART (n=59) who were not treated with an inducing agent. These investigators did not observe any difference in the frequency of latently infected cells among patients treated with ART and valproate over time, or when this group was compared to patients on ART alone[74].
Separate work by Sagot-Lerolle et al.[73], utilized an observational study design in comparing reservoir size in 11 ART-treated patients who were prescribed valproate for a median of 10 years (range 2-14 years) to 13 matched controls. Limiting-dilution viral outgrowth assays using resting CD4+ T cells were performed along with an integrated proviral DNA PCR assay. Three of the 11 valproate-treated subjects were concomitantly enrolled in ongoing structured treatment interruption trials, allowing investigators to determine whether valproate administration influenced the kinetics of viral rebound in the absence of ART. No difference in proviral DNA from peripheral blood mononuclear cells (PBMC) or resting CD4+ T cells was observed, and no change in the frequency of latently infected cells quantified by viral outgrowth was identified between the two groups. All three participants who underwent ART interruption experienced detectable viremia within eight weeks, which did not differ significantly from the median time to rebound observed in these trials.
Two prospective valproate clinical trials were subsequently conducted by the Margolis group in order to determine the role played by ART intensification in the setting of valproate administration[75, 76]. In the first[76], 11 subjects were recruited and valproate 1000mg (Depakote ER) was administered daily for 16 weeks without intensifying antiretroviral agents added to baseline ART regimens. Viral outgrowth was performed twice prior to and twice after the intervention. When the pooled values of each pair of reservoir measurements was compared (ART alone versus ART + valproate), only four of 11 participants experienced a decline in frequency of latently infected cells after the intervention. Single copy assay did not demonstrate any change in low-level viremia throughout the trial. The second trial[75] included three study arms: (1) 16 weeks of valproate with baseline ART, (2) enfuvirtide intensification and valproate or (3) addition of the integrase inhibitor raltegravir to baseline ART and valproate. Three participants in this 11 subject trial had participated in their previous valproate trial and received extended valproate dosing. Decreases in reservoir size observed in these subjects during the initial trial were not sustained when they were evaluated 48-96 weeks from the initial intervention. In the remaining subjects, ART intensification with enfuvirtide or raltegravir administered alongside valproate failed to have an effect on reservoir size.
Perhaps the final word on valproate came from a trial conducted by Routy et al. in which 56 patients on stable ART received 16 or 32 weeks of valproate (500mg twice daily)[77]. No changes in latently infected cell frequency were observed relative to baseline values or between the two arms. The combined results of these prospective and observational studies provide ample evidence that valproate alone was not sufficient to perturb the latent reservoir[78]. These trials did however provide an important framework for the design of future pilot eradication studies and offered a first-hand opportunity to consider the pros and cons of outcome measures that should be employed, as more efficacious HDAC inhibitors were identified.
Disulfiram
The potential for latency reversal induced by disulfiram was identified using a primary cell model of latency[79]. Disulfiram was FDA-approved for use as a deterrent to alcohol ingestion in the 1950s and therefore has decades of clinical safety data supporting its use for novel indications in vivo. The mechanism of action with regard to latency reversal appears to be through depletion of the intracellular protein PTEN, which in turn activates the Akt signaling pathway to initiate proviral transcription in an NFkB-dependent manner[80].
Two pilot clinical trials have tested the hypothesis that disulfiram can perturb the latent reservoir in vivo. Spivak et al. recruited 16 participants who underwent directly observed administration of 500mg disulfiram for 14 days after baseline measurement of latent reservoir size by QVOA[81]. Single copy assay PCR was performed every other day during the intervention and weekly afterward. A second reservoir measurement was performed 10 weeks after disulfiram was discontinued. Disulfiram concentrations were highly variable among participants, with a majority demonstrating no detectable plasma levels at any time during the administration period. Disulfiram was well tolerated without any adverse effects, however, no change in reservoir size was observed. While there was also no clear signal for an increase in low-level viremia, a minority of participants experienced viral blips within hours of disulfiram administration. It is unclear whether metabolism of disulfiram and initiation of viral transcription could lead to a systemic increase in viral load in the time frame in which these blips were observed. A second disulfiram trial has been completed, but not yet published, that may address these outstanding questions. The results to date suggest that disulfiram alone is unlikely to perturb the latent reservoir.
Vorinostat
The initial results and tolerability of valproate were followed closely by the description of ‘next generation’ HDAC inhibitors that have since reached clinical trials. Vorinostat, an FDA-approved chemotherapy agent also known as SAHA, has been tested in three separate trials[82–84]. The first of these[83] enrolled eight participants to undergo a single infusion of vorinostat. Careful measurement of histone acetylation confirmed the bioactivity of the drug in vivo, and rtPCR demonstrated a mean 4.8 fold increase in cell associated HIV-1 RNA six hours after vorinostat dosing (range 1.5-10 fold). No changes in low level viremia or reservoir size were observed. In a follow up study involving some of the same participants, Archin et al.[82] evaluated the effect of 22 doses of vorinostat on intracellular viral RNA levels. A positive signal observed after a single dose in the previous study[83] was not identified in this study[82]. Cell-associated viral RNA showed no changes after doses 11 or 22 in the five participants enrolled. No changes in low level viremia, proviral DNA or reservoir size were observed.
In a separate trial evaluating multiple doses of vorinostat, Elliott et al.[84] demonstrated similar results as the initial single-dose study[83]. 20 participants were administered 400mg vorinostat daily for two weeks. Adverse events were common in this study: 40% of participants experienced diarrhea, lethargy and thrombocytopenia, 20% developed nausea, vomiting, dysgeusia (alteration of taste sensation) and headache, and 10% had changes in liver function testing. No changes were observed in plasma viremia, cell-associated proviral DNA or reservoir size measured by a novel PCR based method, the tat / rev induced limiting dilution assay or TILDA [85]. Cell-associated viral RNA increased 7.4 fold (mean value, inter-quartile range 3.4 to 9.1 fold), and in many participants (9/20) the peak of intracellular RNA production was observed after the dosing interval. This trial made use of high-throughput sequencing techniques to evaluate the transcriptional changes induced during vorinostat administration and those changes that persisted afterward. Interestingly, upregulation of genes associated with protein ubiquitination and MHC Class I antigen presentation was identified ten weeks after vorinostat administration. The lack of change in viremia and reservoir size led these authors to conclude that vorinostat is unlikely to purge the latent reservoir alone.
Panobinostat and Romidepsin
Panobinostat is an FDA-approved chemotherapy agent used in the treatment of multiple myeloma[86, 87]. A pilot eradication clinical trial in Denmark by Rasmussen et al. enrolled 15 patients to receive 20mg of panobinostat three times per week during weeks 1, 3, 5 and 7 of an eight week study[40]. This intermittent dosing schedule allowed investigators to evaluate pharmacodynamics with regard to histone acetylation and cell-associated unspliced viral RNA. Adverse effects were minor, consisting mostly of fatigue (7/15 participants) and diarrhea (2/15). Histone acetylation increased during each dosing interval and returned to baseline during the washout periods, while cell-associated viral RNA increased rapidly after the first dose (mean 2.4 fold increase, range 1.8-3.3) and appeared to increase during each of the dosing intervals. Contemporaneous plasma viremia was measured using a qualitative transcription mediated amplification assay, and the investigators observed an increase in the percentage of participants with assays positive for plasma HIV-1 RNA during panobinostat administration when compared to baseline (54% versus 30%). Total and integrated HIV-1 DNA in CD4+ T cells showed no change during the trial, and QVOA showed no change in the frequency of latently infected cells after panobinostat administration compared to baseline. Despite the lack of significant changes in reservoir size, nine participants elected to undergo ART interruption after panobinostat administration. All participants became viremic with a median time to viral rebound of 17 days (range 14-56 days), which is within the range of viral rebound observed in multiple treatment interruption trials.
Romidepsin is an HDAC inhibitor FDA-approved for the treatment of T cell lymphoma that has shown to be more potent in reversing latency in vitro than vorinostat or panobinostat[88]. However, in a recent trial by Søgaard et al.[89], romidepsin was administered to six patients at a dose of 5mg/m2 intravenously once a week for three weeks. 34 grade I adverse events and 2 grade II events (fatigue and fever) were described. Cell-associated RNA was induced in all participants during the treatment period, and in five of six participants low level viremia became detectable during the trial period. No decrease in proviral DNA was observed. Further in vivo studies would be required to finalize the verdict on Romidepsin.
Future directions
Evidence of low-level viral transcription paired with lack of reservoir perturbation (measured by viral outgrowth, proviral PCR quantitation or viral rebound after ART discontinuation) are all common findings to these early HIV-1 eradication trials. What accounts for the modest in vivo efficacy of LRAs tested to date? A variety of primary T cell latency models, in which uninfected healthy donor T cells are infected in vitro with HIV-1 and progress to a latently infected state, have been developed independently and used to identify potential LRAs. These results in turn have informed the design of various pilot HIV-1 eradication trials described here. A recent comparative analysis of LRA activity across these published in vitro latency models identified significant inter-model discordance[90]. The results of this study indicate that no current in vitro latency model faithfully recapitulates the nature of the HIV-1 latent reservoir. Pre-clinical research on LRA efficacy and toxicity gathered from non-human primate SIV models[91, 92], humanized mouse models[93–96] and purified resting CD4+ T cells obtained from HIV-1 infected, aviremic patients[97, 98] are current strategies addressing this significant knowledge gap (see Boxes 1, 2).
Box 1. The Magic Bullet: characteristics of an ideal anti-latency therapy.
Administered over a finite duration, ideally as a single dose
Well tolerated, with a minimum of adverse effects and drug-drug interactions
Administration should involve minimal complexity
Inexpensive and available worldwide, particularly in areas with the highest HIV-1 prevalence
Active against all clades or sub-types of HIV-1
Lab measurements of clinical efficacy should have excellent performance characteristics, yet be simple enough to implement within existing healthcare infrastructures in both resource-rich and -poor settings
Box 2. Gender Discrepancies in HIV-1 eradication research.
The enrollment demographics of pilot eradication studies reveal a notable gender discrepancy. Women comprised less than 13% of participants in HIV-1 eradication trials (based on trials that included gender among participant demographics). The HIV-1 epidemic in the United States and Europe is skewed toward men: the Centers for Disease Control reports that women comprised 23% of total HIV-1 prevalence in the United States in 2011. However this does not hold true for the worldwide epidemic. World Health Organization HIV-1 statistics identify 17.4 million women living with HIV-1 in 2014, representing 47% of the 36.9 million people estimated to be living with the virus. Further research is required to determine whether gender is a biological variable in regard to the establishment or maintenance of HIV-1 latency. Though not reported, clade B viruses are likely to have been over-represented in these pilot studies as well based on the geographic locations of these trials. The external validity of these assumptions (gender is not a biologic variable; viral persistence is unaffected by viral clade) needs to be addressed when considering eradication strategies to be implemented worldwide.
Pharmacologic strategies for HIV-1 eradication have sought to minimize T cell activation largely in response to the unacceptable toxicity of the OKT3 and cytokine administration trials[65, 66]. HDAC inhibitors have been an attractive choice for pilot eradication trials as they offer an acceptable balance between proviral transcriptional activation and cellular activation. However, the modest outcomes of these clinical trials have led to a re-evaluation of these agents as lead compounds. In two separate ex vivo analyses, LRAs from the protein kinase C family, bryostatin-1[35] and ingenol 3,20 dibenzoate[98], demonstrated significantly higher latency reversal potential than HDAC inhibitors tested. Bryostatin-1 also demonstrated synergy with HDAC inhibitors when used together ex vivo[97]. The accumulation of recent ex vivo results are helping to make a case for PKC agonists, potentially including bryostatin-1 or ingenol derivatives, as candidates for pilot eradication trials. Combination LRA trials making use of multiple anti-latency mechanisms, particularly HDAC inhibition and PKC activation, should also be a priority. Several groups have begun to explore the potential for synergy when LRAs from unique mechanistic classes are combined[97, 99, 100]. In these in vitro studies, PKC agonists appear to hold promise when combined with other agents. Further research is required to understand the potential for off-target effects of these combinations, including the induction of inflammatory cytokine secretion.
PKC agonists act upon T cell activation pathways and up-regulate cell surface markers including CD69. The effect of T cell activation induced by administration of PKC agonists in vivo cannot be predicted from these experiments, though Laird et al. demonstrated that despite upregulation of activation markers, these cells did not produce significant levels of inflammatory cytokines[97]. Of note, in the ex vivo analysis that preceded the clinical trial of romidepsin, CD69 expression increased significantly in resting CD4+ T cells exposed to clinical concentrations of this drug, a finding unique to romidepsin among HDAC inhibitors[88]. Perhaps the efficacy observed with romidepsin in vitro and in vivo reflects an adjunctive mechanism beyond HDAC inhibition that involves T cell activation.
Ongoing trials listed at clinicaltrials.gov at the time of this review include several studies revisiting the use of cytokines as well as toll-like receptor agonists to perturb the reservoir, a second romidepsin trial and the first in vivo study of bryostatin-1 as an LRA (Table 3). Novel gene therapy techniques including the use of CRISPR/Cas9 technology are showing great potential[101], and will hopefully reach clinical trials in the near future. A unique cure strategy has been recently proposed by Mousseau et al.[102, 103] making use of an inhibitor of the HIV-1 accessory protein Tat to suppress proviral transcription in latently infected cells. The mechanistic diversity of these approaches holds promise for the future of HIV-1 eradication research.
Table 3.
ongoing and upcoming eradication studies on clinicaltrials.gov
| Identifier | Title of Study | Status |
|---|---|---|
| NCT02092116 | Safety and Efficacy of the Histone Deacetylase Inhibitor Romidepsin and the Therapeutic Vaccine Vacc-4x for Reduction of the Latent HIV-1 Reservoir (REDUC) | ongoing, not recruiting |
| NCT02269605 | Bryostatin-1 Effect on HIV-1 Latency and Reservoir in HIV-1 Infected Patients Receiving Antiretroviral Treatment (BRYOLAT) | ongoing, not recruiting |
| NCT01935089 | Pilot Peg-Interferon-a2b in Decreasing Viral DNA in HIV | ongoing, not recruiting |
| NCT01069809 | Safety and Efficacy Study of AGS-004 During Analytical Treatment Interruption | ongoing, not recruiting |
| NCT02443935 | Toll-like Receptor 9 Agonist Treatment in Chronic HIV-1 Infection (TEACH) | currently recruiting |
| NCT02475915 | Efficacy of VHM After Treatment Interruption in Subjects Initiating ART During Acute HIV Infection | currently recruiting |
| NCT01295515 | Interferon Alpha 2b Intensification in HIV-Positive Individuals on Antiretroviral Therapy | currently recruiting |
| NCT02071095 | Enhancement by Poly-ICLC During HIV-1 Infection | currently recruiting |
| NCT02227277 | Reducing Proviral HIV DNA With Interferon-a (BEAT-HIV) | currently recruiting |
| NCT02486510 | Effect of Cytoreductive Chemotherapy and a CCR5 Coreceptor Antagonist on HIV-1 Eradication (CHEMOMAR) | currently recruiting |
| NCT02471430 | Reducing the Residual Reservoir of HIV-1 Infected Cells in Patients Receiving Antiretroviral Therapy (ACTIVATE) | not yet open for recruitment |
| NCT02336074 | Research In Viral Eradication of HIV Reservoirs (RIVER) | not yet open for recruitment |
| NCT02440789 | Safety and Efficacy of Sirolimus for HIV Reservoir Reduction in Individuals on Suppressive ART | not yet open for recruitment |
| NCT02191098 | Proof of Principle Study of Pulse Dosing of IL-15 to Deplete the Reservoir in HIV Infected People (ALT-803) | not yet open for recruitment |
| NCT02475655 | Evaluating the Safety and Tolerability of Ruxolitinib in Antiretroviral-Treated HIV-Infected Adults | not yet open for recruitment |
Concluding Remarks
While pilot eradication trials have not achieved the ultimate goal of a durable ART-free virologic remission, a promising trajectory can be observed from early interventions characterized by high toxicity and lack of efficacy to current studies making use of well-tolerated short course therapies that appear to induce proviral transcription. These encouraging early signals offer potential therapeutic building blocks and can serve to highlight the larger unanswered questions in the field of HIV-1 eradication.
In the face of the challenges presented by viral latency, measureable and incremental progress has been made in these early eradication studies. Large questions remain with regard to the ideal agent or combination of agents necessary to perturb the reservoir, as well as to what are the most reliable means of quantifying latency reversal and reservoir depletion (see Outstanding Questions). The collaborative efforts of research groups around the globe and the support of funding agencies committing resources to this critical health problem allow for an optimistic outlook despite the complexity of HIV-1 persistence and the modest gains achieved to date. “It is not the end,” remarked Winston Churchill in his famous 1942 address, balancing cautious optimism with the recognition of a lengthy and costly struggle to come. “It is not even the beginning of the end. But it is, perhaps, the end of the beginning.”
Outstanding questions.
What are the most predictive measurements of reservoir perturbation in vivo? PCR-based methods to detect proviral transcription can be overly sensitive and lead to difficulty in distinguishing between stochastic fluctuations and minor LRA-induced reservoir perturbations. However, the current gold-standard for quantifying reservoir size, quantitative viral outgrowth, lacks the sensitivity to detect small amounts of dormant proviruses in vivo.
Is analytical treatment interruption (ATI) appropriate for HIV-1 eradication pilot trials? If not, what are acceptable surrogate measures of reservoir depletion? ATI represents the most direct measure of the likelihood of a functional cure. However, this exposes participants to the known risks of unchecked viral replication.
Are there adjunctive strategies to attenuate the potential toxicity of LRAs that induce T cell activation in vivo? Early HIV-1 eradication trials demonstrated that activating T cells in vivo led to significant adverse events. However the LRAs that consistently demonstrate the highest efficacy across HIV latency models lead to T cell activation.
Will adjunctive strategies be necessary for clearance of cells harboring proviruses reactivated by LRAs? None of the LRAs that have reached clinical trials have demonstrated perturbation of the latent reservoir. Reservoir depletion is likely to be a pre-requisite for functional cure using the ‘shock and kill’ pharmacologic approach.
Trends.
Antiretroviral therapy (ART) blocks ongoing viral replication but does not cure HIV-1 infection due to the presence of long-lived quiescent proviruses in a minority of resting CD4+ T cells (known as the latent reservoir).
ART administration significantly lowers the risk of viral transmission, and therefore serves as individual treatment and public health prevention. However, worldwide incident infections continue to outpace the number of people started and maintained on ART. Global resource limitations that contribute to this imbalance suggest that ART alone will not be enough to curb the HIV-1 epidemic.
Histone de-acetylase inhibitors (HDACi) are chemotherapeutic agents that can induce transcription of latent proviruses in resting CD4+ T cells. They are the most widely tested class of latency reversing agents (LRAs) at present. Four HDACi have entered pilot clinical trials, but have shown modest efficacy to date. None of the LRAs tested have altered the size of the latent reservoir in vivo.
The optimal LRA class or combination of classes remains unknown. Trials have employed a variety of outcome measures, and the ideal pilot HIV-1 eradication trial design has not been determined.
Acknowledgments
The authors gratefully acknowledge critical input from colleagues Laura Martins and Alberto Bosque in the preparation of this manuscript. This review was supported in part by ongoing funding from the National Center for Advancing Translational Sciences of the National Institutes of Health under award number 1KL2TR001065 (AMS). This work is dedicated to the memory of Dr. Frederick L. Brancati, an inspirational and incomparable mentor.
Glossary Box*
- Latency
Reversibly non-productive state of infection of individual cells
- Reservoir
Infected cell population that allows persistence of replication competent HIV-1 in patients on optimal ART on the order of years
- Sterilizing cure
Complete eradication of all replication-competent forms of the virus
- Functional cure
Permanent viral suppression in the absence of therapy to levels that prevent immunodeficiency and transmission
- Latency reversing agent (LRA)
Small compound capable of inducing de novo viral transcription in latently infected cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
all definitions (except LRA) are from Eisele and Siliciano, Immunity 2012[33]
References
- 1.Hammer SM, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. The New England journal of medicine. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. The New England journal of medicine. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 3.Egger M, et al. Prognosis of HIV-1-infected patients starting highly active antiretroviral therapy: a collaborative analysis of prospective studies. Lancet. 2002;360:119–129. doi: 10.1016/s0140-6736(02)09411-4. [DOI] [PubMed] [Google Scholar]
- 4.Mellors JW, et al. Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Annals of internal medicine. 1995;122:573–579. doi: 10.7326/0003-4819-122-8-199504150-00003. [DOI] [PubMed] [Google Scholar]
- 5.Mellors JW, et al. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 6.Mocroft A, et al. Changing patterns of mortality across Europe in patients infected with HIV-1. EuroSIDA Study Group. Lancet. 1998;352:1725–1730. doi: 10.1016/s0140-6736(98)03201-2. [DOI] [PubMed] [Google Scholar]
- 7.Palella FJ, Jr, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. The New England journal of medicine. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 8.Chun TW, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 10.Wong JK, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 11.Crooks AM, et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. The Journal of infectious diseases. 2015;212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature medicine. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 13.Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell. 2013;155:519–529. doi: 10.1016/j.cell.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davey RT, Jr, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Josefsson L, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4987–4996. doi: 10.1073/pnas.1308313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinoso JB, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9403–9408. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandhi RT, et al. No effect of raltegravir intensification on viral replication markers in the blood of HIV-1-infected patients receiving antiretroviral therapy. Journal of acquired immune deficiency syndromes. 2012;59:229–235. doi: 10.1097/QAI.0b013e31823fd1f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon D, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2010;50:912–919. doi: 10.1086/650749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yukl SA, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. Aids. 2010;24:2451–2460. doi: 10.1097/QAD.0b013e32833ef7bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siliciano JM, Siliciano RF. The Remarkable Stability of the Latent Reservoir for HIV-1 in Resting Memory CD4+ T Cells. The Journal of infectious diseases. 2015;212:1345–1347. doi: 10.1093/infdis/jiv219. [DOI] [PubMed] [Google Scholar]
- 21.Hutter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. The New England journal of medicine. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 22.Durand CM, et al. Developing strategies for HIV-1 eradication. Trends in immunology. 2012;33:554–562. doi: 10.1016/j.it.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasmussen TA, et al. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Human vaccines & immunotherapeutics. 2013;9:993–1001. doi: 10.4161/hv.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Lint C, et al. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cannon P, June C. Chemokine receptor 5 knockout strategies. Current opinion in HIV and AIDS. 2011;6:74–79. doi: 10.1097/COH.0b013e32834122d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. The New England journal of medicine. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deeks SG. HIV: Shock and kill. Nature. 2012;487:439–440. doi: 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- 28.Choudhary SK, Margolis DM. Curing HIV: Pharmacologic approaches to target HIV-1 latency. Annual review of pharmacology and toxicology. 2011;51:397–418. doi: 10.1146/annurev-pharmtox-010510-100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing S, Siliciano RF. Targeting HIV latency: pharmacologic strategies toward eradication. Drug discovery today. 2013;18:541–551. doi: 10.1016/j.drudis.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.International, A.S.S.W.G.o.H.I.V.C. et al. Towards an HIV cure: a global scientific strategy. Nature reviews. Immunology. 2012;12:607–614. doi: 10.1038/nri3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richman DD, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 32.Bruner KM, et al. Towards an HIV-1 cure: measuring the latent reservoir. Trends in microbiology. 2015;23:192–203. doi: 10.1016/j.tim.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37:377–388. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eriksson S, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS pathogens. 2013;9:e1003174. doi: 10.1371/journal.ppat.1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bullen CK, et al. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nature medicine. 2014;20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermankova M, et al. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. Journal of virology. 2003;77:7383–7392. doi: 10.1128/JVI.77.13.7383-7392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laird GM, et al. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS pathogens. 2013;9:e1003398. doi: 10.1371/journal.ppat.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nettles RE, et al. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. Jama. 2005;293:817–829. doi: 10.1001/jama.293.7.817. [DOI] [PubMed] [Google Scholar]
- 39.Palmer S, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. Journal of clinical microbiology. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen TA, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. The lancet. HIV. 2014;1:e13–21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 41.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez G, et al. Accumulation of defective viral genomes in peripheral blood mononuclear cells of human immunodeficiency virus type 1-infected individuals. Journal of virology. 1997;71:2233–2240. doi: 10.1128/jvi.71.3.2233-2240.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill AL, et al. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:13475–13480. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh A, Weinberger LS. Stochastic gene expression as a molecular switch for viral latency. Current opinion in microbiology. 2009;12:460–466. doi: 10.1016/j.mib.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chun TW, et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: implications for eradication. Aids. 2010;24:2803–2808. doi: 10.1097/QAD.0b013e328340a239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaufmann DE, et al. Limited durability of viral control following treated acute HIV infection. PLoS medicine. 2004;1:e36. doi: 10.1371/journal.pmed.0010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lafeuillade A, et al. Predictors of plasma human immunodeficiency virus type 1 RNA control after discontinuation of highly active antiretroviral therapy initiated at acute infection combined with structured treatment interruptions and immune-based therapies. The Journal of infectious diseases. 2003;188:1426–1432. doi: 10.1086/379251. [DOI] [PubMed] [Google Scholar]
- 48.Saez-Cirion A, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS pathogens. 2013;9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steingrover R, et al. HIV-1 viral rebound dynamics after a single treatment interruption depends on time of initiation of highly active antiretroviral therapy. Aids. 2008;22:1583–1588. doi: 10.1097/QAD.0b013e328305bd77. [DOI] [PubMed] [Google Scholar]
- 50.Yerly S, et al. Proviral HIV-DNA predicts viral rebound and viral setpoint after structured treatment interruptions. Aids. 2004;18:1951–1953. doi: 10.1097/00002030-200409240-00011. [DOI] [PubMed] [Google Scholar]
- 51.Ciuffi A, et al. Bioinformatics and HIV latency. Current HIV/AIDS reports. 2015;12:97–106. doi: 10.1007/s11904-014-0240-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henrich TJ, et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. The Journal of infectious diseases. 2013;207:1694–1702. doi: 10.1093/infdis/jit086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Persaud D, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. The New England journal of medicine. 2013;369:1828–1835. doi: 10.1056/NEJMoa1302976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henrich TJ, et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Annals of internal medicine. 2014;161:319–327. doi: 10.7326/M14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luzuriaga K, et al. Viremic relapse after HIV-1 remission in a perinatally infected child. The New England journal of medicine. 2015;372:786–788. doi: 10.1056/NEJMc1413931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strategies for Management of Antiretroviral Therapy Study, G. et al. CD4+ count-guided interruption of antiretroviral treatment. The New England journal of medicine. 2006;355:2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 57.Ghosn J, Delaugerre C. Can we avoid treatment interruption studies in the search for an HIV cure? Aids. 2015;29:1575–1577. doi: 10.1097/QAD.0000000000000763. [DOI] [PubMed] [Google Scholar]
- 58.Li JZ, et al. The need for treatment interruption studies and biomarker identification in the search for an HIV cure. Aids. 2015;29:1429–1432. doi: 10.1097/QAD.0000000000000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho DD, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 60.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 61.Zack JA, et al. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 62.Chun TW, et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nature medicine. 1999;5:651–655. doi: 10.1038/9498. [DOI] [PubMed] [Google Scholar]
- 63.Lafeuillade A, et al. Pilot study of a combination of highly active antiretroviral therapy and cytokines to induce HIV-1 remission. Journal of acquired immune deficiency syndromes. 2001;26:44–55. doi: 10.1097/00126334-200101010-00006. [DOI] [PubMed] [Google Scholar]
- 64.Stellbrink HJ, et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial) Aids. 2002;16:1479–1487. doi: 10.1097/00002030-200207260-00004. [DOI] [PubMed] [Google Scholar]
- 65.Kulkosky J, et al. Intensification and stimulation therapy for human immunodeficiency virus type 1 reservoirs in infected persons receiving virally suppressive highly active antiretroviral therapy. The Journal of infectious diseases. 2002;186:1403–1411. doi: 10.1086/344357. [DOI] [PubMed] [Google Scholar]
- 66.Prins JM, et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. Aids. 1999;13:2405–2410. doi: 10.1097/00002030-199912030-00012. [DOI] [PubMed] [Google Scholar]
- 67.van Laar JM, et al. Reversible neurotoxicity during interleukin-2 therapy for metastatic renal cell carcinoma. European journal of cancer. 1995;31A:1895–1897. doi: 10.1016/0959-8049(95)00297-v. [DOI] [PubMed] [Google Scholar]
- 68.Sosman JA, et al. Phase IB clinical trial of anti-CD3 followed by high-dose bolus interleukin-2 in patients with metastatic melanoma and advanced renal cell carcinoma: clinical and immunologic effects. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1993;11:1496–1505. doi: 10.1200/JCO.1993.11.8.1496. [DOI] [PubMed] [Google Scholar]
- 69.Tyagi M, et al. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. Journal of virology. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455:328–339. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lehrman G, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Routy JP. Valproic acid: a potential role in treating latent HIV infection. Lancet. 2005;366:523–524. doi: 10.1016/S0140-6736(05)67074-2. [DOI] [PubMed] [Google Scholar]
- 73.Sagot-Lerolle N, et al. Prolonged valproic acid treatment does not reduce the size of latent HIV reservoir. Aids. 2008;22:1125–1129. doi: 10.1097/QAD.0b013e3282fd6ddc. [DOI] [PubMed] [Google Scholar]
- 74.Siliciano JD, et al. Stability of the latent reservoir for HIV-1 in patients receiving valproic acid. The Journal of infectious diseases. 2007;195:833–836. doi: 10.1086/511823. [DOI] [PubMed] [Google Scholar]
- 75.Archin NM, et al. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PloS one. 2010;5:e9390. doi: 10.1371/journal.pone.0009390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Archin NM, et al. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. Aids. 2008;22:1131–1135. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Routy JP, et al. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study. HIV medicine. 2012;13:291–296. doi: 10.1111/j.1468-1293.2011.00975.x. [DOI] [PubMed] [Google Scholar]
- 78.Schooley RT, Mellors JW. No cure yet for HIV-1, but therapeutic research presses on. The Journal of infectious diseases. 2007;195:770–772. doi: 10.1086/511830. [DOI] [PubMed] [Google Scholar]
- 79.Xing S, et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. Journal of virology. 2011;85:6060–6064. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doyon G, et al. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. Aids. 2013;27:F7–F11. doi: 10.1097/QAD.0b013e3283570620. [DOI] [PubMed] [Google Scholar]
- 81.Spivak AM, et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2014;58:883–890. doi: 10.1093/cid/cit813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Archin NM, et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. The Journal of infectious diseases. 2014;210:728–735. doi: 10.1093/infdis/jiu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elliott JH, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS pathogens. 2014;10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Procopio FA, et al. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine. 2015;2:872–881. doi: 10.1016/j.ebiom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ocio EM, et al. Evidence of long-term disease control with panobinostat maintenance in patients with relapsed multiple myeloma. Haematologica. 2015;100:e289–291. doi: 10.3324/haematol.2015.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Richardson PG, et al. Panobinostat: a novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert review of anticancer therapy. 2015;15:737–748. doi: 10.1586/14737140.2015.1047770. [DOI] [PubMed] [Google Scholar]
- 88.Wei DG, et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS pathogens. 2014;10:e1004071. doi: 10.1371/journal.ppat.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sogaard OS, et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS pathogens. 2015;11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spina CA, et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS pathogens. 2013;9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dinoso JB, et al. A simian immunodeficiency virus-infected macaque model to study viral reservoirs that persist during highly active antiretroviral therapy. Journal of virology. 2009;83:9247–9257. doi: 10.1128/JVI.00840-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mavigner M, et al. Persistence of virus reservoirs in ART-treated SHIV-infected rhesus macaques after autologous hematopoietic stem cell transplant. PLoS pathogens. 2014;10:e1004406. doi: 10.1371/journal.ppat.1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Denton PW, et al. Generation of HIV latency in humanized BLT mice. Journal of virology. 2012;86:630–634. doi: 10.1128/JVI.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Honeycutt JB, et al. HIV-1 infection, response to treatment and establishment of viral latency in a novel humanized T cell-only mouse (TOM) model. Retrovirology. 2013;10:121. doi: 10.1186/1742-4690-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marsden MD, et al. HIV latency in the humanized BLT mouse. Journal of virology. 2012;86:339–347. doi: 10.1128/JVI.06366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marsden MD, Zack JA. Studies of retroviral infection in humanized mice. Virology. 2015;479–480:297–309. doi: 10.1016/j.virol.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Laird GM, et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. The Journal of clinical investigation. 2015;125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spivak AM, et al. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4+ T Cells from Aviremic Patients. Antimicrobial agents and chemotherapy. 2015;59:5984–5991. doi: 10.1128/AAC.01077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Darcis G, et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS pathogens. 2015;11:e1005063. doi: 10.1371/journal.ppat.1005063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang G, et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS pathogens. 2015;11:e1005066. doi: 10.1371/journal.ppat.1005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dampier W, et al. HIV Excision Utilizing CRISPR/Cas9 Technology: Attacking the Proviral Quasispecies in Reservoirs to Achieve a Cure. MOJ immunology. 2014;1 doi: 10.15406/moji.2014.01.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mousseau G, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell host & microbe. 2012;12:97–108. doi: 10.1016/j.chom.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mousseau G, et al. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio. 2015;6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Agosto LM, et al. Patients on HAART often have an excess of unintegrated HIV DNA: implications for monitoring reservoirs. Virology. 2011;409:46–53. doi: 10.1016/j.virol.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.O’Doherty U, et al. A sensitive, quantitative assay for human immunodeficiency virus type 1 integration. Journal of virology. 2002;76:10942–10950. doi: 10.1128/JVI.76.21.10942-10950.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]