

ABSTRACT
Liver-related autoimmune toxicities triggered by agonistic anti-CD137 antibodies have greatly limited their use in clinical applications. Here, we found that anti-CD137 monoclonal antibody (mAb) treatment in mice induced the infiltration of a large number of S100A4+ macrophages into the liver. Depletion of these cells or deficiency of S100A4 decreased inflammatory cytokine profiles and drastically reduced the number of liver pathogenic CD8+ T cells. Mechanistically, soluble S100A4 directly activated the Akt pathway and specifically prolonged CD8+ T cell survival. Interestingly, one S100A4 neutralizing mAb selectively alleviated liver abnormalities but did not affect the antitumor immunity induced by anti-CD137 mAb therapy. Thus, our study presents a novel molecular link to the liver pathology induced by an immune stimulatory antibody and proposes that combinational immunotherapies targeting those pathways could potentially elicit optimal antitumor immunity with minimal side effects.
KEYWORDS: cancer immunotherapy, CD137, CD8+ T cell, liver pathology, S100A4
Introduction
Cancer immunotherapy has been achieving widespread attention and evolving quickly in recent years.1 Monoclonal antibodies (mAb) targeting immune checkpoint inhibitors such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD-1) pathways have displayed noteworthy clinical benefits in a wide spectrum of human cancers.1-5 However, such checkpoint blockade strategies have only been able to generate durable antitumor immune responses in ∼30% of cancer patients in general with currently unclear biomarkers for the prediction of clinical efficacy.6 Although adoptive cellular transfer (ACT) of engineered autonomous T cells, such as chimeric antigen receptor (CAR) T cells, shows some promise in blood cancers, their effects on late-stage solid cancers are still questionable.1 Immune-stimulatory antibodies (ISAB) intended to provide an agonistic effect on immune co-stimulatory receptors such as CD28, OX40, CD137, CD27, etc., to boost antitumor immunity, differ in mechanism from checkpoint blockade therapy and are becoming attractive modalities for future cancer immunotherapy.7 However, these approaches could cause severe autoimmunity induced by systemic or organ-tropic immune activation, as seen in previous trials with agonistic anti-CD137 mAbs, etc.7-9 Thus, newly designed therapeutic agents carefully balancing antitumor effects and autoimmune toxicity profiles could be of foremost interest for the next generation of ISAB cancer therapies.
CD137 (4-1BB) is an inducible co-signaling receptor of the tumor necrosis factor (TNF) receptor superfamily and is expressed on the surface of activated T cells and on a small population of NK/NKT cells or cells of myeloid origin.10 Engagement of CD137 provides a costimulatory signal to induce T cell expansion, production of interferon-gamma (IFNγ), and prevention of activation-induced death of effector T cells.11 The agonistic anti-CD137 antibody induces regression of established tumors in various animal models12-14 and prevents tumor recurrence in a model of melanoma.15 The signaling domain of CD137 has been fused to the cytoplasmic domain of second-generation CAR T cells, allowing for optimal survival and reducing exhaustion.16,17 Unlike other co-stimulatory receptors such as CD28 and OX40, the tight regulation of CD137 expression by antigen stimulation makes it useful in the isolation of tumor-reactive T cells.18,19 Surprisingly, agonistic CD137 antibodies further display anti-inflammatory properties in a broad spectrum of mouse models for autoimmune diseases such as lupus, experimental autoimmune encephalomyelitis, and arthritis, possibly through the induction of B cell or helper T cell apoptosis.20,21 These profiles of action place anti-CD137 therapy into an ideal category—therapeutic agents with potent antitumor effects but potentially ameliorating autoimmune indications.
However, early clinical trials with an agonistic anti-CD137 antibody (Urelumab, BMS) revealed that it led to severe liver toxicity in humans.22,23 The enhanced CD137 signaling was also found to amplify a subset of memory T cells with unknown specificity, leading to profound pro-inflammatory cytokine production, chronic inflammation, and pathogenesis of liver diseases in a liver-susceptible hepatitis B virus (HBV) transgenic mouse model.24 As CD137-mediated liver pathogenicity and antitumor immunity are both mainly dependent on CD8+ T cells and IFNγ,24,25 there are limited options to dissect these two effects and design an optimal strategy to avoid liver toxicity while maintaining the therapeutic effects on cancers.
S100A4 is a member of the S100 calcium-binding protein family.26,27 It is expressed in various cell types, including fibroblasts, macrophages, and malignant cells.28-30 S100A4 may be intracellular31 or secreted as a normal cytokine.32 Intracellular S100A4 is involved in a wide range of biologic functions, such as the regulation of angiogenesis, cell survival, motility, invasion, or metastasis.33-35 However, S100A4 secreted by tumor and stromal cells is believed to serve as a key player in promoting metastasis of cancer cells or affecting angiogenesis.36-38 Recently, we found an additional mechanism of action (MOA) whereupon S100A4+ cells promote DMBA/TPA-induced skin tumor development by promoting chronic inflammation.39 In addition, we also identified a critical role of S100A4 in promoting liver fibrosis through hepatic stellate cell activation.40 Given the critical role of S100A4 in inflammatory responses and liver pathogenesis, we are interested in understanding the role of this molecule in toxicity induced by CD137 mAb stimulation in vivo. Here, we show that a large number of S100A4+ macrophages infiltrated the liver after anti-CD137 mAb treatment and were responsible for the induction of liver pathology. We also discovered that soluble S100A4 could directly promote CD8+ T cell survival, and CD8+ T cell infiltration into the liver decreased significantly upon S100A4 molecular deficiency or selective depletion of S100A4+ cells. Most interestingly, we that found one S100A4-neutralizing antibody greatly alleviated the liver toxicity induced by anti-CD137 mAb but did not disrupt the role of this antibody in promoting antitumor immunity. Thus, we believe that anti-CD137 mAb treatment combined with S100A4 targeting could become a promising strategy for cancer immunotherapy.
Results
The liver abnormalities induced by agonistic anti-CD137 mAb are independent of the antitumor responses
To access the effect of anti-CD137 mAb on liver toxicity, the agonistic anti-CD137 mAb (2A) or control mAb (RatIg) were intraperitoneally (i.p.) injected into different groups of mice with or without a subcutaneous MC38 tumor cell inoculation. Treatment with anti-CD137 mAb but not the control mAb led to enhanced tumor regression (Fig. 1A) and increased CD8+ T cell infiltration in the tumor (Fig. 1B and Fig. S1A). As expected, we found that anti-CD137 mAb also induced a massive CD8+ T cell infiltration in the liver (Fig. 1B and Fig. S1B) consistent with liver injury, as indicated by serum alanine transaminase (ALT) levels (Fig. 1C). This process was associated with marked infiltration of inflammatory cells with myeloid cell origin into the liver (Fig. 1D and E). This kind of inflammatory response seemed to be localized to the liver as the hematoxylin-eosin (H&E) staining of other organs, including the brain, heart, lung, and kidney, showed only slight damage (Fig. S2). In addition, the liver toxicity induced by anti-CD137 mAb was profound and chronic, as an obvious fibrogenesis indicated by Sirius Red staining was observed on day 30 after the antibody injections (Fig. 1F). Moreover, we found that liver pathogenicity was irrelevant to the tumor as anti-CD137 mAb injection also induced similar levels of liver injury in mice without previous tumor inoculation (Fig. 1G). The results demonstrated that anti-CD137 mAb treatment exhibited a strong antitumor effect but also caused severe liver damage independent of antitumor immunity.
Figure 1.
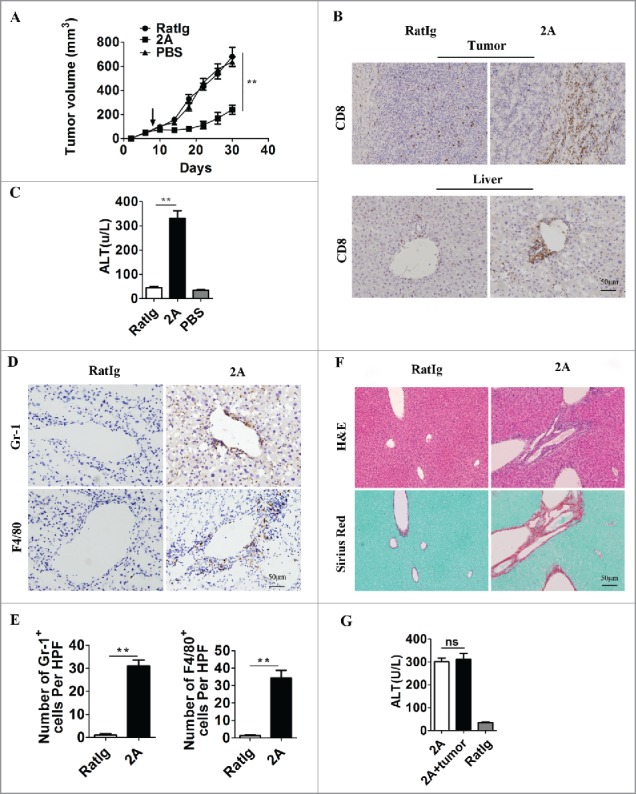
Agonistic anti-CD137 mAb exhibits antitumor activity and induces severe liver injury. Three groups of C57BL/six mice (n = 5 per group) were subcutaneously injected with 5 × 105 MC38 tumor cells at day 0, and then treated with 100 µg 2A, RatIg or PBS for four times on days 8, 11, 14 and 18. (A) The tumor volumes in the three groups were monitored over time. Representative data from three independent experiments are shown. **p < 0.01. (B) Tumor and liver sections were stained for CD8+ on day 21 after tumor inoculation. (C) Serum ALT levels in RatIg, 2A, and PBS-treated mice are shown. **p < 0.01. (D) The liver sections were further stained for Gr-1 and F4/80 and (E) the numbers of Gr-1+ cells and F4/80+ cells in the livers per HPF (×200) are shown. **p < 0.01. (F) Consecutive liver sections were stained with H&E and Sirius Red. Scale bar, 50 μm. (G) One group of C57BL/6 mice without tumors was also treated with 2A four times as a control. Serum ALT levels in 2A-treated mice with or without tumors are shown. ns, not significant.
Anti-CD137 mAb induces infiltration of a large number of S100A4+ macrophages into the liver
In our previous studies, we found that S100A4 could promote liver fibrosis by activating HSCs. To detect the expression of S100A4 after anti-CD137 mAb treatment, we performed quantitative real-time polymerase chain reaction (PCR) (qPCR) analysis for S100A4 on both liver tissue and tumors, together with analysis of another two genes, transforming growth factor-β (TGF-β) and platelet-derived growth factor receptor-β (PDGFR-β), that are closely related to liver injury and fibrosis. Among those genes, we found that S100A4 was significantly upregulated in the liver but remained unchanged in the tumor (Fig. S3A and B). As the liver toxicity of anti-CD137 mAb is irrelevant to tumors, we modified the mouse model to further study the potential role of S100A4, especially in the context of chronic liver diseases.24 We treated 6-week-old male wild-type (WT) mice with anti-CD137 mAb or control RatIg weekly for 5 weeks. Liver tissue was harvested at various time points (Fig. 2A), and tissue sections were subjected to S100A4 staining and Sirius Red staining. As shown in Fig. 2B and C, only a few S100A4+ cells could be detected in the untreated liver; however, anti-CD137 mAb treatment led to a rapid increase of these cells in accordance with the dynamic increase of collagen deposition in the liver. Additionally, we also found that anti-CD137 mAb treatment did not induce significant infiltration of S100A4+ cells in other organs such as brain, heart, lung, and kidney (Fig. S4). These results indicated that the accumulation of S100A4+ cells occurred after anti-CD137 mAb treatment and correlated with the severity of liver fibrosis. To further visualize the features of liver-infiltrating S100A4+ cells, S100A4+/+.GFP transgenic mice expressing green fluorescent protein (GFP) under the control of the S100A4 promoter in one allele41 were treated with anti-CD137 mAb, and the number and phenotype of S100A4+ cells in the liver were examined. As shown in Fig. 2D, the number of S100A4+ cells in the liver after anti-CD137 mAb treatment was much more than that in the untreated liver, and the number increased further after five doses of anti-CD137 mAb. In addition, as shown in Fig. 2E, most of these S100A4+ cells expressed CD11b and F4/80, which are markers of macrophages, but did not express α-smooth muscle actin (α-SMA), the activated fibroblast marker. To further quantify the phenotypes of liver-infiltrating S100A4+ cells, we also performed fluorescence-activated cell sorting (FACS) staining of these cell populations. We found that, among the S100A4-GFP+ cells, 82.3 ± 2.78% were CD11b+, 77 ± 3.35% were Ly6C+, and 76.3 ± 2.2% were F4/80+ (Fig. 2F and G), confirming that most S100A4+ cells that infiltrated the liver were of myeloid origin. These CD11b+ S100A4+ cells could also secrete high concentrations of S100A4 in vitro, as detected by a standard enzyme-linked immunoabsorbent assay (ELISA) (Fig. 2H).
Figure 2.

Anti-CD137 mAb treatment induce the infiltration of a large number of S100A4+ macrophages. (A) Schematic representation of the 2A-induced chronic liver injury model. 6-week-old C57BL/6 mice were treated with 100 µg 2A or RatIg weekly for 5 weeks. Liver tissue was harvested at the indicated time points for further observation. (B) Histological characterization of liver fibrosis and S100A4+ cell accumulation. Consecutive sections were stained with H&E, Sirius Red, or the anti-S100A4 antibody. Scale bar, 50 μm. (C) Quantification of Sirius Red areas in the liver sections. *p < 0.05. (D) To detect the number of S100A4+ cells that infiltrated the liver, S100A4+/+.GFP transgenic mice were treated with 2A as described above. Total numbers of GFP+ cells in the liver (calculated by multiplying the absolute number of liver non-parenchymal cells by the percentage of GFP+ cells) of the untreated (control) or 2A-treated mice at each time point were monitored. Statistical analysis was performed to compare the control group and the 2A-treated groups at different time points (n = 3 per group) after 2A injections. *p < 0.05. (E) Double immunohistochemical (IHC) staining of S100A4 (green) with CD11b, F4/80, or α-SMA in the liver tissue. Nuclei were counter-stained with DAPI (blue). Note that most of the S100A4+ cells were also positive for CD11b and F4/80 (yellow), but not α-SMA. Scale bar, 50 μm. (F-G) Flow cytometry analysis of the phenotypes of S100A4+ cells in the liver of S100A4+/+.GFP mice treated with 2A by staining GFP+ cells with CD11b, Ly6C and F4/80 antibodies. (H) S100A4 concentrations in the cultured supernatant of S100A4+ CD11b+ cells or S100A4− CD11b+ cells as detected by ELISA. **p < 0.01.
Selective depletion of S100A4+ cells attenuates anti-CD137-mAb induced liver injury and fibrosis
To study the contribution of S100A4+ cells during anti-CD137 mAb treatment, we depleted S100A4+ cells by using S100A4-thymidine kinase (TK) transgenic mice treated with ganciclovir (GCV) as indicated before.42,43 S100A4-TK mice were treated with anti-CD137 mAb on day 1 and then injected with GCV or phosphate buffered saline (PBS) on days 1, 3, 4, 6, and 7. The treatment was repeated for 4 weeks. Expression of the Ki67 antigen in S100A4+ cells indicated that these S100A4+ cells were proliferating, rendering them susceptible to depletion by GCV in S100A4-TK transgenic mice (Fig. 3A). As anticipated, the number of infiltrated S100A4+ cells in the liver decreased dramatically in S100A4-TK mice after GCV treatment of 4 weeks, whereas this did not occur in PBS-treated S100A4-TK mice (Fig. 3B and C). After S100A4+ cell depletion, liver injury was decreased as indicated by serum ALT levels (Fig. 3D), and the concentrations of inflammatory cytokines, such as TNF-α, monocyte chemotactic protein-1 (MCP-1), as well as S100A4 decreased significantly in the liver (Fig. 3E). Moreover, the reduction in S100A4+ cells in the liver correlated with an attenuated accumulation of extracellular matrix in the liver tissue (Fig. 3F and G). These results indicated that the selective depletion of S100A4+ cells attenuated liver pathogenesis during anti-CD137 mAb treatment.
Figure 3.

Selective depletion of S100A4+ cells attenuates anti-CD137 mAb-induced liver injury and liver fibrosis. S100A4-TK mice (n = 5) were treated with 100 µg 2A on day 1 and then injected with GCV or PBS on days 1, 3, 4, 6 and 7. The treatment was repeated for 4 weeks. (A) Proliferating S100A4+ cells in 2A-treated livers from S100A4-TK mice were stained for both S100A4 (red) and Ki67 (green). Arrows indicate double-positive cells (yellow). Scale bar, 50 μm. (B) Groups of S100A4-TK+ mice (n = 5 per group) were left untreated or treated with GCV to deplete S100A4+ cells, and liver sections were stained for S100A4. Scale bar, 50 μm. (C) Percentage of S100A4-positive cells. *p < 0.05. (D) Serum ALT levels of S100A4-TK+ mice treated with or without 2A. *p < 0.05. (E) The content of S100A4, MCP-1, and TNF-α protein in the liver homogenates of PBS or GCV-treated TK+ mice was measured by CBA or ELISA. *p < 0.05, **p < 0.01. (F) Groups of S100A4-TK+ mice were left untreated or treated with GCV, and liver sections were stained with Sirius Red. Scale bar, 100μm. (G) Quantification of Sirius Red areas in the liver sections. *p < 0.05.
The liver pathology induced by anti-CD137 mAb is significantly reduced in S100A4−/− or S100A4-TK mice
Previously, we have shown that continuous CD137 stimulation by anti-CD137 mAb led to severe liver pathogenesis from chronic hepatitis to hepatocellular carcinoma in HBV-transgenic mice.24 We were curious to see whether the same treatment could cause chronic liver pathology in S100A4−/− mice, which were constructed by replacing parts of S100A4 exons with in-frame sequences encoding GFP.44 To study the chronic liver pathological effects of anti-CD137 mAb treatment, we treated 15-d-old male S100A4−/− mice and control C57BL/6 mice with one intraperitoneal injection of DEN, followed by weekly anti-CD137 mAb injections for a total of eight doses from day 30. Mice were killed at 8 mo of age to analyze hepatocellular carcinogenesis and histology (Fig. 4A). Liver carcinogenesis under this condition was clearly promoted by anti-CD137 mAb-induced chronic liver inflammation as we seldom observed liver carcinogenesis in either WT or S100A4−/− mice approximately 8 mo after the single DEN treatment (data not shown), similar to that observed in a previous report.45 We found that the level of liver damage was markedly reduced in S100A4−/− mice at week 8 after anti-CD137 mAb treatment (Fig. 4B). Moreover, though all WT mice developed hepatocellular carcinoma (HCC) within 8 mo (Fig. 4C–F), S100A4−/− mice showed profound resistance to liver cancer development. We observed a marked decrease in the total liver tumor numbers in S100A4−/− mice, compared with that in the WT counterparts (19.5 ± 3.86 vs. 1.4 ± 0.37) (Fig. 4D). The maximal tumor diameters were also notably smaller in S100A4−/− mice compared with that in WT controls (8 ± 0.71 mm vs. 1.1 ± 0.29 mm) (Fig. 4E). Consequently, S100A4−/− mice showed reduced liver weight (Fig. 4F). Moreover, plenty of S100A4+ cells had infiltrated into the liver in HCC tissue and the level of collagens was also much higher in WT than S100A4−/− mice (Fig. 4G). In addition, we treated S100A4-TK mice and WT control littermates with DEN/anti-CD137 mAb as mentioned above, and then injected them with GCV or PBS as a control for 2 mo, as illustrated in Fig. 4H. Similarly, GCV treatment in S100A4-TK mice also attenuated HCC development with reduced tumor number and size (Fig. 4I–K) compared with that in the other control groups. The liver weight also decreased in GCV-treated S100A4-TK mice (Fig. 4L). Therefore, these data showed that S100A4 molecular or cellular deficiency significantly decreased the long-term liver pathogenesis induced by anti-CD137 mAb treatment.
Figure 4.

S100A4 deficiency attenuates long-term liver pathological effects of anti-CD137 mAb. (A) Schematic illustration of the 2A/HCC model. 15-day-old S100A4−/− or WT male mice were treated i.p. with a single injection of 50 µg/g DEN and, 1 mo later, treated with 100 µg 2A weekly for 2 mo. All mice were killed 8 mo after DEN treatment of further analysis. (B) Serum ALT levels of 2A-treated WT and S100A4−/− mice. **p < 0.01. (C) Representative liver images of the two groups. Liver tumor nodules are indicated by arrows. (D)–(F) Liver tumor numbers, maximal tumor sizes, and liver weights are shown. **p < 0.01. (G) Liver tissue from both groups was harvested and stained with H&E, anti-S100A4 antibodies, or Sirius Red. Scale bar, 50 μm. (H) Schematic illustration of the 2A/HCC model in S100A4-TK transgenic mice and control littermates. Mice were killed 8 mo after DEN treatment. Representative photographs of the (I) livers (J) liver tumor numbers, (K) maximal tumor sizes, and (L) liver weights. **p < 0.01.
S100A4 deficiency leads to decreased accumulation of CD8+ T cells in the liver
The aberrant accumulation and activation of T cells, especially memory CD8+ T cells, is the major reason for the adverse liver effects induced by anti-CD137 mAb.24 Therefore, we next analyzed the effect of S100A4 on T cell accumulation in the liver. The percentage of CD4+ and CD8+ T cells in intrahepatic lymphocytes in the livers of both S100A4−/− and WT mice was detected by flow cytometry after anti-CD137 mAb treatment (Fig. 5A). Interestingly, on day 28 after antibody treatment, the CD8+ T cell population in S100A4−/− mice was much lower than that in WT mice (29.5 ± 3.86% vs. 10.4 ± 0.37%). We also observed that the CD4+ T cell proportion was also decreased in S100A4−/− mice, but to a much smaller extent (17.7 ± 0.97% vs. 11 ± 0.84%) (Fig. 5B). The total number of liver CD4+ and CD8+ T cells was also significantly decreased (Fig. S5). Immunohistochemical (IHC) staining of liver tissue also showed similar results (Fig. 5C). This effect was anti-CD137-dependent as the T cell populations were normal in naive S100A4−/− mice without any antibody treatment, as well as in a CCl4-induced fibrosis model (data not shown). Moreover, we found that the accumulation of CD8+ T cells decreased significantly, from 33.85 ± 2.03% to 19.8 ± 0.41%, after S100A4+ cell depletion in S100A4-TK mice, whereas CD4+ T cells were seldom influenced (Fig. S6A and B). Thus, our results indicate that S100A4 might play a pathogenic role in modulating hepatic T cells. Given the dominant expression of S100A4 in hepatic macrophages and the potential of this molecule to be secreted, we tested whether soluble S100A4 could directly affect T cell biology in vitro. CFSE-labeled splenic T or liver cells were stimulated with anti-CD3 antibody with or without soluble S100A4, and both CD4+ and CD8+ T cell proliferation were analyzed by FACS. We found that S100A4 had no effect on the proliferation of CD4+ or CD8+ T cells (Fig. 5D). However, CD8+ but not CD4+ T cell apoptosis was greatly inhibited in the presence of S100A4 (Fig. 5E and F), as indicated by 7AAD and Annexin V double staining. Together with the CD8+ T cell data from S100A4−/− and S100A4-TK mice after anti-CD137 mAb treatment, we proposed that S100A4 could promote the accumulation of CD8+ T cells by affecting their survival in the liver.
Figure 5.

S100A4 potentiates CD8+ T cell survival. (A) Representative FACS data and (B) quantification of CD4+ T cells and CD8+ T cells in the liver of WT or S100A4−/− mice (treated with 2A weekly for 4 weeks) were detected by FACS, **p < 0.01. (C) IHC staining for CD4+ and CD8+ in the liver tissue of WT and S100A4−/− mice. Scale bar, 50 μm. (D) CFSE-labeled CD4+ T and CD8+ T cells from the spleen were left unstimulated (left) or stimulated with anti-CD3 antibody (right) with or without soluble S100A4. T cell proliferation was analyzed by CFSE dilution. (E) and (F) CD4+ T and CD8+ T cells from spleen cells were stained with 7-AAD and Annexin V, and cells apoptosis was detected by FACS, **p < 0.01.
S100A4 inhibits CD8+ T cell apoptosis through the Akt pathway
We further tested the signaling pathways in which S100A4 might be involved during T cell apoptosis using a CD8+ murine lymphoid CTLL-2 T cell line. We confirmed that in the presence of S100A4, the spontaneous apoptosis of CTLL-2 T cells caused by IL-2 deprivation was greatly inhibited (Fig. 6A), from 19.47 ± 1.01% to 9.67 ± 0.89%, compared with that in the untreated control condition. Importantly, this inhibitory effect on T cell apoptosis could be reversed by the inclusion of an S100A4 blocking antibody. In accordance with the apoptosis results, cleaved caspase-3 and caspase-9 expression was decreased in CTLL-2 cells after S100A4 treatment (Fig. 6B), and this effect was reversed by the anti-S100A4 antibody. We then investigated the signaling pathways by which S100A4 affected CD8+ T cell apoptosis. CD8+ CTLL-2 cells were stimulated with 1,000 ng/mL S100A4 and collected at different time points for Western Blotting to detect the major survival-related signaling pathways. As shown in Fig. 6C, p-Erk and p-Akt expression were upregulated significantly as early as 5 min after stimulation, demonstrating that S100A4 could directly activate Erk and Akt signaling pathways in CD8+ T cells. We then included Erk and Akt inhibitors to test which signaling pathways were involved in the S100A4-mediated effects. As shown in Fig. 6D and Ethough the application of an Erk inhibitor seemed to have little effect, the inhibitory role on T cell apoptosis by S100A4 could be reversed by the inclusion of Akt inhibitors. Our results, therefore, indicated that S100A4 inhibits the apoptosis of CD8+ T cells through the Akt pathway.
Figure 6.

S100A4 inhibits CD8+ T cell apoptosis though the Akt signaling pathway. (A) CTLL-2 cells, a CD8+ murine lymphoid cell line, were cultured with or without S100A4 (1 µg/mL), or S100A4 combined with anti-S100A4 blocking mAb (6 µg/mL) for 3 d and then subjected to FACS analysis of cell apoptosis. **p < 0.01. (B) Western blots showing the expression of cleaved caspase-3 and caspase-9. (C) CTLL-2 cells were stimulated with S100A4 for varying lengths of time, and the indicated signaling pathways were analyzed by Western blot. (D) An Erk inhibitor or Akt inhibitor was applied to the CTLL-2 culture system supplied with S100A4, and the percentage of apoptotic cells was analyzed via FACS 3 d later. Statistical data are shown (E). **p < 0.01.
Targeting both CD137 and S100A4 in mice elicits strong antitumor responses without severe liver pathology
The data above showed that the liver pathology induced by anti-CD137 mAb is closely associated with the S100A4+ macrophage population inside the liver, which modulates hepatic CD8+ T cell activity. As anti-CD137 mAb has potent antitumor effects but, at the same time, elicits liver toxicity mainly through S100A4, it would be interesting to target both pathways for optimal cancer immunotherapy. We injected WT mice with MC38 tumor cells and then treated them with RatIg, anti-CD137 mAb or anti-CD137 mAb/anti-S100A4 mAb (i.p.) combinational therapy. As shown in Fig. 7A, the anti-CD137/anti-S100A4 combination elicited strong antitumor responses comparable to that of anti-CD137 mAb treatment alone, whereas liver injury was markedly reduced after S100A4 blockage (Fig. 7B). Collagen deposition was significantly reduced at day 7 and day 14 in the liver of the anti-CD137/anti-S100A4 combinational treatment group compared with that in the anti-CD137 mAb group (Fig. 7C). Liver fibrosis was validated by Masson's trichrome staining (data not shown). Both the proportion (Fig. 7D) and the total number (Fig. S7) of CD8+ T cells in the liver were decreased significantly after S100A4 mAb treatment, whereas no effect was observed on the proportion of CD4+ T cells in the liver (Fig. 7D). However, the infiltration of CD8+ T cells into the tumors elicited by anti-CD137 mAb was not affected by the addition of anti-S100A4 mAb, as detected by both FACS (Fig. 7E) and IHC staining (Fig. 7F). The results above demonstrated that targeting both the CD137 and S100A4 pathways could alleviate liver pathology in tumor-bearing mice but maintain an effective antitumor immunity.
Figure 7.
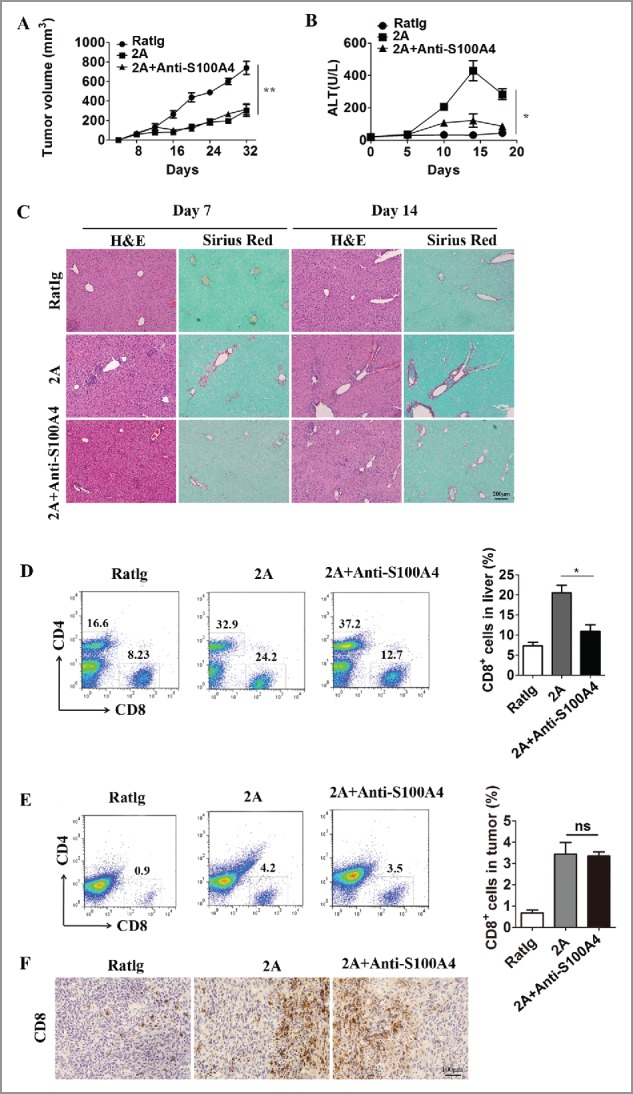
Targeting both the CD137 and S100A4 pathways in mice diminishes liver toxicity but retains antitumor immunity. Three groups of C57BL/six mice (n = 5 per group) were treated with RatIg, 2A, or 2A plus anti-S100A4 mAb 4 times on days 8, 11, 14, and 18. (A) The kinetics of tumor growth in these three groups are shown. **p < 0.01. (B) Serum ALT levels in these three groups are shown. Day 0 represents the first day of 2A injection. *p < 0.05. (C) Liver tissue collected 7 or 14 d after the first injection of 2A were stained with H&E and Sirius red. Representative FACS data showing the proportions of CD4+ T cells and CD8+ T cells and statistical analysis of CD8+ T cells in the (D) tumor and (E) liver. *p < 0.05. (F) Representative IHC staining for CD8+ in tumor sections from different groups.
Discussion
Cancer immunotherapy is becoming an exciting therapeutic option in the treatment of advanced cancers, but the accompanying immune-mediated side effects are still a major hurdle, especially for therapeutic modalities involving agonist antibodies targeting co-stimulatory pathways.7 Given the catastrophic effect of TGN1412 (the super CD28 antagonist antibody)46 and the severe liver toxicity of Urelumab (CD137 agonist antibody developed by BMS) in initial human trials,23 it is imperative that we dissect the MOA of both the toxicity and efficacy to optimize the designs of cancer immunotherapies. In this study, we focused on the toxicity profile of one CD137 agonistic mAb, 2A, and identified a potential target responsible for the adverse effects without disrupting antitumor immunity.
CD137 has been considered one of the most promising targets in the field of cancer immunotherapy since the late 1990s due to its potent function on CD8+ T cell expansion, survival and effector functions, and overexpression in tumor-associated T cells.7 However, this effect of CD137 stimulation is not limited to antitumor responses. Upon treatment, agonist CD137 mAb causes significant accumulation of T cells in multiple sites, including the spleen, lymph-nodes, lung, liver, etc., in normal mice. We confirmed here that significant inflammation could be induced by anti-CD137 mAb injections in several organs of tumor-bearing mice, especially in the liver (Fig. S1). Interestingly, CD137 was previously found to deliver an antigen-independent growth signal for a subpopulation of memory T cells with unknown specificity.47 In the liver-susceptible HBV transgenic mouse model, we observed an expansion of antigen-independent memory CD8+ T cells that are not HBV-specific but cause liver damage and consequent chronic liver diseases in an IFNγ dependent manner.24
The liver has traditionally been considered the “grave yard” of activated/memory T cells and to favor hepatic T cell apoptosis.48 However, whether and how these T cells cause autoimmunity in the liver are still unclear. As most of the intrahepatic CD8+ and CD4+ T cells have an activated phenotype, it is highly likely that they are more reactive to anti-CD137 stimulation. We also found that intrahepatic T cells express higher levels of CD137 (unpublished) and could thus be hyper-reactive to agonistic CD137 mAb treatment. In addition, CD137 signaling is a strong inducer of liver-toxic cytokines, mainly IFNγ and TNF-α, which play critical roles in several liver injury models including ConA, poly I:C, etc. Although the neutralization of IFNγ clearly diminishes the severity of chronic liver diseases induced by anti-CD137 mAb injections, it is not a good target for the reduction of liver toxicity in the case of cancer immunotherapy as it also plays pivotal roles in antitumor immunity, including anti-CD137 mAb therapy.24,25 Thus, whether or not the toxicity/deleterious effects associated with anti-CD137 immunotherapy can be dissociated from its beneficial antitumor activity is becoming an important issue. If this can be achieved, it could lead to the development of new methods of combinatory immunotherapy.
We observed significant S100A4+ macrophage infiltration of the liver after anti-CD137 mAb treatment. These macrophages secreted high concentrations of S100A4 and inhibited the apoptosis of activated CD8+ T cells, a process induced by anti-CD137 mAb through the Akt pathway. Interestingly, both S100A4 and CD137 are highly effective on CD8+ T cells but not CD4+ T cells, although the underlying mechanisms remain unclear. In addition, CD8+ T cell infiltration and spontaneous liver tumor incidence were both decreased in S100A4−/− mice and in S100A4-TK mice selectively depleted of S100A4+ cells. These results indicated that S100A4 maintained the contraction of CD8+ T cells in the liver during the process of anti-CD137 mAb treatment. However, whether this process is parallel to or downstream of IFNγ signaling remains to be discovered. In addition, we found that CD137 engagement combined with an S100A4 blocking antibody could specifically alleviate liver toxicity but maintain antitumor immunity. Thus, our data here further supports the understanding that liver toxicity caused by anti-CD137 mAb treatment is independent of antitumor immunity. Although both toxicity and antitumor immunity may be dependent on the CD8+ T cell population, these two effects could still be separated by the distinct roles that S100A4 plays in the liver and tumor tissues, on the grounds that S100A4 could promote the survival of a subpopulation of myeloid-derived suppressor cells in the tumor and accelerate tumor growth (unpublished). Accordingly, Schwensen et al. recently reported that one S100A4-neutralizing antibody could suppress tumor progression in a model of breast cancer.49 The anti-S100A4 concept may not be limited to anti-CD137 mAb therapy but may also be applied to a wider range of cancer immunotherapies.
S100A4 was initially identified as fibroblast-specific protein (FSP-1).29 However, recent studies have found that it identifies a unique macrophage population, especially within the liver, and is responsible for liver fibrogenesis through the direct activation of hepatic stellate cells.40 Our findings also emphasize a key role of S100A4 in T cell survival and functionality in chronic liver diseases induced by anti-CD137 mAb stimulation. However, several questions remain, including how S100A4 is induced in the liver, what its link with IFNγ is, and how it selectively affects hepatic CD8+ T cell biology but not the antitumor immunity induced by anti-CD137 mAb treatment. Interestingly, we found that anti-S100A4 therapy did not affect the percentage of IFNγ+ CD8+ T cells (Fig. S8A and B), but affected the total number of IFNγ+ CD8+ T cells in the liver (Fig. S8C). As depletion of CD8+ T cells or neutralization of IFNγ totally abolished anti-CD137-induced liver damage, as indicated by our previous studies setting up this model,24 it is possible that IFNγ+ CD8+ T cells play a major role for the anti-S100A4 effect on liver damage. However, because the total number of IFNγ-negative CD8+ T cells was also decreased after anti-S100A4 treatment, IFNγ-negative CD8+ T cells might still contribute to the anti-S100A4 effect, and this still needs to be investigated. According to these results, we proposed a possible role of S100A4 in the control of CD8+ T cell contraction but not the cytokine profile of the T cell that plays a pivotal role in anti-CD137-induced liver toxicity (Fig. 8). Further characterization of its mechanism of action and receptor(s) responsible for its biologic function in the liver will improve our understanding of liver autoimmune toxicity and allow us to design better cancer immunotherapies.
Figure 8.
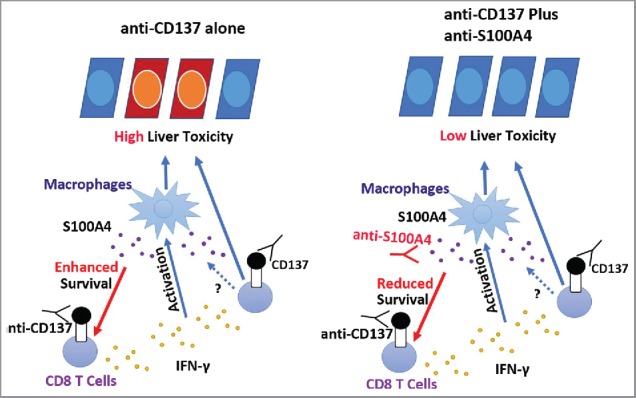
The role of S100A4 in anti-CD137-induced liver toxicity. CD8+ T cells, mainly the memory T cells, are activated by the anti-CD137 antibody in the liver and secrete large amounts of IFNγ, leading to the activation of macrophages and liver toxicity. During this process, large amounts of S100A4 are produced specifically inside of the liver and enhance CD8+ T cell survival, further amplifying the liver damage. Targeting S100A4 with an S100A4 blocking antibody affects CD8+ T cell survival and causes minimal liver toxicity. Similar effects could also be obtained by S100A4 deficiency or depletion of S100A4-positive macrophages.
Materials and methods
Cell lines, mice, and antibodies
The CD8+ murine lymphoid CTLL-2 T cell line (provided by Yijun Wu from the Chinese Academy of Sciences) was cultured in RPMI-1640 (Gibco) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 100 U/mL penicillin/streptomycin, and 100 U/mL murine IL-2. The MC38 murine colon adenocarcinoma cell line (provided by Yangxin Fu from Chinese Academy of Sciences) was cultured in Dulbecco's Modified Eagle Medium (Gibco) supplemented with 10% (vol/vol) FBS and 100 U/mL penicillin/streptomycin.
C57BL/6 and BALB/c mice were purchased from Vital River (Beijing, China). S100A4+/+.GFP and S100A4−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). S100A4-TK transgenic mice were obtained from Dr. Eric G. Neilson (Northwestern University, Feinberg School of Medicine). All mice were bred under specific pathogen-free conditions in the animal facilities at the Institute of Biophysics, Chinese Academy of Sciences. All animal studies were performed with sex- and age-matched mice after being approved by the Institutional Laboratory Animal Care and Use Committee.
Anti-mouse CD137 agonistic mAb (clone 2A, Rat IgG2a) was described previously,13,24 Rat IgG (Sigma-Aldrich, St. Louis, MO) was used as the control.
Monoclonal anti-mouse S100A4 mAb was generated by immunizing BALB/c mice with S100A4 proteins. Using immune spleen cells and the mouse myeloma Sp2/0 cell line as fusion partner,50 hybridomas were generated and selected using standard published techniques.
In vivo tumor growth and anti-CD137 mAb treatment
For the in vivo antitumoral experiments, 6 to 8-week-old C57BL/6 mice received a single subcutaneous injection of MC38 cells on day 0 (5 × 105 cells) and were treated with 2A, control RatIg or PBS at 100 μg per intraperitoneal dose on days 8, 11, 14 and 18. Tumor growth was monitored by using an electronic caliper every 2–3 d and was determined by multiplying perpendicular diameters. For neutralizing S100A4 in vivo, mice were treated i.p. with anti-S100A4 mAb (4 mg/kg) the day before each 2A treatment.
Anti-CD137 mAb induced liver fibrosis
Mice were treated with 100 μg 2A weekly for 5 weeks as described.24
Diethylnitrosamine (DEN)/ anti-CD137 mAb-induced hepatocellular carcinoma
Mice were first treated i.p. with 50 µg/g DEN (Sigma-Aldrich) in 0.1 mL of PBS at the age of 15 d. Mice at the age of 45 d were then treated i.p. with 100 μg 2A or RatIg weekly for 8 weeks. Tumor development was monitored at 8 mo.
Depletion of S100A4-expressing cells
In every week, S100A4-TK mice and control littermates were given 50 mg/kg GCV (HuBeiKeYi Pharmaceutic Corporation, China) dissolved in PBS i.p. on day 1, 3, 4, 6 and 7, while 2A was given i.p. on day 1, the treatment was repeated for 4 weeks.
Histology and immunostaining
Preparation of cryostat or paraffin tissue sections was done as described previously.39 After routine processing, liver sections 7 μm thick were stained with H&E for histological analysis or saturated picric acid containing 0.1% Sirius Red and 0.1% Fast Green for collagen deposition. Paraffin sections were incubated with anti-S100A4 (Abcam, Cambridge, UK), and frozen liver sections were incubated with anti-CD8+, anti-CD4+, anti-F4/80, anti-Gr-1 antibodies (BD PharMingen, San Diego, CA), respectively, and then were incubated with species matched Alexa dye-labeled or horseradish peroxidase (HRP) conjugated secondary antibodies. Frozen liver sections were incubated with anti-S100A4 (Abcam, Cambridge, UK), anti-CD11b (BD PharMingen, San Diego, CA), anti-F4/80 (BD PharMingen, San Diego, CA), anti-α-SMA (Abcam, Cambridge, UK) antibodies, followed by the staining with Alexa Fluor 488 or 555-conjugated secondary antibodies (Invitrogen, Grand Island, NY). Sections were evaluated under the microscope (DP71, OLYMPUS) for bright-field and fluorescence microscopy.
Cytokine and serum biochemical analysis
Serum ALT levels were measured using commercial kits (Biosino, Beijing, China).
For detection of cytokines, livers were homogenized in ice-cold TE buffer. Homogenates were centrifuged at 12,000×g for 15 min. The supernatant was collected and the levels of IL-6, MCP-1, IFNγ, TNF-α, IL-10, and IL-12p70 were assayed by a Mouse Inflammation Cytometric Bead Array (CBA) Kit (BD PharMingen). The data were analyzed using the FCAP array software (BD PharMingen). The relative amount of a target cytokine in one sample = the concentration analyzed by CBA/the weight of the sample. IL-4 and IL-17 were detected by ELISA with paired antibodies (R&D Systems). S100A4 in cell culture supernatant and in the whole liver lysate was detected by a sandwich ELISA as described.40
Isolation of liver non-parenchymal cells
Briefly, cells were isolated from the livers using a two-stage collagenase perfusion technique as described previously.51 Filtered cells were centrifuged at 50×g for 2 min to remove hepatocytes. The remaining non-parenchymal cells (NPC) fraction was collected, washed, and isolated by a 40% and 70% nonlinear Percoll (GE healthcare biosciences, Pittsburgh, PA) gradient system.
Flow cytometry analysis
Single-cell suspensions were prepared directly from bone marrow, and peripheral blood and liver NPCs were stained with the following directly labeled mouse-specific mAbs: Percp/Cy5.5-labeled anti-CD11b (clone M1/70), APC-labeled anti-Ly6C (clone HK1.4), PE-labeled anti-F4/80 (clone BM8), Percp-labeled anti-CD4+ (clone GK1.5), and APC-labeled anti-CD8+ (clone 53–6.7). All antibodies were purchased from Biolegend and used at a 0.2 µg/mL concentration. Cells were collected on a FACS Calibur (BD Biosciences, San Diego, CA) and analyzed by FlowJo software (TreeStar, Ashland, OR).
Apoptosis staining
CTLL-2 cells were deprived of IL-2 and then treated with S100A4 alone or together with an anti-S100A4 antibody, Erk-inhibitor, or Akt-inhibitor. Three days later, cells from each group were collected and stained with PI and Annexin V-fluorescein isothiocyanate (FITC). Cells were tested on an FACS Calibur (BD Biosciences, San Diego, CA) and analyzed by FlowJo software (TreeStar, Ashland, OR).
Western blot analysis
Cell extracts were analyzed by Western blotting as described.52 Briefly, CTLL-2 cells were deprived of IL-2 and cultured in serum-free medium for 6 h. Then, S100A4 was added for 5 min, 15 min, 30 min, 45 min, or 60 min, and cells were harvested, washed in PBS, and lysed. For caspase-9 and caspase-3 detection, CTLL-2 cells were deprived of IL-2 and cultured in medium with or without S100A4 and S100A4 mAb for 48 h or 72 h. Then, the cells were harvested, washed in PBS, and lysed. The primary antibodies used were anti-caspase-9, anti-caspase-3, anti-Erk, anti-p-Erk, anti-Akt, anti-p-Akt, anti-STAT3, anti-p-STAT3, as well as anti-P65 and anti-p-P65 (all from Cell Signaling, Danvers, MA). HRP-conjugated goat anti-mouse or goat anti-rabbit IgG was used as secondary antibody.
Statistical analysis
All data are expressed as mean ± SEM. Differences between two groups were compared using a two-tailed unpaired Student's t-test (GraphPad Prism). The pearson test was used for analysis of correlation between two groups. p-values less than 0.05 were considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by the Ministry of Science and Technology of China (2012CB917103, 2016YF1302305) and the National Natural Science Foundation of China (81630068, 31670881, 91229203 and 81370543).
Abbreviations
- CAR
chimeric antigen receptor
- CCl4
carbon tetrachloride
- CTLA-4
cytotoxic T lymphocyte antigen 4
- DEN
diethylnitrosamine
- EAE
experimental autoimmune encephalomyelitis
- GCV
ganciclovir
- GFP
green fluorescent protein
- IFN
interferon
- mAb
monoclonal antibody
- MCP
monocyte chemotactic protein
- NPCs
non-parenchymal cells
- PD-1
programmed death 1
- PDGFR
platelet-derived growth factor receptor
- TGF
transforming growth factor
- TK
thymidine kinase
- TNF
tumor necrosis factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to the members of our laboratory for discussions and technical help. We thank Professor Yangxin Fu (Chinese Academy of Sciences) and Yijun Wu (Chinese Academy of Sciences) for providing MC38 and CTLL-2 cell lines, and Jiashee Hee, Xu Zhou (Yale University) for critically editing the paper.
Authors contributions
J.Z., K.S., J.W., and Z.Q. were involved in the study design. K.S., J.Z., J.W., Y.L., S.L., and C.D. collected data. K.S., J.Z., J.W., L.C., S.W., and Z.Q. analyzed and interpreted the data. J.W., J.Z., K.S., and Z.Q. wrote the manuscript.
References
- 1.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016; 13(6):394; PMID:27118494; https://doi.org/ 10.1038/nrclinonc.2016.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Eng J Med 2010; 363(8):711-23; PMID:20525992; https://doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Eng J Med 2012; 366(26):2455-65; PMID:22658128; https://doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Eng J Med 2012; 366(26):2443-54; PMID:22658127; https://doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015; 125(9):3384-91; PMID:26325035; https://doi.org/ 10.1172/JCI80011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016; 16(5):275-87; PMID:27079802; https://doi.org/ 10.1038/nrc.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, Melero I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol 2015; 42(4):640-55; PMID:26320067; https://doi.org/ 10.1053/j.seminoncol.2015.05.014 [DOI] [PubMed] [Google Scholar]
- 8.Niu L, Strahotin S, Hewes B, Zhang B, Zhang Y, Archer D et al.. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J Immunol 2007; 178(7):4194-213; PMID:17371976; https://doi.org/ 10.4049/jimmunol.178.7.4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubrot J, Milheiro F, Alfaro C, Palazon A, Martinez-Forero I, Perez-Gracia JL et al.. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol Immunother 2010; 59(8):1223-33; PMID:20336294; https://doi.org/ 10.1007/s00262-010-0846-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol 2003; 3(8):609-20; PMID:12974476; https://doi.org/ 10.1038/nri1148 [DOI] [PubMed] [Google Scholar]
- 11.Myers LM, Vella AT. Interfacing T-cell effector and regulatory function through CD137 (4-1BB) co-stimulation. Trends Immunol 2005; 26(8):440-6; PMID:15979409; https://doi.org/ 10.1016/j.it.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 12.Houot R, Goldstein MJ, Kohrt HE, Myklebust JH, Alizadeh AA, Lin JT, Irish JM, Torchia JA, Kolstad A, Chen L et al.. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 2009; 114(16):3431-8; PMID:19641184; https://doi.org/ 10.1182/blood-2009-05-223958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med 1997; 3(6):682-5; PMID:9176498; https://doi.org/ 10.1038/nm0697-682 [DOI] [PubMed] [Google Scholar]
- 14.Homet Moreno B, Mok S, Comin-Anduix B, Hu-Lieskovan S, Ribas A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 2016; 5(7):e1052212; PMID:27622011; https://doi.org/ 10.1080/2162402X.2015.1052212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narazaki H, Zhu Y, Luo L, Zhu G, Chen L. CD137 agonist antibody prevents cancer recurrence: contribution of CD137 on both hematopoietic and nonhematopoietic cells. Blood 2010; 115(10):1941-8; PMID:20068221; https://doi.org/ 10.1182/blood-2008-12-192591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR et al.. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21(6):581-90; PMID:25939063; https://doi.org/ 10.1038/nm.3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holohan DR, Lee JC, Bluestone JA. Shifting the evolving CAR T cell platform into higher gear. Cancer Cell 2015; 28(4):401-2; PMID:26461084; https://doi.org/ 10.1016/j.ccell.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 18.Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, Powell DJ Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res 2014; 20(1):44-55; PMID:24045181; https://doi.org/ 10.1158/1078-0432.CCR-13-0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chacon JA, Sarnaik AA, Pilon-Thomas S, Radvanyi L. Triggering co-stimulation directly in melanoma tumor fragments drives CD8+ tumor-infiltrating lymphocyte expansion with improved effector-memory properties. Oncoimmunology 2015; 4(12):e1040219; PMID:26587314; https://doi.org/ 10.1080/2162402X.2015.1040219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13(4):227-42; PMID:23470321; https://doi.org/ 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, Fu YX. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med 2002; 8(12):1405-13; PMID:12426559; https://doi.org/ 10.1038/nm1202-796 [DOI] [PubMed] [Google Scholar]
- 22.Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol 2010; 37(5):508-16; PMID:21074066; https://doi.org/ 10.1053/j.seminoncol.2010.09.008 [DOI] [PubMed] [Google Scholar]
- 23.Segal NH, Logan TF, Hodi FS, McDermott DF, Melero I, Hamid O, Schmidt H, Robert C, Chiarion-Sileni V, Ascierto PA et al.. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res 2016; PMID:27756788; https://doi.org/ 10.1158/1078-0432.CCR-16-1272 [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Zhao W, Cheng L, Guo M, Li D, Li X, Tan Y, Ma S, Li S, Yang Y et al.. CD137-mediated pathogenesis from chronic hepatitis to hepatocellular carcinoma in hepatitis B virus-transgenic mice. J Immunol 2010; 185(12):7654-62; PMID:21059892; https://doi.org/ 10.4049/jimmunol.1000927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilcox RA, Flies DB, Wang H, Tamada K, Johnson AJ, Pease LR, Rodriguez M, Guo Y, Chen L. Impaired infiltration of tumor-specific cytolytic T cells in the absence of interferon-gamma despite their normal maturation in lymphoid organs during CD137 monoclonal antibody therapy. Cancer Res 2002; 62(15):4413-8; PMID:12154048 [PubMed] [Google Scholar]
- 26.Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol 2010; 176(2):528-35; PMID:20019188; https://doi.org/ 10.2353/ajpath.2010.090526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra SK, Siddique HR, Saleem M. S100A4 calcium-binding protein is key player in tumor progression and metastasis: preclinical and clinical evidence. Cancer Metastasis Rev 2012; 31(1-2):163-72; PMID:22109080; https://doi.org/ 10.1007/s10555-011-9338-4 [DOI] [PubMed] [Google Scholar]
- 28.Osterreicher CH, Penz-Osterreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, Sasik R, Hardiman G, Karin M, Brenner DA. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A 2011; 108(1):308-13; PMID:21173249; https://doi.org/ 10.1073/pnas.1017547108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 1995; 130(2):393-405; PMID:7615639; https://doi.org/ 10.1083/jcb.130.2.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Chen L, Liu X, Kammertoens T, Blankenstein T, Qin Z. Fibroblast-specific protein 1/S100A4-positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res 2013; 73(9):2770-81; PMID:23539447; https://doi.org/ 10.1158/0008-5472.CAN-12-3022 [DOI] [PubMed] [Google Scholar]
- 31.Kim EJ, Helfman DM. Characterization of the metastasis-associated protein, S100A4. Roles of calcium binding and dimerization in cellular localization and interaction with myosin. J Biol Chem 2003; 278(32):30063-73; PMID:12756252; https://doi.org/ 10.1074/jbc.M304909200 [DOI] [PubMed] [Google Scholar]
- 32.Kiryushko D, Novitskaya V, Soroka V, Klingelhofer J, Lukanidin E, Berezin V, Bock E. Molecular mechanisms of Ca(2+) signaling in neurons induced by the S100A4 protein. Mol Cell Biol 2006; 26(9):3625-38; PMID:16612001; https://doi.org/ 10.1128/MCB.26.9.3625-3638.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, de Groot JF. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res 2014; 20(1):187-98; PMID:24240114; https://doi.org/ 10.1158/1078-0432.CCR-13-1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dmytriyeva O, Pankratova S, Owczarek S, Sonn K, Soroka V, Ridley CM, Marsolais A, Lopez-Hoyos M, Ambartsumian N, Lukanidin E et al.. The metastasis-promoting S100A4 protein confers neuroprotection in brain injury. Nat Commun 2012; 3:1197; PMID:23149742; https://doi.org/ 10.1038/ncomms2202 [DOI] [PubMed] [Google Scholar]
- 35.Fabris L, Cadamuro M, Moserle L, Dziura J, Cong X, Sambado L, Nardo G, Sonzogni A, Colledan M, Furlanetto A et al.. Nuclear expression of S100A4 calcium-binding protein increases cholangiocarcinoma invasiveness and metastasization. Hepatology 2011; 54(3):890-9; PMID:21618579; https://doi.org/ 10.1002/hep.24466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grum-Schwensen B, Klingelhofer J, Grigorian M, Almholt K, Nielsen BS, Lukanidin E, Ambartsumian N. Lung metastasis fails in MMTV-PyMT oncomice lacking S100A4 due to a T-cell deficiency in primary tumors. Cancer Res 2010; 70(3):936-47; PMID:20103644; https://doi.org/ 10.1158/0008-5472.CAN-09-3220 [DOI] [PubMed] [Google Scholar]
- 37.Schmidt-Hansen B, Ornas D, Grigorian M, Klingelhofer J, Tulchinsky E, Lukanidin E, Ambartsumian N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 2004; 23(32):5487-95; PMID:15122322; https://doi.org/ 10.1038/sj.onc.1207720 [DOI] [PubMed] [Google Scholar]
- 38.Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B, Klingelhofer J, Abdul-Al A, Herrmann P, Osterland M et al.. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene 2015; 34(4):424-35; PMID:24469032; https://doi.org/ 10.1038/onc.2013.568 [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Chen L, Xiao M, Wang C, Qin Z. FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Pathol 2011; 178(1):382-90; PMID:21224075; https://doi.org/ 10.1016/j.ajpath.2010.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Li J, Zhang J, Dai C, Liu X, Wang J, Gao Z, Guo H, Wang R, Lu S et al.. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J Hepatol 2015; 62(1):156-64; PMID:25111176; https://doi.org/ 10.1016/j.jhep.2014.07.035 [DOI] [PubMed] [Google Scholar]
- 41.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110(3):341-50; PMID:12163453; https://doi.org/ 10.1172/JCI0215518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, Neilson EG. Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol Ther 2001; 3(2):149-59; PMID:11237671; https://doi.org/ 10.1006/mthe.2000.0251 [DOI] [PubMed] [Google Scholar]
- 43.Salomon B, Maury S, Loubiere L, Caruso M, Onclercq R, Klatzmann D. A truncated herpes simplex virus thymidine kinase phosphorylates thymidine and nucleoside analogs and does not cause sterility in transgenic mice. Mol Cell Biol 1995; 15(10):5322-8; PMID:7565681; https://doi.org/ 10.1128/MCB.15.10.5322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res 2003; 63(12):3386-94; PMID:12810675 [PubMed] [Google Scholar]
- 45.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol 2009; 90(4):367-86; PMID:19659896; https://doi.org/ 10.1111/j.1365-2613.2009.00656.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Eng J Med 2006; 355(10):1018-28; PMID:16908486; https://doi.org/ 10.1056/NEJMoa063842 [DOI] [PubMed] [Google Scholar]
- 47.Zhu Y, Zhu G, Luo L, Flies AS, Chen L. CD137 stimulation delivers an antigen-independent growth signal for T lymphocytes with memory phenotype. Blood 2007; 109(11):4882-9; PMID:17244673; https://doi.org/ 10.1182/blood-2006-10-043463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crispe IN, Dao T, Klugewitz K, Mehal WZ, Metz DP. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol Rev 2000; 174:47-62; PMID:10807506; https://doi.org/ 10.1034/j.1600-0528.2002.017412.x [DOI] [PubMed] [Google Scholar]
- 49.Grum-Schwensen B, Klingelhofer J, Beck M, Bonefeld CM, Hamerlik P, Guldberg P, Grigorian M, Lukanidin E, Ambartsumian N. S100A4-neutralizing antibody suppresses spontaneous tumor progression, pre-metastatic niche formation and alters T-cell polarization balance. BMC Cancer 2015; 15:44; PMID:25884510; https://doi.org/ 10.1186/s12885-015-1034-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McEver RP, Baenziger NL, Majerus PW. Isolation and quantitation of the platelet membrane glycoprotein deficient in thrombasthenia using a monoclonal hybridoma antibody. J Clin Invest 1980; 66(6):1311-8; PMID:6449521; https://doi.org/ 10.1172/JCI109983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawada N, Kuroki T, Kobayashi K, Inoue M, Nakatani K, Kaneda K, Nagata K. Expression of heat-shock protein 47 in mouse liver. Cell Tissue Res 1996; 284(2):341-6; PMID:8625400; https://doi.org/ 10.1007/s004410050594 [DOI] [PubMed] [Google Scholar]
- 52.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P et al.. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 2012; 122(11):4094-104; PMID:23064360; https://doi.org/ 10.1172/JCI64115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.