

Key clinical message
Bilateral enlarged adrenal glands are rare, and as diagnostic delay may have serious consequences for the patient, we recommend a multidisciplinary approach of specialists in the field of endocrinology, oncology, radiology, and clinical chemistry prior to the start of the diagnostic work‐up.
Keywords: Adrenal glands, bilateral, diagnostic delay, multidisciplinary
Introduction
Although bilateral adrenal masses without obvious cause are rare, their incidence is increasing due to the improvement of radiological detection techniques and their frequent use 1, 2. Epidemiological studies focusing on this subject have been mainly performed in Asia, which limits generalization to the Western population. One retrospective analysis carried out in a tertiary center in India included 70 patients with bilateral adrenal masses of which 87.1% had a symptomatic presentation. The remaining 13.9% was incidentally detected. The underlying pathology was found to be a pheochromocytoma (40%), tuberculosis (27.1%), primary adrenal lymphoma (PAL) (10%), metastasis (5.7%), nonfunctioning adenomas (4.3%), and macronodular adrenal hyperplasia (4.3%) 1. Another retrospective study performed in China included eighteen patients of which 78% was symptomatic and reported pheochromocytoma (33%), PAL (22%), nonfunctional cortical adenoma (22%), and metastasis (11%) as the most common causes of bilateral adrenal masses 2. It is generally believed that biochemical screening for pheochromocytoma is crucial to prevent the potentially catastrophic effects of a hypertensive crisis caused by manipulation of the adrenal mass 3, 4. The present case series reviews the diagnostic work‐up of three cases with bilateral massively enlarged adrenal glands caused by rare underlying disorders and discusses the options to reduce diagnostic delay to a minimum.
Case Reports
Case 1
The first patient is an 82‐year‐old woman who was referred to the oncologist because of unintended weight loss of approximately 10 kilogram (kg) over the past three months, progressive fatigue, vertigo, nausea, and aversion to meals. Her medical history was limited to hypothyroidism caused by goiter surgery 45 years ago, which was treated with levothyroxine 100 μg daily. Physical examination revealed hypotension (98/64 mmHg), without other remarkable findings. Computerized tomography (CT) scan showed a homogeneous, hypodense, massive enlargement of both adrenal glands, with a radiological density of 30 Hounsfield Units (HU) after contrast infusion and without any washout. There were no signs of lymphadenopathy or hepatosplenomegaly. Subsequently, a fluorodeoxyglucose (FDG) positron emission tomography (PET) scan showed high FDG uptake in both adrenal glands (left 7.5 × 6.4 × 4.6 cm, right 8.1 × 5.8 × 3.9 cm; Figure 1), without evidence for a primary tumor elsewhere. Laboratory examination revealed a decreased sodium level of 125 mmol/L (normal range (NR): 135–145 mmol/L) and evidence of primary adrenal insufficiency (a decreased fasting cortisol of 107 nmol/L (NR: 171–536 nmol/L) and midday cortisol of 111 nmol/L (NR: 64–327 nmol/L), with elevated ACTH levels of 241 ng/L and 225 ng/L, respectively (NR: <46 ng/L)). Thyroid‐stimulating hormone (TSH) was 1.96 mU/L (NR 0.3–4.0 mU/L) combined with a free thyroxine (FT4) of 22 pmol/L (NR 11–25 pmol/L). Treatment with hydrocortisone (10 mg in the morning, 5 mg at lunch, and 5 mg in the late afternoon) daily was started. Late‐onset bilateral hyperplasia was considered but later discarded as a possibility when plasma 17‐hydroxyprogesterone concentration was found to be normal (1.51 nmol/L, NR <6 nmol/L). Normal values of normetanephrines and metanephrines in 24‐h urine were found (normetanephrine 1.27 μmol/24 h, NR <4.4 μmol/24 h; metanephrine <0.03 μmol/24 h, NR <2.0 μmol/24 h). The interferon‐gamma release assay for mycobacterium tuberculosis was negative, and plasma chromogranine A was within the normal range (29 μg/L, NR 27–94 μg/L). A few weeks after initial presentation, the patient was admitted to the hospital because of an Addison crisis and hypercalcemia (calcium 3.21 mmol/L, NR: 2.20–2.65 mmol/L; albumin 32 g/L, NR: 35–50 g/L). There were no signs of infection on physical and laboratory examination (C‐reactive protein 8 mg/L, NR <10 mg/L; leukocytes 5.0 × 109/L, NR 4.0–11.0 × 109/L). The adrenal crisis was attributed to the erratic use of hydrocortisone because of cognitive deterioration. The combination of hypercalcemia, suppressed PTH levels (<0.3 pmol/L, NR 1.3–6.8 pmol/L) and a normal plasma 25‐hydroxyvitamin D (84 nmol/L, NR >50 nmol/L) raised the suspicion of a lymphoma or bone metastases. The patient was treated with hydration, glucocorticoids, and bisphosphonates. An adrenal biopsy was performed and led to a diagnosis of diffuse large B‐cell lymphoma (DLBCL), classified as stage IVB, with a small B‐cell lymphoma component in the bone marrow specimen. Treatment consisted of six courses rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R‐CHOP21) followed by two courses of rituximab only. Six months later, the PET scan showed complete remission. At this moment, four years after diagnosing PAL, the patient is still in complete remission of her lymphoma. She is currently adequately substituted with hydrocortisone, which was confirmed by repeated early morning cortisol measurements within the normal range. The diagnostic work‐up in this case took over four months.
Figure 1.
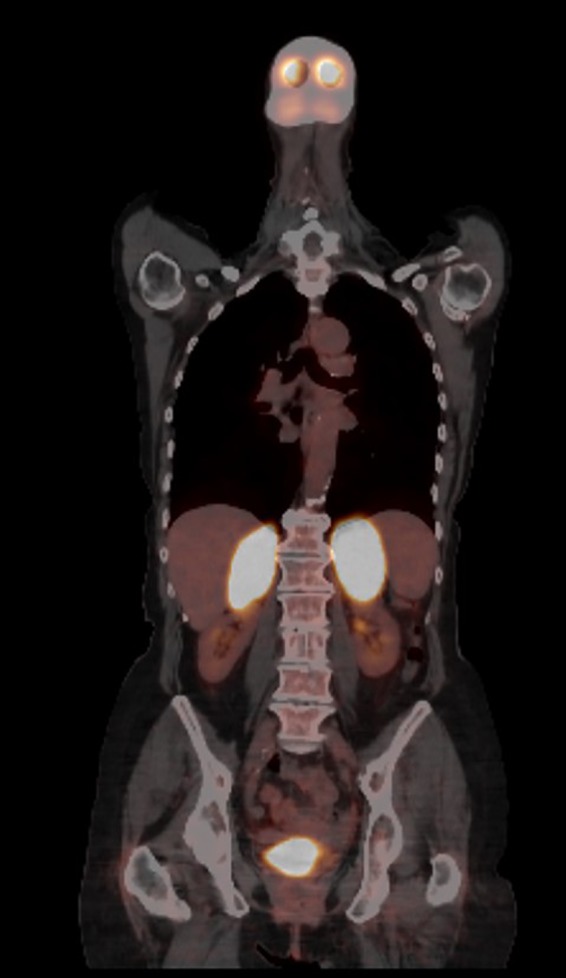
PET scan of patient 1 showing bilateral enlarged adrenal glands (left 7.5 × 6.4 × 4.6 cm, right 8.1 × 5.8 × 3.9 cm) with highfludeoxyglucose uptake.
Case 2
The second patient is an 84‐year‐old woman who was referred to the hematologist because of malaise, progressive fatigue, five kg weight loss, and an elevated erythrocyte sedimentation rate (ESR). Sixteen years previously, she was diagnosed with breast cancer, successfully treated with surgery and radiation therapy. Physical examination at presentation was unremarkable. Blood pressure was 160/88 mmHg. Laboratory examination showed an ESR of 57 mm/h (NR: 1–30 mm/h) and an elevated lactate dehydrogenase of 614 U/L (NR: <250 U/L). No M‐protein was detected. Serum calcium was slightly elevated (2.66 mmol/L) in the face of a normal albumin level (42.2 g/L). Total body CT scan showed homogeneous bilaterally enlarged adrenal glands (left 3.2 × 4.5 cm, right 3.5 × 3.9 cm) with a radiological density of 80 HU after contrast infusion (Figure 2). Adrenal biopsy, performed without prior screening for pheochromocytoma or adrenal insufficiency and without consultation with an endocrinologist, led to the diagnosis of DLBCL, later classified as stage IVB. The patient was treated with chemotherapy consisting of six courses R‐CHOP21 and achieved complete remission. The diagnostic work‐up took over three months. Three and a half years later, the patient died from acute cardiac ischemia.
Figure 2.
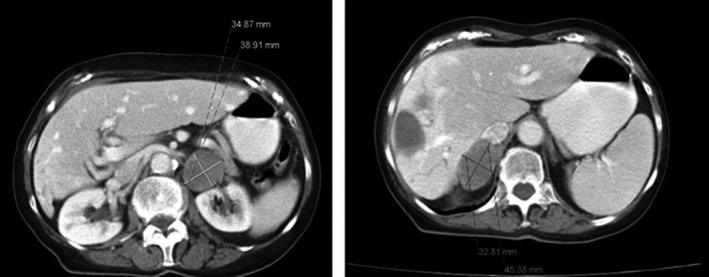
CT scan of patient 2 showing bilateral homogeneous enlarged adrenal glands (left 3.2 × 4.5 cm, right 3.5 × 3.9 cm) and two hepatic cysts.
Case 3
The third patient is a 52‐year‐old, homeless woman with a negative medical history, who was referred to the department of oncology because of weight loss of approximately 30 kg in nine months, generalized abdominal pain, excessive sweating, and hot flushes mainly occurring during the night. She smoked twenty cigarettes a week, but did not use alcohol or drugs. Physical examination only revealed hepatomegaly. Blood pressure was normal (133/87 mmHg), and laboratory examination revealed a severe microcytic anemia (hemoglobin 3.5 mmol/L, NR 7.4–9.9 mmol/L (equal to 5.64 g/dL; NR 12–18 g/dL); MCV 67 fL, NR 80–100 fL), elevated liver enzymes (alkaline phosphatase 405 U/L, aspartate aminotransferase 46 U/L, gamma‐glutamyl transpeptidase 109 U/L; NR: <120 U/l, <30 U/L, and <40 U/L, respectively), and a lactate dehydrogenase of 1757 U/L. Malignant lymphoma was suspected because of the presence of B‐symptoms and the enormously increased lactate dehydrogenase. The CT scan showed a massive enlargement of both adrenal glands, in particular on the left side (15.3 × 10.0 × 15.6 cm) which was a heterogeneous mass with some hypodense areas. The right adrenal gland was a lobed mass of approximately 5 cm. Subsequently, diffuse pulmonary and hepatic lesions were described as well as lymphatic lesions localized left para‐aortal, left para‐iliacal, and in the left renal hilum, suggesting metastatic disease (Figure 3). Adrenal insufficiency was excluded by a fasting plasma cortisol of 588 nmol/L. Despite the fact that the patient was normotensive, it was decided that a pheochromocytoma had to be excluded biochemically before performing a diagnostic adrenal biopsy. An incompletely collected 24‐h urine sample showed a normal normetanephrine level of 3.5 μmol/24 h, with a normal metanephrine (0.65 μmol/24 h). 1.23I‐metaiodobenzylguanidine (1.23I‐MIBG) scan turned out to be negative. The physical condition of the patient deteriorated, and after consultation with the endocrinologist, 24‐h urine was collected again, now using a urinary catheter to ensure a complete sample. This yielded an elevated normetanephrine of 9.17 μmol/24 h and a normal metanephrine level of 0.65 μmol/24 h. 5‐hydroxyindoleacetic (5‐HIAA) proved to be normal (1.1 μmol/mmol creat, NR 0.5–4.0 μmol/mmol creat). In view of the massive tumor load and the relatively mild increases in normetanephrine, it was concluded that metastatic pheochromocytoma was highly unlikely and that a diagnostic biopsy was indicated. This led to a diagnosis of chromophobe renal cell carcinoma (ChRCC). Only limited treatment options with serious adverse effects were available. The patient chose to decline medical treatment targeting her metastatic disease and went to a hospice. The whole diagnostic work‐up covered almost two months.
Figure 3.
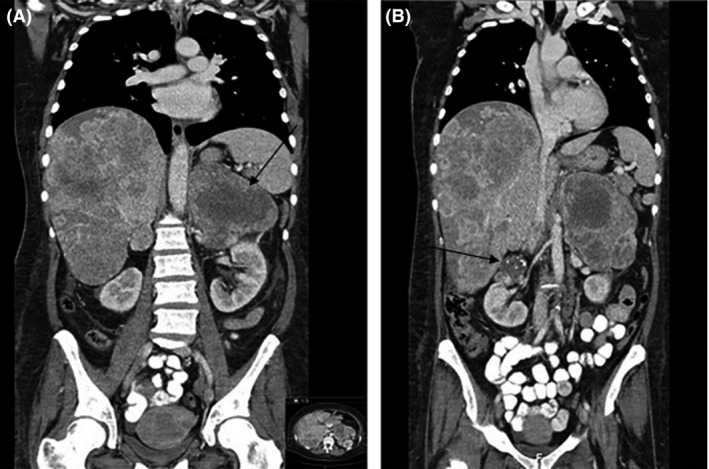
CT scan of patient 3 showing metastatic disease with multiple hepatic lesions, enlargement of both adrenal glands (left 15.3 × 10.0 × 15.6 cm (arrow), right approximately 5 cm; a), and a Bosniak 2F lesion in the right kidney (arrow b). Bosniak 2F lesions are generally benign, with a malignancy rate of 3–12% 25, 26.
Discussion
In this small case series, we describe three cases with bilateral massively enlarged adrenal glands. The time from the first presentation to final diagnosis was two to four months which is much longer than desirable or necessary. Several aspects have played a role in this diagnostic delay, including rareness of the final diagnosis, limited analytic experience with this clinical presentation within a single subspecialism of internal medicine, suboptimal diagnostic choices, and inappropriate timing of diagnostics.
Case 1 and 2 represent two patients diagnosed with a PAL. The pathology of both cases showed a DLBCL, which is with 78% of all cases the most common subtype of PAL 5. Less than 200 cases of PAL have been reported, mainly in studies performed in Asia. PAL is characterized as a highly symptomatic, large (median size 8 cm), hypodense tumor that presents as a bilateral adrenal tumor in approximately 75% of the patients 5. Review of the literature shows that 61% of the patients with PAL have adrenal insufficiency. This may be an overestimation because of publication bias, however, even small size PAL may cause adrenal insufficiency, probably due to diffuse infiltration and complete destruction of the adrenal tissue architecture 5, 6, 7.
Case 3 was diagnosed with a ChRCC that accounts for only 5% of all renal cell carcinomas (RCC). Knowledge regarding ChRCC is scarce. Metastases occur in approximately 6–7% of the cases, most commonly in the liver (33–39%) or lungs (33–36%) 8, 9. The medical literature reports only one case of bilateral adrenal metastases of ChRCC, detected six years after initial nephrectomy 10. Sunitinib, a tyrosine kinase inhibitor, is one of its recommended therapies, although with potentially adverse effects and a limited response rate 8. Bilateral adrenal metastases of a RCC are described in less than twenty cases in the literature and concern in most cases a clear cell RCC 9.
The diagnostic work‐up of the adrenal masses in this case series was delayed, and this may be detrimental to patients with a treatable disease. According to current knowledge, pheochromocytoma must be excluded in case of typical signs or symptoms, when an adrenal incidentaloma is found or when there is a hereditary predisposition. Pheochromocytomas affect only 2–8 per 1,000,000 persons each year and are present in 0.2–0.6% of all hypertensive patients 3, 4, 11. Its prevalence in patients with uni‐ and bilateral adrenal enlargement is 4–5% 4, 11, 12 and 30–40%, respectively 1, 2. This high prevalence of pheochromocytoma found in patients with bilateral adrenal masses is possibly biased as only two epidemiological studies are found in literature, both of which performed in Asia and included mainly patients with a symptomatic presentation. Moreover, Lomte et al. studied patients in a tertiary hospital with two‐thirds of their patients having a familial or syndromic association of pheochromocytoma 1 and Zhou et al. studied only a limited number of eighteen patients 2.
Manipulation of a (unrecognized) pheochromocytoma can result in hypertensive crisis, cardiac arrhythmias, cerebral vascular accident, pulmonary edema, myocardial infarction, and multi‐organ failure 3. This may occur during surgery as well as during adrenal biopsies. Therefore, it is important to recognize the possibility of the tumor and it is recommended to exclude a pheochromocytoma biochemically before executing an adrenal biopsy in patients with adrenal masses 4, 13, 14, 15, 16. About 10% of all pheochromocytomas are malignant and may present as metastatic disease 4, 11. The classic triad of (episodic) palpitations, headache, and sweating is not always present, and hypertension may be absent 3, 4, 11.
Imaging characteristics may serve to raise suspicion of pheochromocytoma in case an adrenal incidentaloma is detected or may be used to localize the tumor in case a pheochromocytoma is suspected biochemically 4. The radiological density of pheochromocytomas on CT is usually greater than 10 HU, and cystic areas, calcifications, necrosis, and hemorrhage are frequently observed 4, 12, 17. Washout of contrast medium is usually delayed 12, 18. High signal intensity on T2‐weighted MRI images is characteristic of pheochromocytoma; however, it may be absent in as much as 35% of the cases 17, 18, 19. Functional imaging with 1,23I‐MIBG scanning may show focal uptake of the norepinephrine analogue 17, 20. However, up to 50% of the normal adrenal glands will also store this analogue physiologically and this may cause a high number of false positives 4. Nevertheless, for the purpose of localizing pheochromocytoma, 1.23I‐MIBG scanning is preferred over FDG‐PET scanning because of its higher sensitivity (85–94% and 76%, respectively 4). However, 68Ga‐DOTANOC PET‐CT scan can also be used because of the expression of somatostatin receptors by pheochromocytoma. It shows a high sensitivity and specificity (92% and 85%, respectively) in patients with the suspicion of pheochromocytoma and may be therefore superior to 1.23I‐MIBG scanning 21. Because the specificity of these radiological characteristics is too limited to provide a definitive diagnosis, biochemical testing is always needed for diagnosing or excluding pheochromocytomas 16.
Pheochromocytoma may be diagnosed by measuring fractionated metanephrines in a 24‐h urine collection or by measurement of plasma‐free metanephrines 4. Plasma‐free metanephrines may have a slightly higher sensitivity but a lower specificity compared to urinary fractionated metanephrines (96–100% relative to 91–98% and 85–89% compared to 91–98%, respectively) 12. One multicenter study confirmed the slightly higher sensitivity of plasma‐free metanephrines (99% relative to 97%), and they also found a higher specificity (89% relative to 69%) 22. Other considerations to prefer plasma measurements over 24‐h urine collections are patient convenience and no risk of incomplete urine collections.
Current consensus is that surgical procedures or biopsies in patients with pheochromocytoma should be preceded by α‐adrenergic blockade with either phenoxybenzamine or doxazosin as the first choice, with no preference for one of the two 4, 16. While phenoxybenzamine is a non‐selective drug with more adverse effects such as reflex tachycardia and postoperative hypotension, the selective drug doxazosin may be less effective in case of large catecholamine excess and often needs the support of other antihypertensive drugs 4, 11, 23.
In conclusion, massive bilateral adrenal masses are rare, and therefore, we recommend a multidisciplinary approach of specialists in the field of endocrinology, oncology, radiology, and clinical chemistry prior to the start of the diagnostic work‐up. Oncologists have to involve an endocrinologist very early in the course of diagnostic work‐up of patients with bilaterally enlarged adrenal glands, on the one hand to exclude adrenal insufficiency and on the other hand to plan endocrine diagnostic work‐up to exclude hormone excess. Adrenal insufficiency always has to be excluded, according to the most recent endocrine society guidelines 24, as it is frequently observed in patients with PAL. According to the current guidelines 4, 22, diagnostic tests for a pheochromocytoma have to be performed, even if the patient seems to be asymptomatic. Measurement of metanephrines should be performed at short notice, and the use of a urinary catheter may be considered to avoid incomplete collection and diagnostic delay.
Conflict of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this study.
Authorship
L.C.A. Drenthen: wrote the manuscript. S.H.P.P. Roerink: supervised the writing of the manuscript and changed the manuscript after review. V. Mattijssen: identified two of the patients and supervised the writing of the manuscript. H. de Boer: provided input in the writing of the manuscript from his expertise in adrenal diseases.
Acknowledgment
We thank Dr. Frank Joosten, radiologist, and dr. Marcel van Borren, clinical chemist, for their expert advice and comments.
Clinical Case Reports 2018; 6(4): 729–734
References
- 1. Lomte, N. , Bandgar T., Khare S., Jadhav S., Lila A., Goroshi M., et al. 2016. Bilateral adrenal masses: a single‐centre experience. Endocrine Connections 5:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou, J. , Ye D., Wu M., Zheng F., Wu F., Wang Z., et al. 2009. Bilateral adrenal tumor: causes and clinical features in eighteen cases. International Urology and Nephrology 41:547–551. [DOI] [PubMed] [Google Scholar]
- 3. Harari, A. , and Inabnet W. B. 3rd. 2011. Malignant pheochromocytoma: a review. The American Journal of Surgery 201:700–708. [DOI] [PubMed] [Google Scholar]
- 4. Lenders, J. W. , Duh Q. Y., Eisenhofer G., Gimenez‐Roqueplo A. P., Grebe S. K., Murad M. H., et al. 2014. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. The Journal of Clinical Endocrinology and Metabolism 99:1915–1942. [DOI] [PubMed] [Google Scholar]
- 5. Rashidi, A. , and Fisher S. I.. 2013. Primary adrenal lymphoma: a systematic review. Annals of Hematology 92:1583–1593. [DOI] [PubMed] [Google Scholar]
- 6. Diamanti‐Kandarakis, E. , Chatzismalis P., Economou F., Lazarides S., Androulaki A., and Kouraklis G.. 2004. Primary adrenal lymphoma presented with adrenal insufficiency. Hormones 3:68–73. [DOI] [PubMed] [Google Scholar]
- 7. Lam, K. Y. , and Lo C. Y.. 2002. Metastatic tumours of the adrenal glands: a 30‐year experience in a teaching hospital. Clinical Endocrinology 56:95–101. [DOI] [PubMed] [Google Scholar]
- 8. Vera‐Badillo, F. E. , Conde E., and Duran I.. 2012. Chromophobe renal cell carcinoma: a review of an uncommon entity. International Journal of Urology: Official Journal of the Japanese Urological Association 19:894–900. [DOI] [PubMed] [Google Scholar]
- 9. Hoffmann, N. E. , Gillett M. D., Cheville J. C., Lohse C. M., Leibovich B. C., and Blute M. L.. 2008. Differences in organ system of distant metastasis by renal cell carcinoma subtype. The Journal of Urology 179:474–477. [DOI] [PubMed] [Google Scholar]
- 10. Wu, H. Y. , Xu L. W., Zhang Y. Y., Yu Y. L., Li X. D., and Li G. H.. 2010. Metachronous contralateral testicular and bilateral adrenal metastasis of chromophobe renal cell carcinoma: a case report and review of the literature. Journal of Zhejiang University Science B 11:386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adler, J. T. , Meyer‐Rochow G. Y., Chen H., Benn D. E., Robinson B. G., Sippel R. S., et al. 2008. Pheochromocytoma: current approaches and future directions. Oncologist 13:779–793. [DOI] [PubMed] [Google Scholar]
- 12. Young, W. F. Jr . 2007. Clinical practice. The incidentally discovered adrenal mass. The New England Journal of Medicine 356:601–610. [DOI] [PubMed] [Google Scholar]
- 13. Vanderveen, K. A. , Thompson S. M., Callstrom M. R., Young W. F. Jr, Grant C. S., Farley D. R., et al. 2009. Biopsy of pheochromocytomas and paragangliomas: potential for disaster. Surgery 146:1158–1166. [DOI] [PubMed] [Google Scholar]
- 14. Delivanis, D. A. , Erickson D., Atwell T. D., Natt N., Maraka S., Schmit G. D., et al. 2016. Procedural and clinical outcomes of percutaneous adrenal biopsy in a high‐risk population for adrenal malignancy. Clinical Endocrinology 85:710–716. [DOI] [PubMed] [Google Scholar]
- 15. Paulsen, S. D. , Nghiem H. V., Korobkin M., Caoili E. M., and Higgins E. J.. 2004. Changing role of imaging‐guided percutaneous biopsy of adrenal masses: evaluation of 50 adrenal biopsies. AJR. American Journal of Roentgenology 182:1033–1037. [DOI] [PubMed] [Google Scholar]
- 16. Chen, H. , Sippel R. S., O'Dorisio M. S., Vinik A. I., Lloyd R. V., Pacak K., et al. 2010. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 39:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leung, K. , Stamm M., Raja A., and Low G.. 2013. Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging. AJR. American Journal of Roentgenology 200:370–378. [DOI] [PubMed] [Google Scholar]
- 18. Baez, J. C. , Jagannathan J. P., Krajewski K., O'Regan K., Zukotynski K., Kulke M., et al. 2012. Pheochromocytoma and paraganglioma: imaging characteristics. Cancer Imaging : The Official Publication of the International Cancer Imaging Society 12:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeiger, M. A. , Thompson G. B., Duh Q. Y., Hamrahian A. H., Angelos P., Elaraj D., et al. 2009. American association of clinical endocrinologists and American association of endocrine surgeons medical guidelines for the management of adrenal incidentalomas: executive summary of recommendations. Endocrine Practice: Official Journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists 15:450–453. [DOI] [PubMed] [Google Scholar]
- 20. McDermott, S. , McCarthy C. J., and Blake M. A.. 2015. Images of pheochromocytoma in adrenal glands. Gland Surgery 4:350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma, P. , Dhull V. S., Arora S., Gupta P., Kumar R., Durgapal P., et al. 2014. Diagnostic accuracy of (68)Ga‐DOTANOC PET/CT imaging in pheochromocytoma. European Journal of Nuclear Medicine and Molecular Imaging 41:494–504. [DOI] [PubMed] [Google Scholar]
- 22. Lenders, J. W. , Pacak K., Walther M. M., Linehan W. M., Mannelli M., Friberg P., et al. 2002. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287:1427–1434. [DOI] [PubMed] [Google Scholar]
- 23. van der Zee, P. A. , and de Boer A.. 2014. Pheochromocytoma: a review on preoperative treatment with phenoxybenzamine or doxazosin. The Netherlands Journal of Medicine 72:190–201. [PubMed] [Google Scholar]
- 24. Bornstein, S. R. , Allolio B., Arlt W., Barthel A., Don‐Wauchope A., Hammer G. D., et al. 2016. Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. The Journal of Clinical Endocrinology and Metabolism 101:364–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graumann, O. , Osther S. S., Karstoft J., Horlyck A., and Osther P. J.. 2013. Evaluation of Bosniak category IIF complex renal cysts. Insights Into Imaging 4:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muglia, V. F. , and Westphalen A. C.. 2014. Bosniak classification for complex renal cysts: history and critical analysis. Radiologia Brasileira 47:368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]