

Key Clinical Message
Peripheral lymphocyte subsets may be less time‐consuming and are a prognostic tool for managing thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly (TAFRO) syndrome. Here, we report a superelderly case of plasma cell type TAFRO syndrome treated effectively using corticosteroid hormones.
Keywords: Corticosteroid therapy, flow cytometry, superelderly patient, TAFRO syndrome
Introduction
Castleman disease (CD) is a rare systemic disorder classified under atypical lymphoproliferative disorders. Multicentric CD (MCD) is its subtype wherein patients develop multiple lesions and systemic symptoms 1.
Thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly (TAFRO) syndrome is a new disease concept, which has been named based on the thrombocytopenia, anasarca, myelofibrosis, renal failure, and organomegaly 2, 3. Architectural features typical of unicentric hyaline vascular CD are not observed in patients with TAFRO syndrome; furthermore, enlarged nuclei are found in proliferating endothelial cells at germinal centers and the interfollicular zone of such patients 2. The median age of disease onset is 50 years (range, 23–72) 4.
Complete remission rates following corticosteroid therapy with/without interleukin (IL)‐6‐targeting strategies are lower in TAFRO syndrome than those in classical MCD 2. Here, we report a superelderly patient with TAFRO syndrome who developed high‐grade fever and an acute disease that could have been fatal within weeks. Her clinical symptoms markedly improved on treatment with prednisolone (PSL) alone.
Case History
An 85‐year‐old woman with a fever of 38°C was admitted to our hospital for 2 weeks during a follow‐up for previous cerebral infarction and aortic aneurysm. A physical examination revealed mild bilateral cervical and axillary lymph node edema. Laboratory findings were white blood cells, 18,500/μL (normal, 4000–9000); hemoglobin, 9.6 g/dL; platelets, 11.4 × 104/μL (normal, 12.5–17.0); C‐reactive protein (CRP), 6.83 mg/dL (normal, ≤0.30); alkaline phosphatase (ALP), 244 IU/L (normal, 115–359); lactate dehydrogenase (LDH), 198 IU/L (normal, 120–240); blood urea nitrogen, 18.9 mg/dL (normal, 8.0–22.0); creatinine, 1.11 mg/dL (normal, 0.60–1.10); immunoglobulin (Ig)G, 1632 mg/dL (normal, 870–1700); IgM, 38 mg/dL (normal, 46–260); antinuclear antibody (ANA), ×320 (normal, <40); and soluble interleukin (sIL)‐2 receptor, 6690 U/mL (normal, 124–466). The study of tumor markers revealed negative results. Examinations of urine were protein‐positive (1+) 0.4 g/gcr: normal, <0.15), but Bence‐Jones protein test result was negative.
She tested negative for human immunodeficiency virus and human herpesvirus 8. Repeated blood cultures were sterile. Both QuantiFERON‐TB and β‐D‐glucan test results were negative. A computed tomography (CT) scan revealed small bilateral cervical, axillary, and inguinal lymph nodes. She was treated with antibiotics for a suspected infection but gradually developed abdominal distension, pyrexia, and dyspnea. Moreover, her serum IL‐2R of 30,200 U/mL and IL‐6 of 39.3 pg/mL were increased. She subsequently developed severe thrombocytopenia. Compared with the findings of a 2‐week‐old CT scan, larger bilateral cervical, axillary, and inguinal lymph nodes as well as bilateral pleural effusion and anasarca were observed in the scan performed during this follow‐up (Fig. 1Aa, b). A biopsy of the inguinal lymph nodes revealed that the expanded and atrophic germinal centers were penetrated by highly dense endothelial venules (Fig. 1Aa, b). Numerous CD138‐positive plasma cells were seen in the interfollicular zone (Figs. 1B and 2C). CD21 immunostaining revealed disrupted patterns of follicular dendritic cell networks (Figs. 1B, 2D). And also the dominant cell type was lambda rather than kappa without monoclonality (data not shown). This case involved three major categories: anasarca, thrombocytopenia, and systemic inflammation, and two minor categories: Castleman disease‐like on lymph node biopsy and progressive renal insufficiency(5). We diagnosed the patient with TAFRO syndrome. Her TAFRO activity score was 7(severe) 5.
Figure 1.
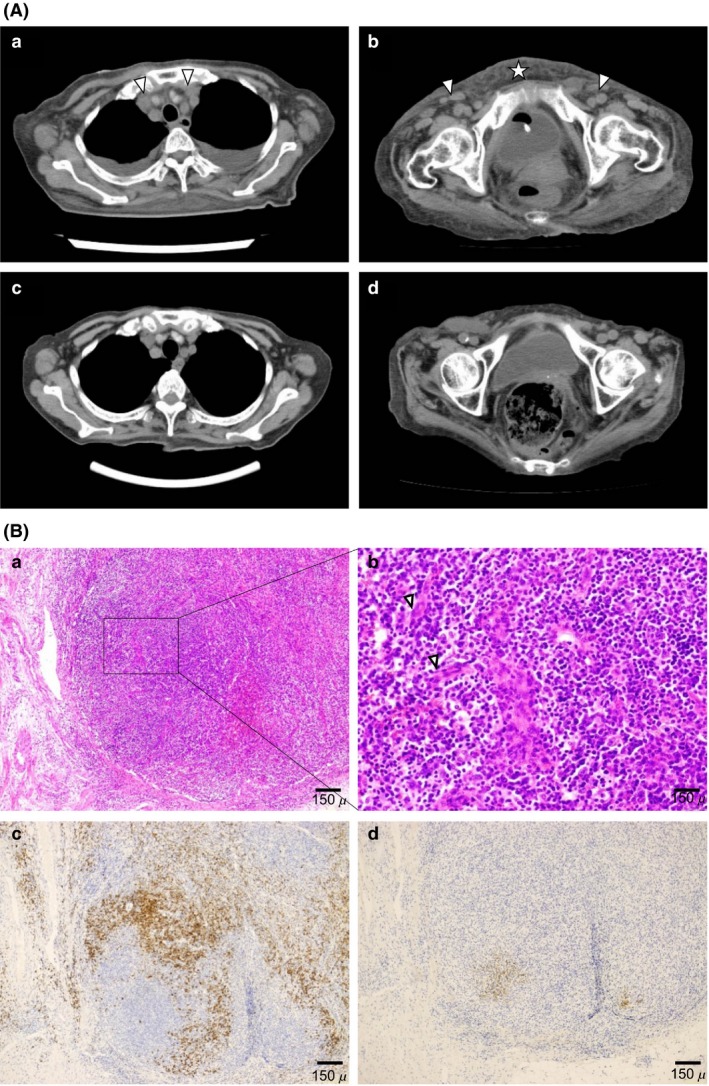
Computed tomography findings (A) and microscopic findings (B). (A) Computed tomography (CT) findings. Thoracic and pelvic CT scans before (a, b) and after (c, d) 1 month of prednisolone therapy showing resolution of pleural effusion and anasarca and shrinking of mediastinal and inguinal lymph nodes (c, d). Arrowhead denotes lymph node. Star denotes anasarca. (B) Biopsy specimen from a small lymph node showing atrophic germinal centers and intact sinuses. Hematoxylin and eosin (H&E) stain. (a) Interfollicular zone was expanded, and atrophic germinal centers were penetrated by highly dense endothelial venules with enlarged nuclei. H&E stain. (b) The interfollicular zone was characterized by the proliferation of highly dense endothelial venules and a small number of mature plasma cells. H&E stain. Arrowhead denotes endothelial cell. (c) A large number of CD138‐positive plasma cells in the interfollicular zone. (d) CD21 immunostaining shows disrupted pattern of follicular dendritic cell networks.
Figure 2.
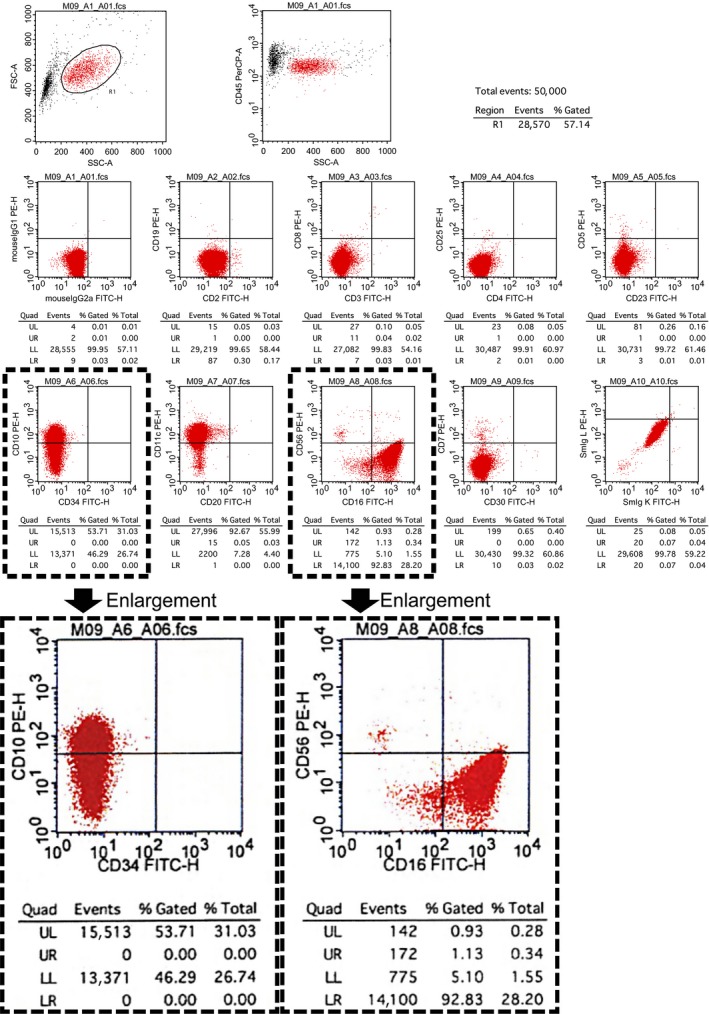
Flow cytometry analysis. A gate is set for the lymphocyte subset in peripheral blood. High power: CD16 expression versus CD56 expression on NK cells is shown. Immunophenotype of NK cells is 81.9%; CD56dim CD16+ and CD10p+ expression cells is shown.
Furthermore, flow cytometric immunophenotyping of NK cells in peripheral blood revealed aberrant expression of CD56(dim)CD16(+)‐associated antigens (Fig. 2). TAFRO syndrome was preliminarily diagnosed on day 18 of hospitalization. Hemoglobin was 9.0 g/dL, white blood cells were 7200/μL (neut. 82.5%, lympho. 5.7%, mono. 6.6%, eosino. 5.0%, baso. 0.2%), platelets were 4.2 × 104/μL, alkaline phosphatase was 349 U/L, gamma‐glutamyl transferase was 72 U/L, creatinine was 1.25 mg/dL, CRP was 7.21 mg/L, sIL‐2R was 3020 U/mL, and IL‐6 was 39.4 pg/mL. Subsequently, she developed severe thrombocytopenia and renal dysfunction. Almost all lymph nodes obtained at the diagnosis were only slightly wider in the greatest diameter than the normal measure (median, 9 mm; range, 6–14) 5. She was started on 50 mg PSL due to the critical condition. PSL adequately controlled fever and pleural effusion (Figs. 1A, 2C, D); her platelet count increased after 10 days of PSL therapy and serum IL‐2R and IL‐6 levels improved. She maintained a remission state for 9 months (Fig. 3).
Figure 3.
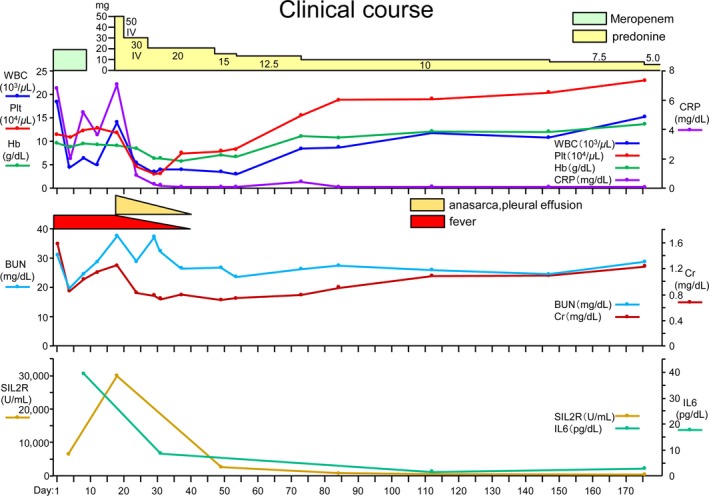
Clinical course.
Discussion
We report a superelderly female case of TAFRO syndrome wherein PSL alone markedly improved the clinical symptoms. The elderly female patient underwent PSL15 mg therapy, and his inflammation including CRP IL‐6 showed immediate improvement. However, his condition including pleural effusion and ascites worsened 6. In our case, the female patient treated with PSL50 mg maintained a remission state. Because currently TAFRO syndrome is diagnosed based on a combination of clinical conditions, this syndrome may be heterogeneous, and it is important to diagnose and treat the syndrome as soon as possible. The diagnostic criteria for TAFRO syndrome are unclear; this can lead to delays in diagnosis and treatment that may affect outcome.
Here, the symptoms were high fever and a high serum sIL2R level at admission. We considered autoimmune inflammatory disorders and hematopoietic malignancies, including Hodgkin's and non‐Hodgkin's lymphoma. She gradually developed abdominal distension and dyspnea, and a CT scan revealed anasarca, pleural effusion, and systemic lymphadenopathy Moreover, serum IL‐2R and IL‐6 levels were increased. Subsequently, she developed severe thrombocytopenia. Twenty days transpired between hospital admission and the start of therapy, wherein she developed multiple organ failure, requiring critical care support, and published the largest case series (25 cases) on this disease, with majority being from Japan 4. Almost all patients had anasarca, organomegaly, bone marrow fibrosis, and fever. Based on this series, the authors proposed a set of diagnostic criteria that could improve recognition of this entity in clinical settings. This case showed atrophic germinal centers, expansion of the interfollicular zone, proliferation of highly dense endothelial venules, and relatively few mature plasma cells (Fig. 2A and B). Hyaline vascular‐ or mixed‐type MCD disease features were observed, such as an expanded mantle zone with lymphocytes arranged concentrically in an “onion‐peel” manner, resembling a lollipop and hyperplastic follicles of varying sizes with mildly increased interfollicular vascularity. No architectural features typical of unicentric hyaline vascular CD were observed 4.
The optimal treatment for patients with TAFRO syndrome remains unknown 4, 5. The first cases described in Japan were responsive to steroids alone, but improvements in thrombocytopenia and anasarca were not consistently achieved 2, 3, 4, 6.
Patients with TAFRO syndrome have been treated alone or in combination with other immunosuppressants or IL‐6 suppressants 4, 5. Other patients have died of the disease despite treatment. Moreover, corticosteroids are effective in treating middle‐aged patients 4. In one series, a single corticosteroid was able to control the disease in 47.8% of patients 4. Moreover, serum IL‐6 levels of responders and nonresponders suggested little evidence for an association between serum IL‐6 levels 2.
In peripheral blood, human NK cells normally constitute approximately 15% of lymphocytes and can be subdivided into different populations based on the relative expression of the surface markers CD16 and CD56. The consequence of these different repertoires of chemokine receptors and adhesion molecules are divergent migratory properties 7. Two major subsets are defined based on CD56 expression: CD56bright and CD56dim cells, the latter comprising >90% of NK cells 7. With regard to NK cell subsets, studies have shown that although the proportions and/or number of CD56dim NK cells increase with age 8, there might be a shift to increased proportions of CD56dimCD16− cells. From flow cytometry, it might be important to characterize TAFRO syndrome in the elderly, and her inflammation including CRP and IL‐6 immediately improved, but are currently unknown for specific pathological findings.
The CD56bright subset preferentially migrates to secondary lymphoid organs, whereas the CD56dim cells migrate to acute inflammatory sites 7. Gating peripheral blood cells except mature‐naïve cells in our case showed acute phenomenon including progressive thrombocytopenia and pleural effusion during the course of disease.
Because TAFRO syndrome is currently diagnosed based on a combination of clinical conditions, this syndrome may be heterogeneous. Thus, a review of the literature was performed. Both PubMed and Embase databases were searched, with “tafro” [All Fields] and “superelderly” and “corticosteroid hormone therapy,” but only three studies were found (last update June 2017). Elderly patients satisfying the diagnostic criteria can maintain remission for 6 months after corticosteroid therapy. Elderly patients satisfying the diagnostic criteria can maintain remission for 6 months after corticosteroid therapy.
Conclusions
This is the first reported superelderly case of TAFRO syndrome treated with corticosteroid monotherapy. It is crucial that TAFRO syndrome be suspected in patients with multiple lymphadenopathy without any known autoimmune diseases or other well‐defined lymphoproliferative disorders 3, 8, particularly if anasarca, pleural effusion, and ascites are present. Flow cytometry may be less time‐consuming and is prognostic tool for managing TAFRO syndrome.
The pathophysiology of TAFRO syndrome must be clarified to develop a more rationalistic treatment strategy.
Consent
Written informed consent was obtained from the patient for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor‐in‐Chief of Clinical Case Reports.
Authorship
YS: conducted the experiments and wrote the manuscript. MA: wrote the manuscript. KN: wrote the manuscript and suggested flow cytometry findings. TO: wrote the manuscript and suggested radiographic findings. HYa: conducted the experiments, wrote the manuscript, and provided pathological findings. YN: wrote the manuscript. HYo: conducted the experiments and wrote the manuscript. All authors have read and approved the final manuscript. The authors declare that they have no competing interests.
Conflict of Interest
None declared.
Clinical Case Reports 2018; 6(4): 638–643
References
- 1. Fajgenbaum, D. C. , Uldrick T. S., Bagg A., Frank D., Wu D., Srkalovic G., et al. 2017. International, evidence‐based consensus diagnostic criteria for HHV‐8–negative/idiopathic multicentric Castleman disease. Blood 129:1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kawabata, H. , Takai K., Kojima M., Nakamura N., Aoki S., Nakamura S., et al. 2013. Castleman‐Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J. Clin. Exp. Hematop. 53:57–61. [DOI] [PubMed] [Google Scholar]
- 3. Takai, K. , Nikkuni K., Momoi A., Nagai K., Igarashi N., and Saeki T.. 2013. Thrombocytopenia with reticulin fibrosis accompanied by fever, anasarca and hepatosplenomegaly: a clinical report of five cases. J. Clin. Exp. Hematop. 53:63–68. [DOI] [PubMed] [Google Scholar]
- 4. Iwaki, N. , Fajgenbaum D. C., Nabel C. S., Gion Y., Kondo E., Kawano M., et al. 2016. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV‐8‐negative multicentric Castleman disease. Am. J. Hematol. 91:220–226. [DOI] [PubMed] [Google Scholar]
- 5. Masaki, Y. , Kawabata H., Takai K., Kojima M., Tsukamoto N., Ishigaki Y., et al. 2016. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int. J. Hematol. 103:686–692. [DOI] [PubMed] [Google Scholar]
- 6. Masaki, Y. , Nakajima A., Iwao H., Kurose N., Sato T., Nakamura T., et al. 2013. Japanese variant of multicentric castleman's disease associated with serositis and thrombocytopenia–a report of two cases: is TAFRO syndrome (Castleman‐ Kojima disease) a distinct clinicopathological entity? J. Clin. Exp. Hematop. 53:79–85. [DOI] [PubMed] [Google Scholar]
- 7. Béziat, V. , Duffy D., Quoc S. N., Le Garff‐Tavernier M., Decocq J., Combadière B., et al. 2011. CD56brightCD16+ NK cells: a functional intermediate stage of NK cell differentiation. J. Immunol. 186:6753–6761. [DOI] [PubMed] [Google Scholar]
- 8. Hazeldine, J. , and Lord J. M.. 2013. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res. Rev. 4:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]