

ABSTRACT
Tumor infiltrating lymphocytes have been associated with a better prognostic and with higher response rates in patients treated with checkpoint inhibiting antibodies, suggesting that strategies promoting tumor inflammation may enhance the efficacy of these currently available therapies. Our aim was thus to develop a new vaccination platform based on cold-inducible RNA binding protein (CIRP), an endogenous TLR4 ligand generated during inflammatory processes, and characterize whether it was amenable to combination with checkpoint inhibitors. In vitro, CIRP induced dendritic cell activation, migration and enhanced presentation of CIRP-bound antigens to T-cells. Accordingly, antigen conjugation to CIRP conferred immunogenicity, dependent on immunostimulatory and antigen-targeting capacities of CIRP. When applied in a therapeutic setting, vaccination led to CD8-dependent tumor rejection in several tumor models. Moreover, immunogenicity of this vaccination platform was enhanced not only by combination with additional adjuvants, but also with antibodies blocking PD-1/PD-L1, CTLA-4 and IL-10, immunosuppressive molecules usually present in the tumor environment and also induced by the vaccine. Therefore, priming with a CIRP-based vaccine combined with immune checkpoint-inhibiting antibodies rejected established B16-OVA tumors. Finally, equivalent activation and T-cell stimulatory effects were observed when using CIRP in vitro with human cells, suggesting that CIRP-based vaccination strategies could be a valuable clinical tool to include in combinatorial immunotherapeutic strategies in cancer patients.
KEYWORDS: checkpoint inhibitor, dendritic cells, targeting, therapeutic vaccination, TLR4 ligand
Introduction
CD8 T-cells play an important role in tumor recognition and rejection.1-3 Indeed, tumor infiltration by CD8 T-cells is not only associated with a better prognostic,2 but also with a higher response rate to newly developed therapies based on checkpoint inhibiting antibodies.4 Therefore, immunotherapies aimed at enhancing the number and properties of tumor-infiltrating T-cells are needed. Vaccines as monotherapies have achieved limited results in the clinic,5,6 but with the advent of clinically available immune checkpoint inhibitors, they are gaining new options in combination therapies.7-12 Thus, it is of interest to develop potent immunization strategies amenable to combination with checkpoint inhibitors. Pathogen-associated molecules13 have been used to activate professional antigen presenting cells, namely dendritic cells (DC), and prime thus T-cell responses through the provision of antigenic stimuli, costimulatory molecules and T-cell polarizing cytokines.14 In addition, there are also endogenous danger signals expressed or released by eukaryotic cells during stress or inflammatory conditions15 which induce DC maturation. Accordingly, vaccination strategies have been developed based on the administration of antigens of interest together with the DC-activating molecules (adjuvants), avoiding thus the use of microorganisms or cells. In addition to DC activation, efficient antigen capture is a requisite to prime functional T-cell responses which greatly improves vaccine efficacy.16-18 Therefore, besides immunostimulatory properties, targeting of antigen to DC has also been pursued through its covalent binding to the adjuvant molecules.19 With this aim, Toll-like receptor (TLR) ligands have been used as adjuvants, not only for their immunostimulatory properties, but also for their ability to target antigens to DC.20-22 Due to the need of inflammation-inducing molecules for antitumor strategies, we focussed our interest in cold-inducible RNA-binding protein (CIRP), whose expression is upregulated during mild hypothermia,23 ultraviolet irradiation24 and hypoxia.25 It has been recently reported that CIRP has important pro-inflammatory properties, it is released as a mediator that triggers inflammatory responses during hemorrhagic shock and sepsis, activating macrophage inflammatory properties in a TLR4-dependent manner.26 Since we had previously demonstrated that other protein and peptidic TLR ligands can be used as adjuvants due to their ability to target antigens to DC and induce their activation,22,27 we hypothesized that CIRP may also behave as an adjuvant with targeting and immunostimulatory capacity. We have thus explored these properties in CIRP, the capacity of CIRP-containing constructs as anti-tumor vaccines and their effect when combined with checkpoint inhibitors, demonstrating the efficacy of this vaccination platform.
Results
SIIN-CIRP protein induces DC maturation, cytokine production and migration
CIRP has been described as a TLR4-binding protein which activates macrophages and induces the production of inflammatory cytokines.26 To study its effect on DC as well as its antigen targeting capacity, which would result in presentation to T-cells, we designed a fusion protein containing amino acids 254–276 (QLESIINFEKLTEW) from ovalbumin, which includes the T-cell epitope OVA(257–264) (SIINFEKL) (recognized by CD8 T-cells from H-2b mice) and three amino-acid flanking residues, linked to the N-terminus of murine CIRP (from now on SIIN-CIRP). A 6-His tail was also added to the N-terminus for protein purification (Fig. 1A). Protein was expressed in bacteria and highly purified (Fig. 1B) until endotoxin levels were below 10 ng endotoxin/mg of protein. Analyses of the capacity of SIIN-CIRP to induce TLR4-mediated activation carried out in HEK-293 cells confirmed that in those cells transfected with TLR4-MD2-CD14, IL-8 was induced by SIIN-CIRP and by LPS, whereas in control cells expressing lacZ, neither SIIN-CIRP nor LPS, induced cytokine production (Fig. 1C).
Figure 1.
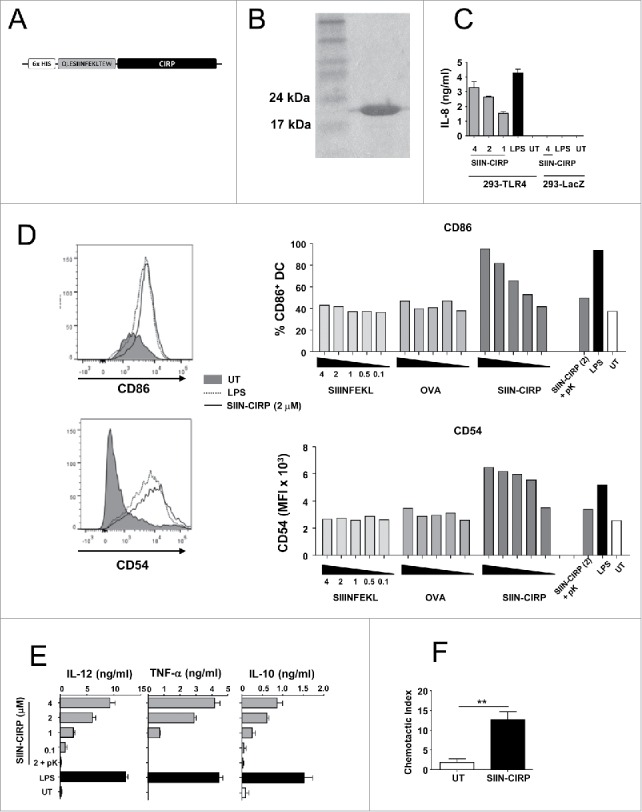
SIIN-CIRP protein induces DC maturation, cytokine production and migration. (A) Schematic representation of protein construct containing the CIRP moiety bound to the antigenic CD8 epitope SIINFEKL plus flanking sequences and a 6 His tail. (B) Representative gel with Coomassie staining corresponding to SIIN-CIRP after protein purification. (C) 293 cells expressing TLR4 or lacZ were incubated for one day with different doses of SIIN-CIRP or LPS and cell activation was determined by the production of IL-8. (D) DC were incubated with graded concentrations of SIIN-CIRP, recombinant OVA, untargeted SIIN. As controls LPS (1 μg/ml), SIIN-CIRP (2 μM) treated with proteinase K or untreated cells were also included. One day later expression of CD86 and CD54 was analyzed by flow cytometry. (E) Supernatants of DC treated as in D were harvested and cytokine content was measured by ELISA. (F) DC untreated or treated with 2 μM SIIN-CIRP were incubated in transwell plates and their in vitro migration towards CCL21 was determined. Results are representative of 2–3 independent experiments.
Once demonstrated the TLR4 binding capacity of SIIN-CIRP, its effect on murine DC maturation was analyzed. SIIN-CIRP and LPS, but not untargeted CD8 epitope or the original antigen OVA, upregulated expression of maturation markers CD86 and CD54 (Fig. 1D). Importantly, treatment of SIIN-CIRP with proteinase K completely abolished marker upregulation, indicating that the stimulatory effect observed was due to the protein and not to potential bacterial endotoxin.
To demonstrate that linking of SIIN peptide to a protein moiety was not sufficient for DC activation we included SIIN in a new truncated construct (SIIN-ΔCIRP). This protein contains only amino acids 1–100 from CIRP (Supplementary Fig. 1 A) and lacks CIRP region 100–125, known to bind the TLR4 partner protein MD2,26 presumably involved in CIRP inflammatory properties. SIIN-ΔCIRP induced much lower activation of TLR4-MD2-CD14-expressing HEK-293 cells than SIIN-CIRP (Supplementary Fig. 1B). Accordingly, SIIN-ΔCIRP did not induce DC maturation, measured as up-regulation of CD86 (Supplementary Fig. 1C).
Regarding cytokine production, DC treated with graded doses of SIIN-CIRP showed a dose-dependent response (in the range of that induced by 1 μg/ml of LPS) in the production of IL-12, TNF-α and IL-10 (Fig. 1E), confirming the stimulatory capacity of the protein. However SIIN-ΔCIRP, as occurred with phenotypic maturation, was inactive (Supplementary Fig. 1D). Finally, we also demonstrated in migration assays that incubation of DC with SIIN-CIRP greatly increased their migratory capacity in migration assays (Fig. 1F).
Linkage to CIRP improves DC presentation of SIIN peptide to T-cells
After characterizing its DC activating capacity, SIIN-CIRP was tested in antigen presentation assays. DC pre-incubated with equimolar amounts of SIIN peptide, OVA, CIRP or SIIN-CIRP, were co-cultured with OT-I CD8 T-cells, specific for SIIN peptide, and DC/T-cell interactions visualized by electron microscopy. Strong interactions were observed in SIIN-treated DC, with higher numbers of T-cells in close contact with DC, followed by DC treated with SIIN-CIRP or OVA. Poor interactions were seen in CIRP-treated and in untreated DC (Fig. 2A). This was confirmed by quantifying the mean cleft size, the fraction of the synapses occupied by close to intermediate appositions and the mean size of continuous region of close apposition (Fig. 2B). In a next series of experiments analyzing functional DC/T-cell interactions, we found that SIIN-CIRP-incubated DC strongly stimulated OT-I T-cell proliferation in a dose-dependent manner (Fig. 2C), as opposed to untreated DC or mature DC incubated with LPS but without antigen. Due to the importance of IFN-γ production by CD8 T-cells in antitumor responses, we measured the production of this cytokine in equivalent experiments. Contrary to image analyses, where SIIN peptide showed the most intense interactions, a high production of IFN-γ was induced by DC treated with SIIN-CIRP, when compared with DC incubated with peptide SIIN or the whole OVA protein, which induced lower or no production of this cytokine by CD8 OT-I T-cells (Fig. 2D). All together, these in vitro results indicate that SIIN-CIRP is able to induce DC activation in a TLR4-dependent manner, favouring antigen presentation to T-cells.
Figure 2.
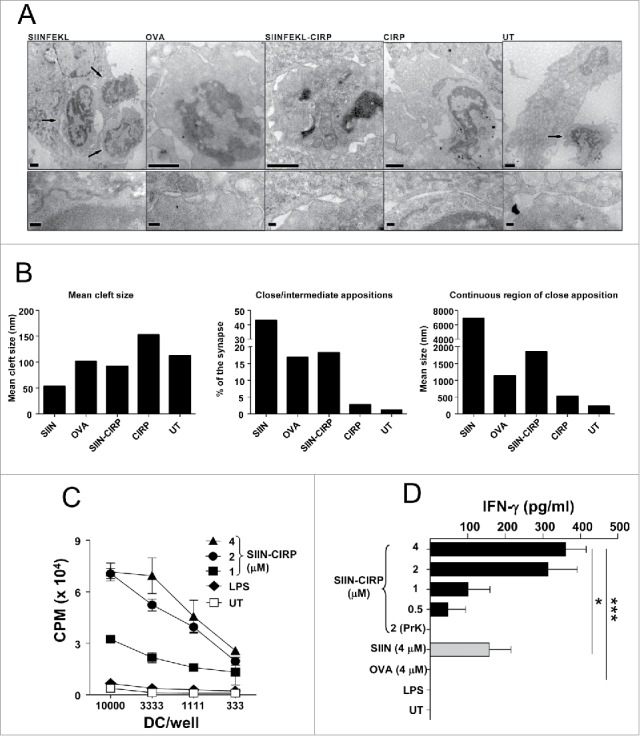
Linkage of SIIN CD8 epitope to CIRP enhances antigen presentation. DC incubated with equimolar amounts of SIIN, OVA, SIIN-CIRP, CIRP or left untreated, were co-cultured with purified CD8 T cell from OT-I mice. Then cells were fixed and visualized by electron microscopy. (A) Representative micrographs from each group (upper row) and details of contacting areas or synapses (lower row). Arrows indicate T-cells. Magnification bars in upper row correspond to 1 µm, and in lower row correspond to 200 nm. (B) Analyses of mean clef size, the fraction of the synapses occupied by close to intermediate appositions and the mean size of continuous region of close apposition corresponding to 9–15 conjugates/group, measured in electron micrographs from thin sections of cells. (C) T-cell proliferation of purified CD8 OT-I T-cells co-cultured with DC pre-incubated with graded doses of SIIN-CIRP, LPS (1 μg/ml) or left untreated. (D) IFN-γ released to the supernatants by OT-I CD8 cells co-cultured with DC pre-incubated with SIIN-CIRP, SIIN or OVA. Results are representative of two independent experiments.
Antigen targeting by CIRP enhances in vivo induction of T-cell responses
Once demonstrated in vitro the activation and targeting properties of CIRP, we next tested the in vivo immunogenicity of SIIN-CIRP. Mice received a single immunization with SIIN-CIRP in saline in the absence of any additional adjuvant. One week later CD8 responses induced against SIIN were compared with those induced by immunization with the same molar amounts of untargeted SIIN or the SIIN-containing protein OVA. While SIIN-CIRP clearly primed SIIN-specific IFN-γ-producing cells, untargeted SIIN or OVA did not induce appreciable T-cell responses (Fig. 3A). Neither did coupling of SIIN to the truncated CIRP protein (SIIN-ΔCIRP construct) (Fig. 3B) nor co-administration of SIIN and CIRP as separated molecules (Fig. 3C) led to T-cell responses as strong as those induced by SIIN-CIRP, demonstrating that responses induced by SIIN-CIRP depend on DC activating and targeting properties of CIRP.
Figure 3.
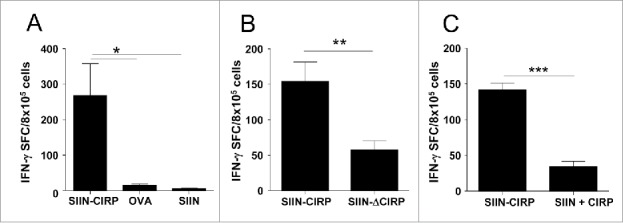
Antigen targeting by CIRP enhances in vivo induction of T-cell responses. C57 BL/6 mice (n = 4/group) were immunized with 2 nmoles of SIIN-CIRP, untargeted SIIN or OVA protein (A), 2 nmoles of SIIN-CIRP or SIIN-ΔCIRP (B) or 2 nmoles of SIIN-CIRP or a mixture of equimolar amounts of SIIN and CIRP (C). In all cases, one week later animals were sacrificed, splenocytes stimulated with 1 μg/ml SIIN and the number of IFN-γ-producing cells measured by ELISPOT. Values obtained in the absence of stimulation with SIIN were always below 20 SFC for all groups. Results are representative of 2–3 independent experiments.
CIRP activates MyD88 and TRIF pathways and induces type I IFN-dependent T-cell responses
Since TLR4 ligands may signal through MyD88- and/or TRIF-dependent elements28,29 we analyzed which pathways were activated by SIIN-CIRP in DC. Time course experiments showed that both genes dependent on MyD88 (Fig. 4 A, left panels) and TRIF (Fig. 4A, right panels), were upregulated upon incubation of DC with SIIN-CIRP. Moreover, since IFN-beta upregulation by CIRP was maintained in DC, and type I IFN has emerged as an important pathway in T-cell-dependent tumor rejection,30,31 we tested the IFN dependency of the CIRP-induced T-cell responses. SIIN-CIRP induced a lower phenotypic maturation (Fig. 4B) and cytokine production (Fig. 4C) in DC from mice lacking type I IFN receptor. Moreover, immunization experiments showed that poorer responses were induced in these animals than in WT mice (Fig. 4D), suggesting that T-cell responses induced by CIRP-targeted antigens depend on type I IFN signalling pathway.
Figure 4.
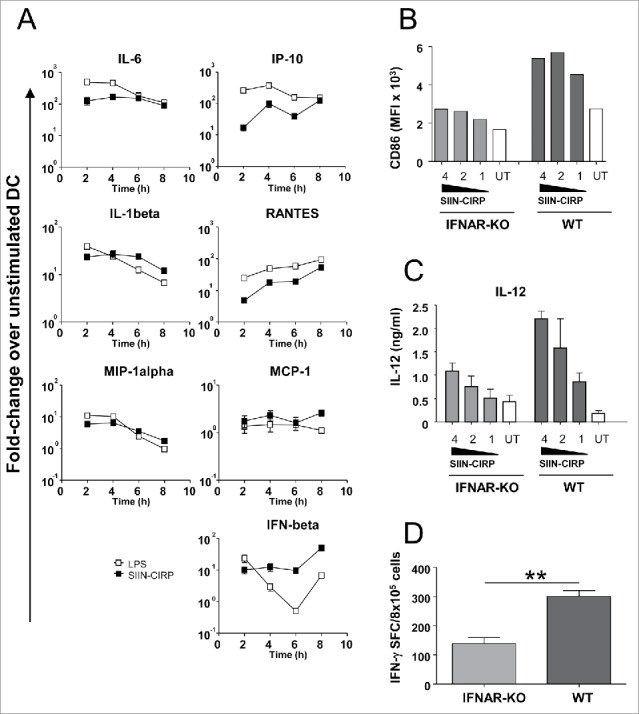
SIIN-CIRP activates MyD88- and TRIF-dependent pathways and induces type I IFN-dependent T-cell responses. (A) DC incubated with SIIN-CIRP (2 μM), LPS (1 μg/ml) or left untreated were harvested at different time-points. Expression of representative MyD88-dependent (left column) and TRIF-dependent (right column) genes was measured by RT-PCR. Results are expressed as fold-change with respect untreated DC. DC from IFNAR KO or WT mice were stimulated with graded doses of SIIN-CIRP and phenotypic maturation (B) and IL-12 production (C) were determined. (D) C57 BL/6 and IFNAR KO mice (n = 4/group) were immunized with 2 nmoles of SIIN-CIRP and responses against SIIN were determined by ELISPOT. Results are representative of two independent experiments.
Vaccination with a CIRP-containing immunogen has CD8-dependent therapeutic anti-tumor effect
To test the capacity of SIIN-CIRP as an anti-tumor therapeutic vaccine, mice with 5 mm established E.G7-OVA tumors were vaccinated with SIIN-CIRP, CIRP without any co-expressed antigen, untargeted peptide SIIN, a mixture of SIIN and CIRP or left untreated. SIIN-CIRP-vaccinated mice, but not control groups with SIIN or CIRP, had a delay in tumor growth, as compared with untreated mice (Fig. 5A). Moreover, in accordance with results shown in Fig. 3C, co-administration of SIIN + CIRP did not demonstrate antitumor effect. Moreover, 60% of mice vaccinated with SIIN-CIRP survived after treatment, whereas survival rates in remaining groups ranged around 15–25%. These results were confirmed in the MC-38-OVA tumor model, with SIIN-CIRP vaccination inducing tumor rejection also in this setting (Fig. 5B).
Figure 5.
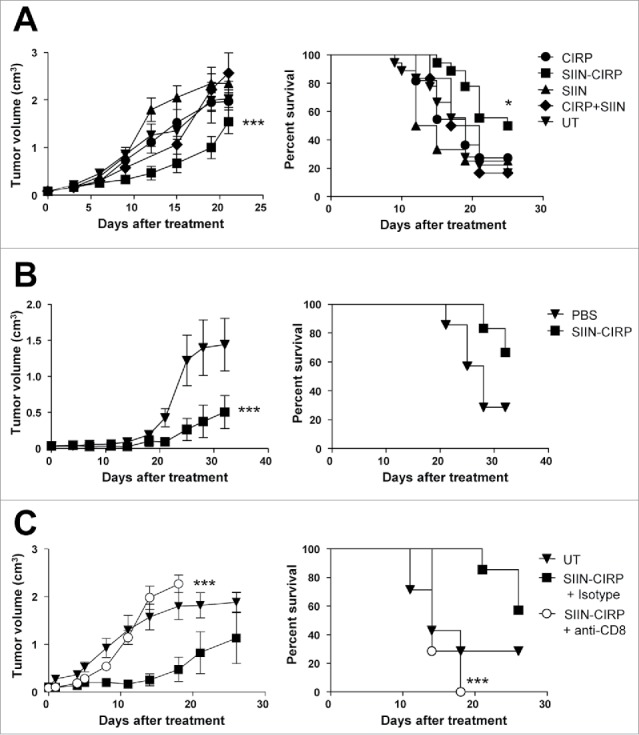
Vaccination with SIIN-CIRP has therapeutic anti-tumor effect. (A) C57 BL/6 mice bearing 5 mm E.G7-OVA tumors were treated for 3 weeks with 2 weekly i.t. injections of SIIN-CIRP, CIRP, peptide SIIN, SIIN + CIRP as a mixture or PBS. (B) Mice bearing 5 mm MC38-OVA tumors were treated as in A with SIIN-CIRP or PBS. (C) Mice with E.G7-OVA tumors were depleted of CD8 cells or given control antibodies and then treated with the SIIN-CIRP vaccine. Tumor volume (left) and animal survival (right) were evaluated in all cases. Results correspond to 2 independent experiments with 6–8 mice/group in each experiment.
Depletion experiments carried out in combination with the SIIN-CIRP vaccine in mice bearing E.G7-OVA tumors showed that depletion of CD8 cells completely abolished the therapeutic effect of vaccination (Fig. 5C), as expected by the presence of CD8 T-cell epitope SIIN peptide as the only antigen in the vaccine, and confirming the essential role of CD8 cells in our vaccination protocol.
CIRP as a vaccination platform for combination with other immune enhancing strategies
Since we have shown that vaccine efficacy can be improved by adjuvant combination,32 to enhance responses induced by CIRP-based vaccines, we immunized mice with SIIN-CIRP and adjuvants signalling through different DC receptors and activation routes. Combination with non-TLR adjuvants (agonistic anti-CD40 antibodies), as well as with poly(I:C) (TLR3) and CpG oligonucleotides (TLR9) clearly enhanced CIRP-induced T-cell responses, whereas no enhancement was observed with Imiquimod (TLR7). Finally, a multiple adjuvant combination (MAC)32 also enhanced CIRP-induced responses (Fig. 6A).
Figure 6.
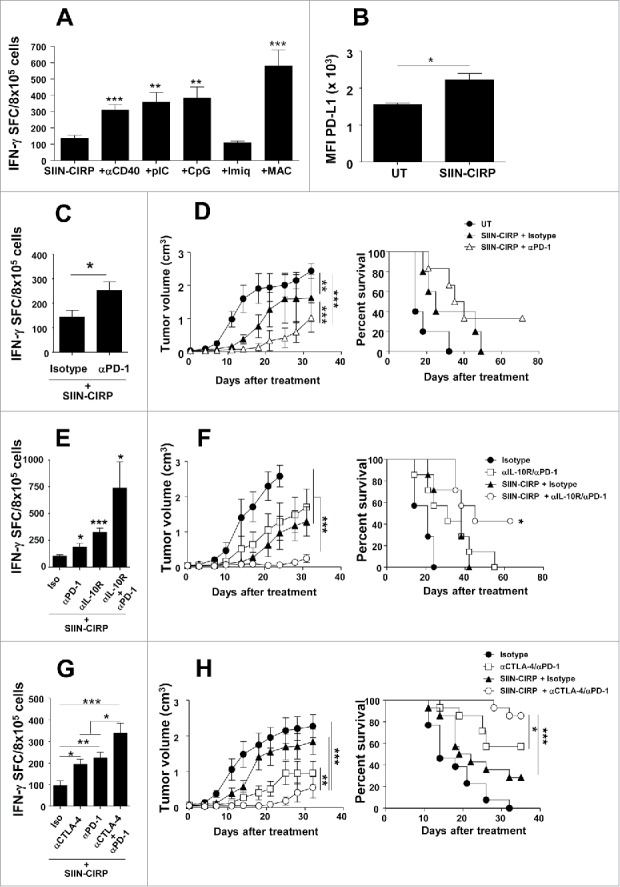
Combination with additional adjuvants or blockade of immunosuppressive elements enhances CIRP-dependent vaccination resulting in higher antitumor effect. (A) C57 BL/6 mice (n = 4/group) were immunized with 2 nmoles of SIIN-CIRP alone or combined with adjuvants anti-CD40 antibodies, poly(I:C), CpG, Imiquimod or a multiple adjuvant combination (MAC) containing poly(I:C), Imiquimod and anti-CD40. One week later immune responses against SIIN were determined by ELISPOT. (B) DC were incubated with SIIN-CIRP or left untreated and next day PD-L1 expression was determined by flow cytometry. Results are expressed as mean fluorescence index (MFI) of PD-L1. (C, E and G) C57 BL/6 mice (n = 4/group) were immunized with 2 nmoles of SIIN-CIRP plus control antibodies or antibodies against PD-1 (C) IL-10R, PD-1 or both (E) or CTLA-4, PD-1 or both (G). Responses against SIIN were measured as above. (D, F and H) C57 BL/6 mice (n = 6–8/group) bearing 5 mm B16-OVA tumors were vaccinated for 3 weeks with 2 weekly i.t. injections of SIIN-CIRP in combination with weekly administration of isotype control or antibodies against PD-1 (D) IL-10R and PD-1 (F) or CTLA-4 and PD-1 (H). Results are representative of 2 independent experiments.
Together with the presence of tumor-infiltrating lymphocytes, expression of PD-L1 has been intensively studied as a biomarker of efficacy in anti-PD-1/PD-L1 therapies.33. Interestingly, PD-L1 was upregulated in DC after incubation with SIIN-CIRP (Fig. 6B), suggesting that CIRP-based vaccines would benefit from inhibition of this pathway. Indeed, combination of SIIN-CIRP with PD-1-blocking antibodies enhanced T-cell responses (Fig. 6C). To test the therapeutic efficacy of combined strategies using CIRP-based vaccines in a more challenging tumor model, we carried out experiments in mice bearing 5 mm B16-OVA tumors. SIIN-CIRP vaccination in the presence of control antibodies delayed tumor growth in this poorly immunogenic tumor model, although no animal survived at the end of the experiment. However, combination of the SIIN-CIRP vaccine with antibodies against PD-1 almost abolished tumor growth during treatment period, which was later strongly delayed, resulting in above 30% long term survival (Fig. 6D).
We have recently demonstrated that blockade of adjuvant-induced IL-10 (a cytokine induced by CIRP; Fig. 1E) greatly improves anti-tumor efficacy of therapeutic vaccines.34. Although vaccination with SIIN-CIRP combined with single blockade of IL-10R or PD-1 enhanced vaccine potency, simultaneous blockade of these molecules in combination with vaccine induced the strongest T-cell responses (Fig. 6E). Accordingly, therapeutic vaccination plus double blockade resulted in greatly delayed tumor growth in parallel with tumor rejection in more than 40% of mice (Fig. 6F).
Finally, we also considered blockade of CTLA-4, another immune checkpoint clinically targeted. As occurred with IL-10R/PD-1 blockade, combined CTLA-4/PD-1 blockade also enhanced T-cell responses (Fig. 6G), which resulted in a strong antitumor effect, clearly improving the therapeutic efficacy over that induced by the vaccine or by antibodies (Fig. 6H).
CIRP induces efficient human DC maturation
Homology between human and murine CIRP is above 95% at the amino acid level. Moreover, we had seen that murine CIRP could activate HEK293 cells expressing human TLR4 (Fig. 1 C). We thus took advantage of this high similarity across species and tested the effect of murine CIRP on human DC. Incubation of human monocyte-derived DC with CIRP induced clear phenotypic upregulation of maturation-associated markers CD86 and CD54 (Fig. 7A) as well as production of IL-12, TNF-α and IL-10 (Fig. 7B). Regarding the T-cell stimulatory capacity of CIRP-treated human DC, we observed in MLR assays that human DC treated with CIRP stimulated allogeneic T-cell proliferation more efficiently than untreated DC, confirming the mature phenotype of these cells (Fig. 7C).
Figure 7.
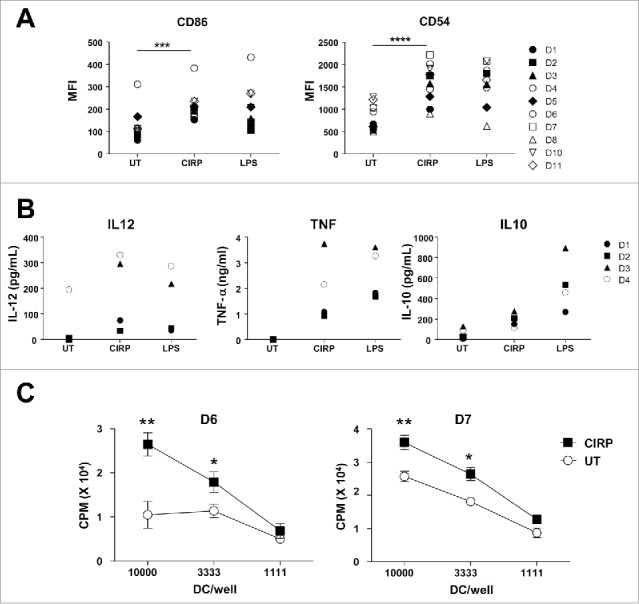
CIRP induces human DC maturation and enhances T-cell activation. (A) Human monocyte-derived DC from healthy donors (D1 to D11) were incubated with 2 μM CIRP, LPS (1 μg/ml) or left untreated. Next day expression of maturation markers CD86 and CD54 was measured by flow cytometry. (B) Cytokine production by DC corresponding to representative donors #1 to #4 was measured by ELISA. (C) Human DC were differentiated from two individuals and treated as above. They were incubated for four days with T-cells from allogeneic donors and T-cell proliferation was measured.
Discussion
Increased levels of tumor-infiltrating lymphocytes have been associated to higher responses to checkpoint inhibitors,4 suggesting that strategies promoting tumor inflammation may enhance response rates to these inhibitors. In this setting, although therapeutic vaccination clinical trials have yield limited clinical results,5,6 combination of both strategies may offer better results. Indeed, tumor PD-L1 expression, which is associated to higher therapeutic efficacy, may result as a consequence of adaptive resistance induced by IFN-γ produced by infiltrating T-cells. Conversely, PD-L1 may be absent in tumors lacking lymphocytes, suggesting that vaccination combined with checkpoint blockade would be a suitable strategy for these patients.35 Thus, it is of paramount importance the design of new vaccines containing molecules with capacity to stimulate DC and target antigens to this cell population, criteria needed for proper T-cell activation.36 With this aim, we tested in vitro and in vivo properties of CIRP, a TLR4-binding endogenous danger molecule released during different stress conditions, with inflammatory properties.26 In vitro characterization of a fusion protein containing CIRP and a CD8 T-cell epitope demonstrated its immunostimulatory activity on DC as well as its antigen targeting properties, which improved in vitro antigen presentation and resulted in stronger in vivo T-cell responses. These experiments showed that both properties were needed for CIRP activity, since co-administration of antigen with uncoupled CIRP or antigen linkage to a truncated CIRP molecule lacking inflammatory properties yielded much poorer responses. Important efforts have been carried out to characterize DC receptors suitable for antigen targeting.19 However, in some cases, despite efficient antigen capture and payload delivery to DC, these pathways do not result in DC maturation, requiring additional signals for efficient activation and proper antigen presentation.16 In the case of CIRP, both properties are present in the same molecule.
As previously reported and confirmed in our work, CIRP-induced cell activation is related to TLR4 binding. Peptides belonging to amino acids 100 to 125 have been reported to bind the TLR4-partner protein MD2.26 However, the full CIRP sequence seems to be required for their functional properties, since as characterized here, the truncated CIRP(1–100) protein lacks the capacity displayed by full CIRP. Moreover, SIIN antigen bound to the CIRP region 100–125, responsible for MD2-binding, is unable to induce T-cell responses (our unpublished results). TLR4 is a receptor whose ligation leads to activation of two signalling pathways, mediated by TRIF and MyD88 molecules, related to its capacity to induce type I IFN and pro-inflammatory mediators.28 There are strong adjuvants which induce both pathways, whereas in other cases, molecules and microorganisms with selectivity for one of them have been described.29,37,38 We show here that CIRP induces the expression of genes corresponding to both pathways, although our results do not formally demonstrate that both are actually needed. In this sense, we show that IFN-beta, a TRIF-dependent molecule, plays an important role on the DC activating capacity of CIRP and the ensuing T-cell responses. In this regard, as an endogenous danger molecule which induces TLR4-dependent DC activation, migration and release of type I IFN, CIRP induces some of the features of immunogenic cell death,39 a death modality associated with the immunogenic properties of many chemotherapeutic agents used in the clinic.
Due to their effects on DC, vaccination with CIRP-containing immunogens induced antitumor responses able to reject established tumors, as demonstrated in several models. These experiments showed again that both properties (DC stimulation and antigen targeting) are responsible for its antitumor efficacy, since the single administration of CIRP into the tumor, even in the presence of tumor-provided endogenous antigens, does not have any meaningful effect. Although we do not discard the potential capacity of CIRP-based immunogens to activate CD4 T-cell responses which would support activation of CD8 T-cells, we show here that even in the absence of CD4 epitopes, CIRP-based vaccines may induce functionally competent CD8 T-cells which are responsible for tumor rejection.
Therapeutic vaccination may bypass the lack of immunogenicity of tumor cells, resulting in the induction of potent T-cell responses. However, there are other mechanisms which preclude tumor rejection, such as the immunosuppressive tumor microenvironment and immunomodulatory molecules induced by the vaccine to avoid excessive T-cell activation. PD-L1, IL-10 and CTLA-4 are examples of these elements which may operate at the level of T-cell activation during antigen presentation by DC and at the effector phase of tumor cell recognition.40,41 Indeed, preclinical41 and recent clinical results (in the case of PD-1/PD-L1)42 obtained after blocking these pathways have shown the pertinence of this approach. Thus, the expression of these molecules induced by vaccination suggests that those effects observed when using monotherapies based on inhibitory antibodies could be improved if combined with CIRP-based vaccines. Accordingly, we have demonstrated that combinations of CIRP-based vaccines with blocking antibodies result in higher therapeutic effect, due to superior effect at the induction phase (as we have demonstrated) and enhanced activity at the effector phase. Indeed, monotherapies based on checkpoint inhibiting antibodies seem to display a superior activity in those tumors with already primed immune responses, whereas “cold” tumors lacking immune effectors show poor responses to these therapies.35 Thus, already available anti-PD-1 therapies may benefit from priming of antitumor immunity with CIRP-based vaccines.
Finally, an important step to consider when translating these findings to the clinical setting is the activity of this molecule in human cells. Although not specifically tested with the human CIRP protein, the high sequence homology of the murine version at the amino acid level (> 95%) allowed us to demonstrate that this approach is also feasible with human cells. Hence, human DC were efficiently activated when incubated with CIRP, results which demand further characterization before being used in humans.
In summary, we have shown that the endogenous danger molecule CIRP has DC activating and targeting properties, which facilitates antigen presentation for efficient T-cell activation. These properties allow the use of CIRP as a vaccination platform which can be included in combinatorial strategies containing other immunostimulatory approaches, such as antibodies blocking inhibitory pathways. These new combinations will help to provide better results than those currently obtained by using immunological monotherapies.
Materials and methods
Recombinant protein and peptide antigens
pET14b plasmids encoding CIRP and CIRP-bound antigens and a 6x His tail (Genscript) were used to transform either BL21(DE3) (Novagen) or ClearColi BL21 (DE3) E. coli cells (Lucigen). After overnight induction with 0.4 mM IPTG and bacterial lysis, proteins were harvested from inclusion bodies and resuspended in buffer containing 8 M urea and 20 mM HEPES pH 7.2. They were purified by affinity chromatography (HisTrap; Pharmacia) using a fast protein liquid chromatography platform (AKTA; Pharmacia). Before protein elution, endotoxin was removed by extensive washing with buffer UTT (8 M urea, 20 mM HEPES, 0.4% Tween 20, 0.4% Triton X-100, pH 7.2), and then eluted with 500 mM imidazol. The eluted protein was desalted using HiTrap desalting columns (Pharmacia). Endotoxin levels were always below 10 ng endotoxin/mg of protein as tested by Quantitative Chromogenic Limulus Amebocyte Lysate assay (Lonza). Recombinant endotoxin-free OVA protein (Endograde) and peptide OVA(257–264) SIINFEKL were purchased from Hyglos (Germany) and from Genecust (Luxemburg), respectively.
Mice
Experimental work with C57 BL/6 (Harlan), OT-I (C57 BL/6-Tg(TcraTcrb)1100Mjb/J; Jackson Laboratories) and IFNAR KO mice (C57 BL/6-IFN-a/bRo/o; a gift from Mathew Albert, Institute Pasteur, Paris, France) was conducted according to relevant national and international guidelines, after approval by the institutional review board.
Cell lines
HEK293/human TLR4 (hTLR4)-MD2-CD14- or HEK293/LacZ-expressing cells (Invivogen) were grown in complete DMEM medium (supplemented with 10% FCS, 2 mM glutamine, 1% Penicillin/Streptomycin) plus 5 μg/ml blasticidin and 25 μg/ml hygromycin. E.G7-OVA thymoma cells (ATCC), MC38-OVA colorrectal carcinoma cells (a kind gift of Dr. I Melero; Pamplona, Spain) and B16-OVA melanoma cells (a kind gift of Dr. G. Kroemer; Paris, France) were cultured in complete RPMI medium (RPMI 1640 supplemented with 10% FCS, 2 mM glutamine and 1% Penicillin/Streptomycin).
Dendritic cell stimulation experiments
Bone marrow-derived DC were generated as described.43 Human DC were differentiated from monocytes44 obtained from buffy coats from the Blood and Tissue Bank of Navarra. Samples were obtained after informed consent and all investigation was conducted according to the principles of the Declaration of Helsinki after approval by the institutional ethical review board. In both cases, DC were collected and cultured in 96-well plates (2 × 105 cells/well) with different concentrations of CIRP-containing protein, LPS (1 μg/ml) or left unstimulated. One day later supernatants were harvested to determine cytokine production and DC were analyzed by flow cytometry. In some experiments CIRP-containing proteins were previously digested for 30 min with proteinase K (20 mg/ml).
Flow cytometry
Murine DC were stained with FITC-labelled anti-CD54, PE-anti-CD11c, PE-Cy5-anti-CD86 and PE-anti-PD-L1 antibodies, whereas for human DC, FITC-labelled anti-CD86 and PE-labelled anti-CD54 antibodies and their corresponding control isotypes (all from BD-Biosciences; San Diego, CA) were used. After 30 min, cells were washed and surface expression of the different molecules was analyzed by using a FACScanto flow cytometer (BD-Biosciences).
DC migration assays
DC migration assays were carried out in 24-well plates with transwell inserts of 5 μm pore size using a Transwell chamber (Costar Corning, Cambridge, MA). DC (2 × 105) harvested one day after CIRP stimulation were cultured in the upper compartment, whereas the lower compartment contained culture medium with or without 30 ng/ml of CCL21 (Peprotech). After 2 hours, inserts were removed and migrated cells were counted as described.45 Results are expressed as chemotactic index, fold increase of migrated cells in the presence vs. absence of CCL21.
DC/T-cell co-cultures
For analyses of DC/T-cell interactions by electron microscopy, 106 murine DC previously incubated for 12 h with antigens (proteins or peptides at 1 μM) were co-cultured on coverslips for 30 min with 5 × 105 murine splenic CD8 T-cells from OT-I mice positively selected (Miltenyi). Next, cells were fixed and processed as described.46 For in vitro T-cell stimulation assays, purified murine CD8 OT-I T-cells mice (104 cells/well) were cultured in 96-well plates with graded numbers of DC previously incubated for 12 h with antigens. After 24 h, supernatants were harvested to measure IFN-γ production and cells pulsed overnight with 0.5 μCi of tritiated thymidine to determine cell proliferation. For human DC, MLR were carried out as described44 by co-cultivating allogeneic lymphocytes (105 cells/well) with graded numbers of monocyte-derived DC previously incubated with CIRP.
Sample processing for Transmission Electron Microscopy (TEM)
For ultrastructural studies, cells from each treatment were adhered to poly-L-lysine-coated coverslips. Then, the samples were processed following the protocol described previously with modifications.46 Briefly, the cells were treated with a mixture of 2% formaldehyde (Ultra Pure EM Grade, Polysciences Inc., Philadelphia, USA) and 2.5% glutaraldehyde (EM Grade, TAAB Laboratories Equipment Ltd., Berks, UK) in PBS for 1 h at room temperature. The cell monolayer on the coverslips was then washed with PBS and distilled water, post-fixed for 45 minutes with 1% osmium tetroxide (TAAB Laboratories Equipment Ltd.) in PBS, washed with distilled water, treated during 45 minutes with 1% aqueous uranyl acetate (Electron Microscopy Sciences, Hatfield, USA), washed again and dehydrated with increasing quantities (50%, 75%, 95% and 100%) of ethanol seccosolv (Merck KGaA, Darmstadt, Germany). The samples were maintained in coverslips throughout the process and finally embedded in epoxy resin 812 (TAAB Laboratories Equipment Ltd.) contained in gelatine capsules (Electron Microscopy Sciences). The epoxy resin was polymerized for 2 d at 60 °C. Resin was detached from the coverslips by successive immersions in liquid nitrogen and hot water. Ultrathin, 70-nm-thick sections were obtained with an Ultracut UCT ultramicrotome (Leica Microsystems), transferred to 200 mesh Nickel EM grids (Gilder, Lincolnshire, UK) and stained with 3% aqueous uranyl acetate (10 minutes) and lead citrate (2 minutes) (Electron Microscopy Science). Sections were visualized on a JEOL JEM 1200 EXII electron microscope operating at 100 kV (JEOL Ltd., Tokyo, Japan).
Cell activation experiments with HEK293 cells
Control LacZ-expressing HEK293 cells or hTLR4-MD2-CD14-transduced cells (5 × 104 cells/well) were cultured in 96 well plates with different protein antigens or LPS. Next day supernatants were harvested and cell activation was determined by measuring IL-8 production.
Analysis by real-time PCR
Total RNA extraction from DC and real-time PCR were performed as described,32 using primers shown in Supplementary Table 1. Results were normalized according to β-actin. The amount of each transcript was expressed by the formula: 2ΔCt (ΔCt = Ct(β-actin)-Ct(gene)).
Cytokine determination by ELISA
Cytokines produced by murine or human DC (IL-12, TNF-α and IL-10), HEK293 cells (IL-8) or CD8 T-cells (IFN-γ) were determined by ELISA sets (BD-Biosciences).
Immunization of mice and analysis of T-cell responses
C57 BL/6 or IFNAR KO mice were immunized s.c. with equimolar amounts (2 nmoles) of CIRP-containing protein antigens, OVA protein, peptides or unbound mixtures of CIRP protein and antigenic peptide. In some experiments, mice received i.p. 500 μg of anti-IL-10R (1B1.3 A; BioXcell), 50 μg of anti-PD-1 (RMP1-14; BioXcell), 100 μg of anti-CTLA4 (9D9; BioXcell) or the corresponding isotype control antibodies (BioXcell) on day 0. In adjuvant combination experiments mice also received Imiquimod cream (Meda Aldara™; topical application; 2.5 mg/mouse), poly(I:C) (Amersham; 50 μg/mouse s.c.), CpG 1668 (Sigma; 50 μg/mouse s.c.), agonistic FGK45.5 anti-CD40 antibodies (BioXcell; 50 μg/mouse s.c.) or a multiple adjuvant combination (MAC)32 containing Imiquimod, poly(I:C) and anti-CD40. One week later animals were sacrificed and T-cell responses were measured by enumerating IFN-γ-producing cells by ELISPOT as described32 using a kit from BD-Biosciences. For these experiments splenocytes were stimulated with 1 μg/ml of OVA(257–264) or left unstimulated.
Tumor treatment experiments
C57 BL/6 mice were injected s.c. with 105 tumor cells (E.G7-OVA, MC38-OVA) or intradermically (B16-OVA cells). One week later, when the tumor diameter was about 5 mm, they were treated for 3 weeks with 2 weekly i.t. injections of CIRP-containing immunogens, CIRP, peptide antigen, the unbound mixture or PBS. In combination experiments, vaccine was accompanied by three weekly i.p. injections of antibodies (500 μg of anti-IL-10R, 100 μg of anti-PD-1, 100 μg of antiCTLA4) or the corresponding isotype control. Tumor volume was calculated according to the formula: V = (length x width2)/2. For depletion experiments, mice received i.p. injections of 200 μg of anti-CD8β (H35.17.2; a kindly gift of Dr. C. Leclerc; Institute Pasteur, Paris, France) or isotype control (BE0088, BioXcell) antibodies on days -1, 0, 1 and 6, being 0 the day when treatment starts. Mice were killed when tumor diameter reached 17 mm.
Statistical analysis
Survival curves of animals treated with different protocols were plotted according to the Kaplan–Meier method and were compared using the log-rank test. Immune responses were analyzed using nonparametric Kruskal-Wallis and Mann-Whitney U tests. P<0.05 was taken to represent statistical significance.
Supplementary Material
Funding Statement
Ministerio de Economía y Competitividad/Insituto de Salud Carlos III (grant number. PI14/00343).
Conflicts of interest
The authors report no conflict of interest.
Acknowledgments
The authors thank the Navarra Blood and Tissue Bank for providing human samples, M. Albert for IFNAR-KO mice, G. Kroemer, I. Melero and C. Leclerc for tumor cell lines and reagents, and Edurne Elizalde for her help with PCR assays.
Financial support
This work was supported by Ministerio de Economia y Competitividad/Instituto de Salud Carlos III (grant PI14/00343), Gobierno de Navarra (PI038), “Murchante contra el cáncer” and Territorios Solidarios-BBVA to PS.
References
- 1.Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–65.doi: 10.1146/annurev.iy.12.040194.002005. PMID:8011285. [DOI] [PubMed] [Google Scholar]
- 2.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al.. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science.2006;313:1960–4.doi: 10.1126/science.1129139. PMID:17008531. [DOI] [PubMed] [Google Scholar]
- 3.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer.2012;12:298–306.doi: 10.1038/nrc3245. PMID:22419253. [DOI] [PubMed] [Google Scholar]
- 4.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature.2014;515:568–71.doi: 10.1038/nature13954. PMID:25428505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al.. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–24.doi: 10.1038/nrclinonc.2014.111. PMID:25001465. [DOI] [PubMed] [Google Scholar]
- 6.Butterfield LH. Cancer vaccines. Bmj. 2015;350:h988.doi: 10.1136/bmj.h988. PMID:25904595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–66. doi: 10.1084/jem.190.3.355. PMID:10430624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curran MA, Allison JP. Tumor vaccines expressing flt3 ligand synergize with ctla-4 blockade to reject preimplanted tumors. Cancer Res. 2009;69:7747–55. doi: 10.1158/0008-5472.CAN-08-3289. PMID:19738077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, et al.. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:501–8.doi: 10.1016/S1470-2045(12)70006-2. PMID:22326924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, et al.. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–8.doi: 10.1200/JCO.2013.51.4802. PMID:24145345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA Jr, Donehower RC, Jaffee EM, et al.. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36:382–9.doi: 10.1097/CJI.0b013e31829fb7a2. PMID:23924790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti L, Morgan C, Sandler AD. Combination of Id2 Knockdown Whole Tumor Cells and Checkpoint Blockade: A Potent Vaccine Strategy in a Mouse Neuroblastoma Model. PLoS One.2015;10:e0129237.doi: 10.1371/journal.pone.0129237. PMID:26079374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemmi H, Akira S. TLR signalling and the function of dendritic cells. Chem Immunol Allergy.2005;86:120–35.doi: 10.1159/000086657. PMID:15976491. [DOI] [PubMed] [Google Scholar]
- 14.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811.doi: 10.1146/annurev.immunol.18.1.767. PMID:10837075. [DOI] [PubMed] [Google Scholar]
- 15.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. PMID:18340345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199:815–24.doi: 10.1084/jem.20032220. PMID:15024047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boscardin SB, Hafalla JC, Masilamani RF, Kamphorst AO, Zebroski HA, Rai U, Morrot A, Zavala F, Steinman RM, Nussenzweig RS, et al.. Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J Exp Med. 2006;203:599–606.doi: 10.1084/jem.20051639. PMID:16505139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, et al.. Differential antigen processing by dendritic cell subsets in vivo. Science.2007;315:107–11.doi: 10.1126/science.1136080. PMID:17204652. [DOI] [PubMed] [Google Scholar]
- 19.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802.doi: 10.1038/nri2173. PMID:17853902. [DOI] [PubMed] [Google Scholar]
- 20.Wille-Reece U, Flynn BJ, Lore K, Koup RA, Kedl RM, Mattapallil JJ, Weiss WR, Roederer M, Seder RA. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc Natl Acad Sci U S A. 2005;102:15190–4.doi: 10.1073/pnas.0507484102. PMID:16219698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, Drijfhout JW, Melief CJ, Overkleeft HS, van der Marel GA, et al.. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem. 2007;282:21145–59.doi: 10.1074/jbc.M701705200. PMID:17462991. [DOI] [PubMed] [Google Scholar]
- 22.Lasarte JJ, Casares N, Gorraiz M, Hervas-Stubbs S, Arribillaga L, Mansilla C, Durantez M, Llopiz D, Sarobe P, Borras-Cuesta F, et al.. The extra domain A from fibronectin targets antigens to TLR4-expressing cells and induces cytotoxic T cell responses in vivo. J Immunol. 2007;178:748–56.doi: 10.4049/jimmunol.178.2.748. PMID:17202335. [DOI] [PubMed] [Google Scholar]
- 23.Xue JH, Nonoguchi K, Fukumoto M, Sato T, Nishiyama H, Higashitsuji H, Itoh K, Fujita J. Effects of ischemia and H2O2 on the cold stress protein CIRP expression in rat neuronal cells. Free Radic Biol Med. 1999;27:1238–44. doi: 10.1016/S0891-5849(99)00158-6. PMID:10641716. [DOI] [PubMed] [Google Scholar]
- 24.Sheikh MS, Carrier F, Papathanasiou MA, Hollander MC, Zhan Q, Yu K, Fornace AJ Jr. Identification of several human homologs of hamster DNA damage-inducible transcripts. Cloning and characterization of a novel UV-inducible cDNA that codes for a putative RNA-binding protein. J Biol Chem. 1997;272:26720–6.doi: 10.1074/jbc.272.42.26720. PMID:9334257. [DOI] [PubMed] [Google Scholar]
- 25.Wellmann S, Buhrer C, Moderegger E, Zelmer A, Kirschner R, Koehne P, Fujita J, Seeger K. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117:1785–94.doi: 10.1242/jcs.01026. PMID:15075239. [DOI] [PubMed] [Google Scholar]
- 26.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, et al.. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–95.doi: 10.1038/nm.3368. PMID:24097189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durantez M, Fayolle C, Casares N, Belsue V, Riezu-Boj JI, Sarobe P, Prieto J, Borras-Cuesta F, Leclerc C, Lasarte JJ. Tumor therapy in mice by using a tumor antigen linked to modulin peptides from Staphylococcus epidermidis. Vaccine. 2010;28:7146–54.doi: 10.1016/j.vaccine.2010.08.070. PMID:20817012. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al.. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science.2003;301:640–3. doi: 10.1126/science.1087262. PMID:12855817. [DOI] [PubMed] [Google Scholar]
- 29.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science.2007;316:1628–32.doi: 10.1126/science.1138963. PMID:17569868. [DOI] [PubMed] [Google Scholar]
- 30.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al.. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003.doi: 10.1084/jem.20101158. PMID:21930769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16.doi: 10.1084/jem.20101159. PMID:21930765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aranda F, Llopiz D, Diaz-Valdes N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martinez M, Durantez M, Mansilla C, Prieto J, et al.. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res. 2011;71:3214–24.doi: 10.1158/0008-5472.CAN-10-3259. PMID:21402711. [DOI] [PubMed] [Google Scholar]
- 33.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al.. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280 A in cancer patients. Nature.2014;515:563–7.doi: 10.1038/nature14011. PMID:25428504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llopiz D, Aranda F, Díaz-Valdés N, Ruiz M, Infante S, Belsúe V, Lasarte JJ, Sarobe P. Vaccine-induced but not tumor-derived Interleukin-10 dictates the efficacy of Interleukin-10 blockade in therapeutic vaccination. Oncoimmunology.2016;5:e1075113. doi: 10.1080/2162402X.2015.1075113. PMID:27057445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139–45.doi: 10.1158/0008-5472.CAN-15-0255. PMID:25977340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zom GG, Khan S, Filippov DV, Ossendorp F. TLR ligand-peptide conjugate vaccines: toward clinical application. Adv Immunol. 2012;114:177–201. doi: 10.1016/B978-0-12-396548-6.00007-X. PMID:22449782. [DOI] [PubMed] [Google Scholar]
- 37.Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS. Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 2005;73:2940–50. doi: 10.1128/IAI.73.5.2940-2950.2005. PMID:15845500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT. Selective TRIF-dependent signaling by a synthetic toll-like receptor 4 agonist. Sci Signal. 2012;5:ra13.doi: 10.1126/scisignal.2001963. PMID:22337809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell.2015;28:690–714. doi: 10.1016/j.ccell.2015.10.012. PMID:26678337. [DOI] [PubMed] [Google Scholar]
- 40.Fu C, Liang X, Cui W, Ober-Blobaum JL, Vazzana J, Shrikant PA, Lee KP, Clausen BE, Mellman I, Jiang A. beta-Catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc Natl Acad Sci U S A.2015;112:2823–8.doi: 10.1073/pnas.1414167112. PMID:25730849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulko V, Liu X, Krco CJ, Harris KJ, Frigola X, Kwon ED, Dong H. TLR3-stimulated dendritic cells up-regulate B7-H1 expression and influence the magnitude of CD8 T cell responses to tumor vaccination. J Immunol. 2009;183:3634–41.doi: 10.4049/jimmunol.0900974. PMID:19710456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, Eysmans C, Richards A, Schell MJ, Fisher KJ, et al.. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21:712–20.doi: 10.1158/1078-0432.CCR-14-2468. PMID:25524312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zabaleta A, Llopiz D, Arribillaga L, Silva L, Riezu-Boj JI, Lasarte JJ, Borras-Cuesta F, Prieto J, Sarobe P. Vaccination against hepatitis C virus with dendritic cells transduced with an adenovirus encoding NS3 protein. Mol Ther. 2008;16:210–7.doi: 10.1038/sj.mt.6300333. PMID:17923840. [DOI] [PubMed] [Google Scholar]
- 44.Echeverria I, Pereboev A, Silva L, Zabaleta A, Riezu-Boj JI, Bes M, Cubero M, Borras-Cuesta F, Lasarte JJ, Esteban JI, et al.. Enhanced T cell responses against hepatitis C virus by ex vivo targeting of adenoviral particles to dendritic cells. Hepatology.2011;54:28–37.doi: 10.1002/hep.24325. PMID:21452282. [DOI] [PubMed] [Google Scholar]
- 45.Riezu-Boj JI, Larrea E, Aldabe R, Guembe L, Casares N, Galeano E, Echeverria I, Sarobe P, Herrero I, Sangro B, et al.. Hepatitis C virus induces the expression of CCL17 and CCL22 chemokines that attract regulatory T cells to the site of infection. J Hepatol.2011;54:422–31.doi: 10.1016/j.jhep.2010.07.014. PMID:21129807. [DOI] [PubMed] [Google Scholar]
- 46.Chiappi M, Conesa JJ, Pereiro E, Sorzano CO, Rodriguez MJ, Henzler K, Schneider G, Chichon FJ, Carrascosa JL. Cryo-soft X-ray tomography as a quantitative three-dimensional tool to model nanoparticle:cell interaction. J Nanobiotechnology.2016;14:15.doi: 10.1186/s12951-016-0170-4. PMID:26939942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.