

ABSTRACT
Anti-PD-1 immunotherapy is the standard of care for treating many patients with non-small cell lung cancer (NSCLC), yet mechanisms of treatment failure are emerging. We present a case of NSCLC, who rapidly progressed during a trial (NCT02318771) combining palliative radiotherapy and pembrolizumab. Planned tumor biopsy demonstrated PD-1 expression by NSCLC cells. We validated this observation by detecting PD-1 transcript in lung cancer cells and by co-localizing PD-1 and lung cancer-specific markers in resected lung cancer tissues. We further investigated the biological role of cancer-intrinsic PD-1 in a mouse lung cancer cell line, M109. Knockout or antibody blockade of PD-1 enhanced M109 viability in-vitro, while PD-1 overexpression and exposure to recombinant PD-L1 diminished viability. PD-1 blockade accelerated growth of M109-xenograft tumors with increased proliferation and decreased apoptosis in immune-deficient mice. This represents a first-time report of NSCLC-intrinsic PD-1 expression and a potential mechanism by which PD-1 blockade may promote cancer growth.
KEYWORDS: PD-1, Therapeutic Resistance, Immunotherapy, Lung Cancer
Introduction
Programmed death-1 (PD-1) and programmed death-ligand 1(PD-L1) immuno-checkpoint inhibitors represent one of the most significant breakthroughs in the treatment of advanced malignancies. Clinical trials demonstrating improved, durable response with anti-PD-1 or anti-PD-L1 drugs have led to FDA approval in their treatment of advanced melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), head and neck cancer, and refractory Hodgkin's lymphoma, to name a few.1–6 Despite their successes, response rates to PD-1/PD-L1 inhibitors hover around 20–40%, and attempts to reliably predict a response to these drugs have been largely imprecise despite widespread use of PD-L1 as a biomarker for favorable response.7,8,9,10 Indeed, several retrospective studies have even observed a subset of patients who experience unusually rapid progression upon initiation of PD-1/PD-L1 axis blockade, a phenomenon referred to as hyperprogression.11–14
PD-1 is a transmembrane protein receptor, found on T-, B-, natural killer cells, and monocytes. Ligation of PD-1 to its ligand on cancer cells, PD-L1, induces apoptosis, cell cycle arrest, and anergy.7,15,16 Interestingly, PD-1 expression has also been detected on melanoma-initiating stem cells.17 In a series of novel experiments, Kleffel et al. recently demonstrated that PD-1 expressed by melanoma cells has intrinsic tumor-promoting effects and that treatment with anti-PD-1 mAb (a-PD-1) suppressed this effect.18
Here, we present a first-time report of cancer-intrinsic PD-1 in a patient with NSCLC whose cancer rapidly progressed upon initiation of anti-PD-1 therapy. We present data on a PD-1 expressing murine NSCLC cell line that implicates the blockade of cancer-intrinsic PD-1 as a mechanism by which cancer survival may be enhanced in-vitro and in-vivo.
Results & discussion
Rapid progression in a patient with nsclc treated with pembrolizumab
A lung cancer patient who failed several chemotherapy regimens experienced rapid progression after being enrolled on clinical trial NCT02318771, a Phase I study of radiotherapy (RT) plus pembrolizumab for metastatic or recurrent head and neck cancer, RCC, or NSCLC. The trial design is outlined in Fig. 1A. This was a 61-year old female with 35 pack-year smoking history who presented with Stage IV T2N2M1 adenocarcinoma of the lung without EGFR mutations or ALK-translocation in May 2013. Between May 2013 and February 2015, the patient had progressed on pemetrexed and carboplatin, docetaxel, navelabine, and gemcitabine. Additionally, the patient was treated with whole-brain RT in October 2014 and palliative RT to a supraclavicular metastasis in November 2014. In March 2015, the patient was randomized to arm A1 on clinical trial NCT02318771. Per study protocol (Fig. 1A), a posterior left lower lobe lung lesion, designated on axial CT in Fig. 1B, was biopsied; treated with 8 Gy in one fraction; and then re-biopsied five days after radiation. The patient was then started on 200 mg of pembrolizumab once every three weeks (Fig. 1A). After just three cycles of pembrolizumab, clinical progression was noted from right bronchus obstruction, multiple new pulmonary nodules, and interval increase in intracranial lesions. Notably, the lesion treated with RT had nearly doubled in size in just two months.
Figure 1.
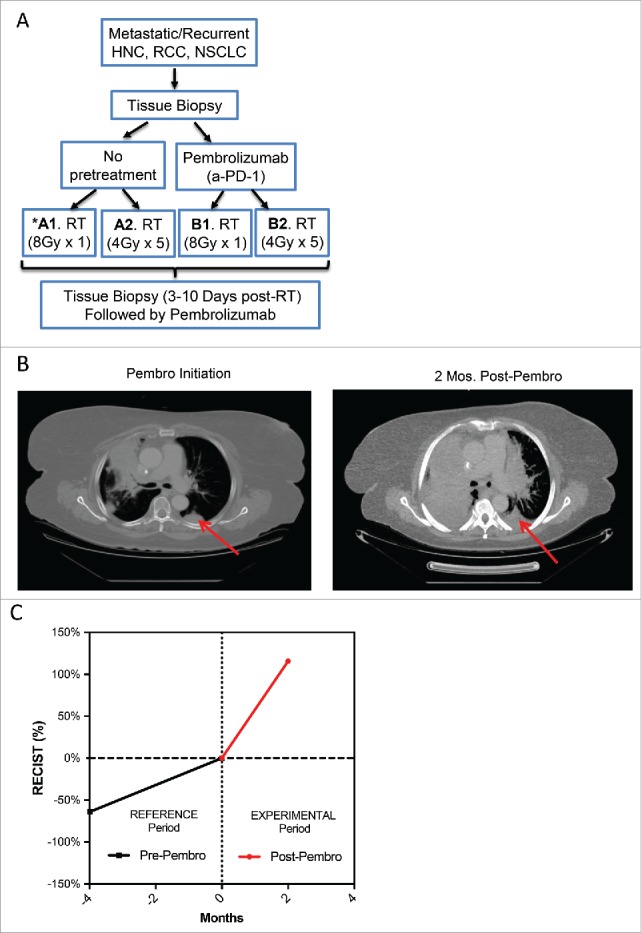
Rapid Progression in a Patient with NSCLC Treated with Pembrolizumab. A patient with NSCLC was enrolled on a clinical trial of RT and pembrolizumab. (A) An outline of the study schema is shown. The patient was randomized to Arm A1 and received 8 Gy in 1 fraction followed by pembrolizumab. (B) CT scans of the chest are shown (left) at the time of pembrolizumab initiation and (right) at two months after pembrolizumab initiation, at which time clinical progression was noted. The red arrow indicates the target lesion, which was treated with 8 Gy in one fraction. (C) The sum of the diameters of RECIST target lesions are plotted in reference to their sum at the time of pembrolizumab initiation. The reference and experimental time periods are the time periods before and after pembrolizumab therapy, respectively. The increase in tumor growth following pembrolizumab corresponded to a 2.4-fold increase in tumor growth rate, confirming hyperprogression.11
Several retrospective series have described hyperprogression after the initiation of anti-PD-1 therapy, defined by a two-fold increase in tumor growth rate (TGR) during the experimental period of therapy compared with a reference period before PD-1/PD-L1 blockade, as determined by RECIST 1.1 criteria.11–14 We calculated the TGRs during the experimental and reference periods as previously described,11 using the sum of the diameter of non-irradiated RECIST 1.1 target lesions. This sum, at multiple time points, is expressed as a percentage of their size immediately prior to the pembrolizumab initiation in Fig. 1C. During just three cycles of pembrolizumab, this patient's RECIST target lesions had grown by 114% (Fig. 1C), corresponding to a 2.4-fold increase in TGR after pembrolizumab.
NSCLC cancer-intrinsic PD-1 expression
PD-1-expressing tumor-infiltrating lymphocytes (TILs) as well as PD-L1-expressing tumor cells are relative, though imperfect predictors of favorable response to anti-PD-1 or PD-L1 therapy.1,2,19–21 As a part of planned biomarker study under this clinical trial, biopsies of the irradiated target lesion, obtained after RT and prior to pembrolizumab, were assessed for PD-1-expressing TILs and tumor PD-L1 expression with PD-L1, CD45, and PD-1 immunohistochemistry stains. Although the tumor cells avidly expressed PD-L1 (Fig. 2A), there were no detectable CD45 positive cells. The absence of TILs predicts lack of efficacy from anti-PD-1 therapy.21 Unexpectedly, cancer cells were stained diffusely positive for PD-1, indicating cancer-intrinsic PD-1 expression, a finding only previously reported in melanoma (Fig. 2A).17,18 Notably, we recently reported a case of renal cell carcinoma treated on the arm A2 of the same clinical trial, who experienced a similar clinical course.20 Similar to the NSCLC case, the tumor tissue was void of TILs with high PD-L1 expression as previously reported.20 It, as well, stained diffusely positive for PD-1 (Supplementary Data, Fig. 1.) To date, cancer-intrinsic PD-1 expression has only been described in melanoma, and its biologic role has never been studied in NSCLC.17,18 Following this finding of PD-1 expression in the NSCLC tumor, we sought to validate NSCLC-intrinsic PD-1 expression and establish an experimental model to assess its biologic role.
Figure 2.
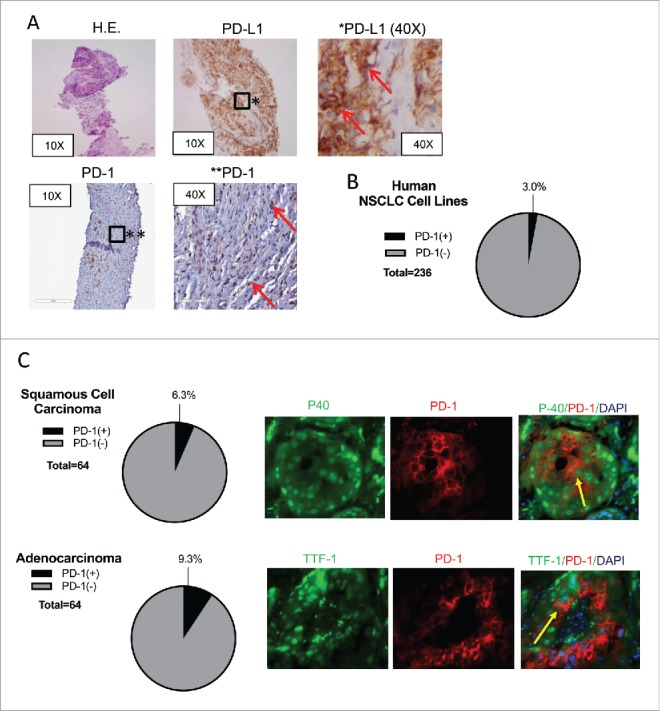
Human NSCLC Expresses PD-1. (A) Tumor biopsy of the target lesion stained with H.E., anti-PD-L1, and anti-PD-1 is shown at low and high power magnification. Red arrows designate cells staining positive for PD-L1 or PD-1. (B) The EMBL-EBI Expression Atlas was queried for human NSCLC cell lines that express PD-1 and RNA-sequencing data were found from four different experiments. The relative percentage of PD-1 expressing cell lines is shown in the pie chart in black. (C) Tissue microarray slides from 128 resected NSCLC specimens (64 squamous cell carcinoma and 64 adenocarcinoma) assessed for coexpression of PD-1 and P40 and TTF-1, respectively are shown. Yellow arrows designate cells expressing PD-1 and P40 or TTF-1.
PD-1 expression among human NSCLC
To validate our findings of cancer-intrinsic PD-1 expression in human NSCLC, we queried the EMBL-EBI Expression Atlas for human NSCLC cell lines in culture rather than lung cancer tissues, which are likely to be infiltrated with PD-1-expressing lymphocytes.22 Four RNA-sequenced data sets were available, which showed 3% of sequenced lung cancer cell lines (7 of 236) expressing detectable (>0.5 FPKM) PD-1 transcript (Fig. 2B). To establish PD-1 expression in lung cancer tissue, tissue microarray slides that consist of 64 human squamous cell carcinoma and 64 human adenocarcinoma were analyzed by immunofluorescence for simultaneous expression of PD-1 and either P40 or TTF-1, respectively. Dual fluorescence was observed in four (6.3%) of the squamous cell carcinoma and 6 (9.3%) of the adenocarcinoma samples (Fig. 2C).
Collectively, this represents the first report of PD-1 expression in human NSCLC. Should these data be representative of larger cohorts of patients, a smaller, yet significant fraction of NSCLC would express PD-1 in comparison to melanoma.18 However, T cells only express PD-1 upon cytokine stimulation and activation, while their naïve counterparts lack PD-1 expression.23 As it relates to the NSCLC case in Fig. 1, it is possible that the radiation prior to pembrolizumab increased type I interferon production by either tumor cells or immune cells,24 which, consequently, may have increased PD-1 expression on tumor cells as it does in T cells.23 Future studies should examine whether cancer cells are subject to the same interferon-mediated induction of PD-1 expression as T cells.23
Murine NSCLC cell line, M109, expresses PD-1
To identify a mouse model to study the biological role of NSCLC-intrinsic PD-1, we interrogated murine NSCLC lines for PD-1 expression by RT-PCR (Fig. 3A). PD-1 transcript was only detected in M109 cell line, and its corresponding protein was detected by immunofluorescence (Fig. 3A) and by flow cytometry (data not shown). To assess whether PD-1 is expressed by M109 in-vivo, M109 tumors were grown in PD-1 knockout mice to eliminate any infiltrating PD-1 positive immune cells from the host. As shown, tumor-expressed PD-1 was again detected in tissue by immunofluorescence (Fig. 3A).
Figure 3.
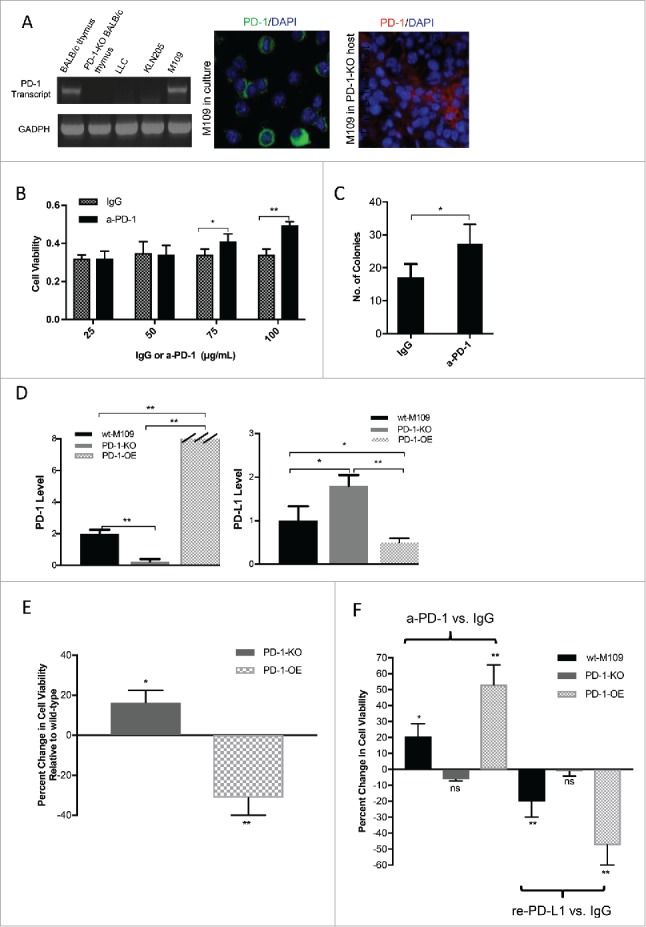
Increased Cell Viability of M109 following PD-1 Blockade in-vitro. (A) Three murine NSCLC cell lines were interrogated for PD-1 transcript by RT-PCR, as shown. Thymic tissue from wild-type and PD-1-knockout BALB/c mice served as positive and negative controls. PD-1 expression by the M109 cell line was assessed in cell culture (green) and in-vivo in a PD-1 knockout BALB/c mouse (red). (B) Increasing doses of a-PD-1 (25, 50, 75, or 100 μg/mL) or IgG isotype control were added to M109 cells, and viability was measured by MTT assay. (C) 100 μg/mL of a-PD-1 or IgG was added to M109 cells, and effects on cell survival and clonogenicity were measured by clonogenic assay. (D) Relative PD-1 and PD-L1 transcript levels determined by qRT-PCR between the wt-M109, PD-1 knockout (KO) and PD-1-overexpressing (OE) are shown. GADPH served as the internal control. (E) MTT assays assess the viability of the PD-1-KO and PD-1-OE cell lines in reference to the wt-M109 cell line. (F) MTT viability assays were performed after the addition of 100 μg/mL a-PD-1 vs. isotype control (left) or 5 μg/mL recombinant PD-L1 vs. isotype control (right) to the three cell lines are shown. Changes in viability from these assays are plotted in reference to viability after the addition of isotype control. All values are listed as mean +/- SD. (*p<0.05, **p<0.001, unpaired t-test).
PD-1/PD-L1 mediates cell viability of M109 in-vitro
To determine the role of PD-1 expressed by M109 cells, we assessed cell viability of M109 cells by MTT assay in the presence of increasing doses of anti-PD-1 antibody or IgG isotype control. a-PD-1 increased M109 cell viability in a dose-dependent manner, as shown in Fig. 3B. To validate the findings of this assay, we performed clonogenic assays with M109 cells in the presence of 100 μg/mL of a-PD-1 or isotype control. Increased clonogenicity was detected in cells treated with a-PD-1 (Fig. 3C).
We next generated two M109 cell line variants: a PD-1 knockout (KO) cell line by the CRIPSR-cas9 technique and a PD-1 overexpressing (OE) cell line using a lentiviral particle transfection system. The triplet cell model was assessed for PD-1 transcript by RT-PCR to confirm successful manipulation of PD-1 expression. An abundance of PD-1 transcript was detected in the PD-1-OE cell line, while PD-1 was barely detectable in the PD-1-KO cell line, confirming successful knockout (Fig. 3D). To completely characterize the PD-1/PD-L1 axis, we assessed the three cell lines for PD-L1 expression by RT-PCR. All three cell lines were found to express PD-L1 as well, though there appeared to be an inverse relationship between PD-1 and PD-L1 expression (Fig. 3D).
To assess whether manipulation of PD-1 expression affected viability, MTT assays comparing the three cell lines were performed. PD-1-KO cells were more viable relative to the wt-M109 cell line, and PD-1-OE cells were markedly less viable (Fig. 3E). To study the effects of PD-1 blockade on the triplet cell model in-vitro, we performed MTT assays after the addition of a-PD-1 or isotype control. a-PD-1 increased viability of the wt (P<0.05) and PD-1-OE cells (P<0.001) (Fig. 3F). However, PD-1-KO cells were unaffected, confirming M109 PD-1 as the target of a-PD-1-mediated increases in cell viability. To determine the effects of PD-1 activation on cell viability, MTT assays were performed after the addition of recombinant PD-L1 or IgG in serum-free media in each of the three cell lines. PD-L1 decreased viability of the wt (P<0.05) and, to a greater degree, the PD-1-OE cell line (P<0.001), while the PD-1-KO cell line was unaltered, as shown in Fig. 3F. Collectively, these data suggest that PD-1 axis activation decreases cancer cell survival, similar to the biological functions of PD-1 well-characterized in T-cells.1,7,21,25
The biological significance of tumor-expressed PD-1 in-vivo following anti-PD-1 therapy
Tumors may acquire resistance to PD-1/PD-L1 blockade by upregulating of alternative immunosuppressive pathways that disable anti-tumor immunity regardless of PD-1/PD-L1 interactions.2 As such, the degree to which TILs populate the pre-treatment TME is directly correlated with response to PD-1 blockade.21 To determine the effects of a-PD-1 in a model comparable to the aforementioned case in which the direct effects of a-PD-1 on cancer cells could be isolated, wt-M109 tumors grown in NSG mice were treated with a-PD-1 or isotype control. Similar to our results in-vitro, tumors treated with a-PD-1 were significantly larger at the end of the experiment (Fig. 4A). M109 xenografts were collected and analyzed for proliferative and apoptotic markers by immunohistochemistry. Tumors treated with a-PD-1 displayed increased staining for Ki-67 and decreased staining for cleaved caspace-3, indicating increased proliferation and decreased apoptosis analogous to the effects of PD-1 blockade in T-cells (Fig. 4B).1,7,21,25 In a separate experiment, we grew wt-M109 tumors in PD-1-KO BALB/c mice to determine whether the observed increases in tumor growth following PD-1 blockade could be sustained in an immunocompetent host without changing T-cell activation as the TILs are PD-1 negative. Tumors treated with a-PD-1 again were larger after three weeks. Further characterization of this model is underway.
Figure 4.
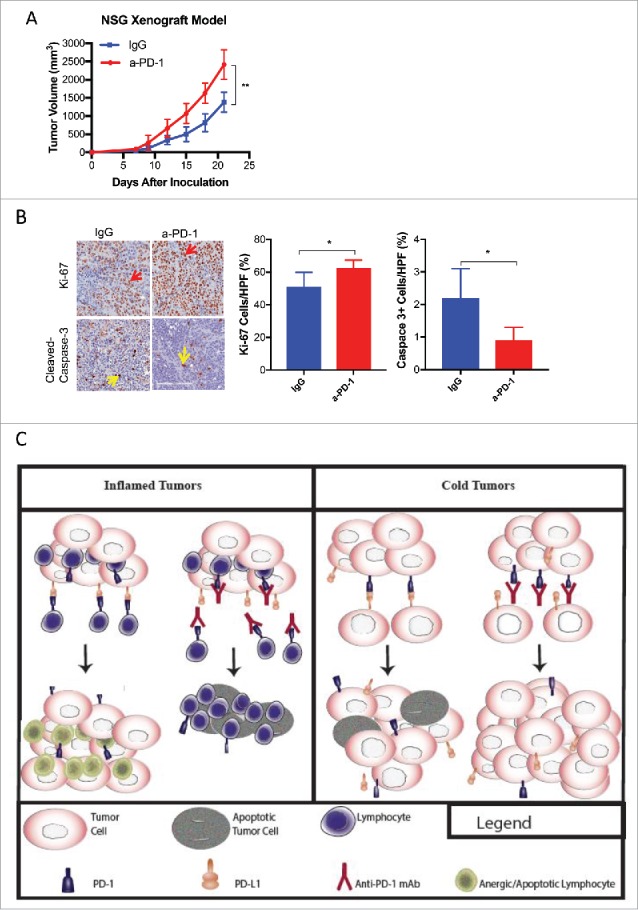
The Effects of Anti-PD-1 on M109 in-vivo. (A) A growth curve of subcutaneous wt-M109 tumors grown in NSG mice treated with a-PD-1 (N = 6) or isotype control (N = 6) is shown. (B) Tumors were harvested from sacrificed mice and assessed for Ki-67 and cleaved-caspase-3 by immunohistochemistry, as shown. Average percentages of Ki-67(+) and Cleaved-caspase-3(+) cells were calculated per high power field (HPF). All values are listed as mean +/- SD (*p<0.05, **p<0.001, unpaired t-test). Statistical differences between two groups were determined by the unpaired t-test. (C) An illustration showing the mechanism by blockade of NSCLC-intrinsic PD-1 may contribute to therapeutic resistance or even promote growth is shown. The left panel of the illustration summarizes prior understanding of PD-1/PD-L1 inhibition in tumors void of PD-1 expression and illustrates the effect of PD-1 blockade in inflamed tumors that do not express PD-1. T-lymphocytes proliferate and generate an anti-tumor immune response. PD-1, expressed by T- cells is activated by PD-L1 on tumor cells, causing T-cell anergy and apoptosis. The right panel illustration demonstrates the effect of anti-PD-1 therapy in a non-inflamed, or cold, tumor that expresses PD-1. Blockade the growth suppressive interaction between PD-1/PD-L1 results in increased viability.
Several studies have recently described a “tumor flare” or hyperprogression, associated with PD-1/PD-L1 axis blockade that is distinct from pseudoprogression in some patients.11–14 Interestingly, in Checkmate 057, a phase III randomized control trial comparing the PD-1 inhibitor nivolumab to docetaxel in NSCLC, the progression free survival curve favored docetaxel for the first six months, leading some to speculate that early hyperprogression as the cause.9,11 The notion of anti-PD-1 therapy promoting tumor progression in some patients has been limited by simplification of tumor growth kinetics, lack of a total washout period of prior therapy in some patients, assumptions of a total lack of efficacy of prior therapy, and a lack of mechanistic explanation. Our study, however, suggests blockade of cancer-intrinsic PD-1 can release NSCLC from anti-survival effects of its interaction with its ligand, providing the first biologic proof of principle that PD-1 blockade may, albeit rarely, enhance cancer viability.
Importantly, we are unable to definitively prove that pembrolizumab directly induced hyperprogression in the NSCLC case. Nevertheless, we uncovered a biologic role of NSCLC-intrinsic PD-1 that parallels this patient's clinical course. The biopsy from the NSCLC case and the lymphocyte-deplete NSG tumors were void of TILs, both in contrast to inflamed tumors likely to favorably respond to PD-1 blockade, as illustrated in Fig. 4C.21 Both the M109 cell line and the tumor tissue from the NSCLC case simultaneously expressed PD-1 and PD-L1. Given the growth-suppressive effects of PD-1 activation seen in M109 cells, we hypothesize that inhibition of this signaling accounts for the increased viability observed with PD-1 blockade (Fig. 4C), which is supported by the changes in apoptotic and proliferative markers in the NSG model. Moreover, hyperprogression may be a result of two independent, co-occurring events: cancer-intrinsic PD-1 expression and inherent resistance to PD-1 blockade (i.e., depletion of TILs in the tumor microenvironment). In the absence of this resistance, perhaps the blockade NSCLC-intrinsic PD-1 serves to only partially mitigate the efficacy of anti-PD-1 immunotherapy. Regardless, the relevance of NSCLC-intrinsic PD-1 in predicting responses to PD-1 inhibitors must be further validated in large cohorts of patients.
While our data indicate that inactivation of PD-1 with a-PD-1 has diverse effects on cell death and growth (Fig. 4B), it remains unanswered what precise downstream mechanisms contribute to this. One possibility is that PD-1 activation upregulates pro-apoptotic proteins, including BIM, as it does in CD8+ T cells.16,26 In human CD4+ T cells, PD-1 activation impedes cell cycle progression at the G1-S checkpoint through multiple complex mechanisms, including the upregulation of the G1 phase inhibitor p15INK4 and indirectly increasing inhibition of cyclin-dependent kinase 2.15 Further studies are needed to determine whether increases in cancer cell survival in-vitro and in-vivo are related to cycle progression or altering apoptotic regulation.
Possibly of more importance, however, will be determining the complex consequences of the phosphatases that interact with activated PD-1, which may account for the apparent duality in the role of cancer-intrinsic PD-1 signaling in our study compared to that of Kleffel et al. They reported suppression, rather than promotion, of growth with PD-1 blockade in PD-1-expressing melanoma.18 The SHP1 and SHP2 phosphatases are the mediators of PD-1 signaling, leading to T-cell anergy.27 Kleffel et. al. hypothesized that the differential effects of PD-1 blockade on T-lymphocytes vs. melanoma were due to the differences in SHP2 signaling (mostly oncogenic in cancer models) in two cell types.18 Though largely recognized as an oncogenic phosphatase, SHP2 does not uniformly function as a proto-oncogene across all cancer histologic types or mutational landscapes.28–30 EGFR mutations, for example, sequester SHP2 and prevent its ability to promote downstream Erk activation.31 Contrary to SHP2, SHP1, also activated upon PD-1/PD-L1 ligation in both T cells and neurons,32 functions as a tumor suppressor by degrading JAK kinases33 and dephosphorylating STAT3.34 Therefore, the partnering phosphatase following PD-1 activation may vary, leading to either pro-survival or death signaling in PD-1-expressing cancer cells and variable biological consequences of PD-1 blockade.
In conclusion, this study provides the first report of PD-1 expression in NSCLC. It reveals a mechanism by which anti-PD-1 therapy will be rendered less efficacious or even deleterious. Further studies are needed to elucidate the mechanism behind which PD-1 blockade can promote tumor growth and to assess the role of intrinsic tumor expression in larger cohorts of patients treated with PD-1/PD-L1 inhibitors.
Methods
Patient data and informed consent
We report data on two patients enrolled on clinical trial NCT02318771, a phase I exploratory study to investigate the immunomodulatory activity of radiation therapy (RT) in combination with MK-3475 (pembrolizumab) in patients with recurrent/metastatic head and neck, renal cell cancer, melanoma and lung cancer. This study was approved by the institutional review board at Thomas Jefferson University, and informed written consent was obtained prior to randomization.
EMBL-EBI expression atlas
The EMBL-EBI Expression Atlas website (https://www.ebi.ac.uk/gxa/home) was queried for PD-1 gene transcript in NSCLC as determined by RNA sequencing by searching for the PDCD1 gene in Homo Sapiens, using sample properties keyword “LUNG.22” The expression cutoff for PDCD1 in the EMBL-EBI expression atlas was 0.5 FPKM.
Tissue microarray
Human NSCLC (64 adenocarcinoma and 64 squamous cell carcinoma) tissue microarray (TMA) slides were purchased from US BioMax, Inc. TMA slides were deparaffinized and rehydrated followed by antigen retrieval, where slides were immersed in Antigen Unmasking Solution (H-3300, Vector Laboratories). The sections were blocked in 0.5% casein in PBS for 1 hour at room temperature then incubated with primary antibodies overnight at 4°C. Slides were washed and incubated with appropriate fluorochrome-conjugated secondary antibodies for 1 hour, washed, and nuclei were counterstained with DAPI (Sigma). Images were acquired using a Nikon Ae1R with High Speed Resonant Scanner Confocal Microscopy.
Cell culture
The Madison 109 (M109) tumor cells were grown in RPMI 1640 Medium (ThermoFisher, #11875-093) supplemented with 10% FBS Medium (ThermoFisher Scientific) under standard cell culture conditions. Cell lines were maintained in humidified incubators with 5% CO2 at 37°C. EL4 murine T lymphocytes were obtained from American Type Culture Collection.
RT-PCR
Total RNA was isolated from murine NSCLC cell lines, murine PD-1(+/+) or PD-1(-/-) thymus using RNeasy Plus Mini Kit® (Qiagen, #74136). Standard cDNA synthesis reactions were carried out using the Superscript III FirstStrand Synthesis System® for RT-PCR (Invitrogen, #18080051). reverse transcribed products were amplified with the Platinum PCR SuperMix High Fidelity Kit (Invitrogen, #12532024). Full-length PD-1 and PD-L1 were amplified and sequenced following reverse transcription of total mRNA using specific primer pairs (PD-1: forward: 5′-ATGCAGATCCCACAGGCGCC-3′ and reverse: 5′-TCAGAGGGGCCAAGAGCAGTG-3′; PD-L1: forward: 5′-TTGCTACGGGCGTTTACTATC-3′ and reverse: 5′-TCCCGTTCTACAGGGAATCT-3′; Beta-actin: forward 5′-TCCTTCGTTGCCGGTCCACCA-3′ and reverse 5′-ACCAGCGCAGCGATATCGTCTC-3′). Murine GADPH served as the loading control. Samples were assayed as described by Kleffel et. al.18
Cell viability assay
MTT assays were performed as previously described.35 In brief, cells were seeded into 96-well plates with 2,500 cells per well, 12 replicates per group, in 1640 RPMI medium (Sigma-Aldrich). Anti-mouse PD-1 (Bio XCell, #BE0146) or IgG were added 24 hours after implantation. For the recombinant PD-L1 assay, after 24 hours, cells were washed with PBS three times and then replaced with serum replacement medium (SRM) with 5 μg/mL of recombinant IgG (R&D Systems), or SRM with 5 μg/mL recombinant mouse PD-L1 (R&D Systems, #Q9NZQ7). After 72 hours, cells were incubated with MTT (Sigma-Aldrich, #57360-69-7). The absorbance was measured at wavelength of 570 nm with a Flexstation 3 Molecular Device (Sunnydale, CA). At the time of collection, OD was between 0.25 and 0.80.
Clonogenic assay
Exponentially growing M109 cells were seeded in 60-mm dishes and treated with 100 μg/mL anti-PD-1 antibody or IgG with three replicates per group. Cells were incubated in CO2 at 37°C, and colony formation was measured as previously described after adequate growth.36
Immunohistochemistry and immunofluorescence
Immunohistochemistry analysis of PD-1 expression in tumor biopsies obtained from the NSCLC case as well as Ki-67 and Cleaved-Caspase 3 were done as described previously.37 For immunofluorescence, sections were fixed using 2% paraformaldehyde for 20 minutes at room temperature. Specimens were blocked with the supernatant of 0.5% casein/phosphate-buffered saline, stirred for 1 hour, and incubated with unconjugated goat anti-mouse PD-1(Abcam, #Ab137132), anti-human PD-1(R&D Systems, #AF-1086), or anti-human CD45 (R&D Systems, #MAB-1430) overnight at 4°C followed by washes and incubation with FITC labeled anti-goat secondary antibodies (Molecular Probes, #A-11034) for 1 hour at room temperature. Specimens were counterstained with DAPI, washed with PBS-Tween20 0.1%, mounted with Vectashield mounting medium (Vector Labs, #H-1000), and imaged using a 40× objective with Nikon C2 Microscope (Molecular Devices, Inc.).
Generation of M109 PD-1 knockout and overexpressing cell lines
Stable PD-1 knockout (KO) M109 lines were generated using the CRISPR/Cas9 system from Santa Cruz Biotechnology, murine PDCD1 Double Nickase Plasmid (#sc-422150-NIC) or Control CRISPR/Cas9 Plasmid (#sc-418922) using lipofectamine transfection agent (Thermofisher, #11668019) and 1 μg/mL puromycin (Thermofisher, #A113802) for selection, as described by Shlyakhtina et al.38 Effective knockout of PD-1 was confirmed by qRT-PCR.
PD-1-OE cells were generated by transfecting M109 cells with a PD-1-overexpression mGFP-tagged lentivector particles purchased from Origene (#MR227347L2 V) according to the manufacturer's protocol using polybrene (Origene) to enhance transfection. Stably transduced cells were propagated and flow-sorted for PD-1-overexpressing subpopulations. Cells were stored in liquid nitrogen and re-cultured at the time of MTT assay, and qRT-PCR was used to confirm PD-1 overexpression.
M109 mouse models
All in-vivo experiments were carried out in accordance with the Thomas Jefferson University Institutional Animal Care & Use Committee. 250 μg of anti-PD-1 antibody (Bio XCell, #BE0146) or IgG control were given by IP three days before tumor inoculation to NSG mice. 0.5× 105 wt-M109 cells were inoculated subcutaneously on the right flank of the twelve mice (six per group), and twice weekly IP treatments with anti-PD-1 mAb or IgG were given thereafter.
Statistics
Statistical comparisons were performed using the unpaired t-test. Data are shown as the mean ± SD. Statistically significant differences were designated by P-value <0.05. All reported P-values are two-sided. GraphPad Prism 5 (GraphPad Software Inc.) was used for statistical analyses.
Supplementary Material
Funding Statement
This work was supported by NCI 1R21CA178229-01 and DOD LC150118.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015;27(1):39–46. doi: 10.1093/intimm/dxu095. PMID:25323844. [DOI] [PubMed] [Google Scholar]
- 2.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23. doi: 10.1016/j.cell.2017.01.017. PMID:28187290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH, Balmanoukian A, Loriot Y, et al.. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet North Am Ed. 2016;387(10031):1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, Radford J, Ribrag V, Molin D, Vassilakopoulos TP, et al.. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J Clin Oncol. 2017;35(19):2125–32. doi: 10.1200/JCO.2016.72.1316. PMID:28441111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow LQ, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, Berger R, Eder JP, Burtness B, Lee SH, et al.. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J Clin Oncol. 2016;34(32):3838–45. doi: 10.1200/JCO.2016.68.1478. PMID:27646946.28081914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plimack ER, Bellmunt J, Gupta S, Berger R, Chow LQM, Juco J, Lunceford J, Saraf S, Perini RF, O'Donnell PH. Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): a non-randomised, open-label, phase 1b study. Lancet Oncol. 2017;18(2):212–20. doi: 10.1016/S1470-2045(17)30007-4. PMID:28081914. [DOI] [PubMed] [Google Scholar]
- 7.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015;5(1):13110. doi: 10.1038/srep13110. PMID:26279307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gridelli C, Ardizzoni A, Barberis M, Cappuzzo F, Casaluce F, Danesi R, Troncone G, De Marinis F. Predictive biomarkers of immunotherapy for non-small cell lung cancer: results from an Experts Panel Meeting of the Italian Association of Thoracic Oncology. Transl Lung Cancer Res. 2017;6(3):373–86. doi: 10.21037/tlcr.2017.05.09. PMID:28713682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al.. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. PMID:26412456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharon E. Can an immune checkpoint inhibitor (sometimes) make things worse? Clin Cancer Res. 2017;23(8):1879–81. doi: 10.1158/1078-0432.CCR-16-2926. PMID:28258060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria J-C, et al.. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res. 2017;23(8):1920–8. doi: 10.1158/1078-0432.CCR-16-1741. PMID:27827313. [DOI] [PubMed] [Google Scholar]
- 12.Saâda-Bouzid E, Defaucheux C, Karabajakian A, Palomar Coloma V, Servois V, Paoletti X, Even C, Fayette J, Guigay J, Loirat D, et al.. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2017;28(7):605–1611. doi: 10.1093/annonc/mdx178. PMID:27646946.28351930 [DOI] [PubMed] [Google Scholar]
- 13.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res. 2017;23(15):4242–50. doi: 10.1158/1078-0432.CCR-16-3133. PMID:28351930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss GJ, Beck J, Braun DP, Bornemann-Kolatzki K, Barilla H, Cubello R, Quan W, Sangal A, Khemka V, Waypa J, et al.. Tumor Cell-Free DNA Copy Number Instability Predicts Therapeutic Response to Immunotherapy. Clin Cancer Res. 2017;28(7);605–1611. doi: 10.1158/1078-0432.CCR-17-0231. PMID:28320758.22740686 [DOI] [PubMed] [Google Scholar]
- 15.Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5(230):ra46. doi: 10.1126/scisignal.2002796. PMID:22740686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larrubia JR, Benito-Martínez S, Miquel J, Calvino M, Sanz-de-Villalobos E, González-Praetorius A, Albertos S, García-Garzón S, Lokhande M, Parra-Cid T. Bim-mediated apoptosis and PD-1/PD-L1 pathway impair reactivity of PD1(+)/CD127(−) HCV-specific CD8(+) cells targeting the virus in chronic hepatitis C virus infection. Cell Immunol. 2011;269(2):104–14. doi: 10.1016/j.cellimm.2011.03.011. PMID:21481848. [DOI] [PubMed] [Google Scholar]
- 17.Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, Zhou J, Hodi FS, Spagnoli GC, Murphy GF, et al.. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010;70(2):697–708. doi: 10.1158/0008-5472.CAN-09-1592. PMID:20068175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al.. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell. 2015;162(6):1242–56. doi: 10.1016/j.cell.2015.08.052. PMID:26359984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somasundaram A, Burns TF. Pembrolizumab in the treatment of metastatic non-small-cell lung cancer: patient selection and perspectives. Lung Cancer (Auckl). 2017;8:1–11. PMID:28293123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander GS, Palmer JD, Tuluc M, Lin J, Dicker AP, Bar-Ad V, Harshyne LA, Louie J, Shaw CM, Hooper DC, et al.. Immune biomarkers of treatment failure for a patient on a phase I clinical trial of pembrolizumab plus radiotherapy. J Hematol Oncol. 2016;9(1):96. doi: 10.1186/s13045-016-0328-4. PMID:27663515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. PMID:25428505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Füllgrabe A, Fuentes AM-P, Jupp S, Koskinen S, et al.. Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016;44(D1):D746–52. doi: 10.1093/nar/gkv1045. PMID:26481351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186(5):2772–9. doi: 10.4049/jimmunol.1003208. PMID:21263073. [DOI] [PubMed] [Google Scholar]
- 24.Lim JYH, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol Immunother. 2014;63(3):259–71. doi: 10.1007/s00262-013-1506-7. PMID:24357146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20(4):265–71. doi: 10.1097/PPO.0000000000000059. PMID:25098287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons RM, Liu X, Pulko V, Harrington SM, Krco CJ, Kwon ED, Dong H. B7-H1 limits the entry of effector CD8(+) T cells to the memory pool by upregulating Bim. Oncoimmunology. 2012;1(7):1061–73. doi: 10.4161/onci.20850. PMID:23170254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228(1):342–59. doi: 10.1111/j.1600-065X.2008.00760.x. PMID:19290938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi C, Han T, Tang H, Huang K, Min J, Li J, Ding X, Xu Z. Shp2 inhibits proliferation of esophageal squamous cell cancer via dephosphorylation of stat3. Int J Mol Sci. 2017;18(1). doi: 10.3390/ijms18010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, Hsu DD, Wang H, Feng G-S. Dual faces of SH2-containing protein-tyrosine phosphatase Shp2/PTPN11 in tumorigenesis. Front Med. 2012;6(3):275–9. doi: 10.1007/s11684-012-0216-4. PMID:22869052. [DOI] [PubMed] [Google Scholar]
- 30.Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr Pharm Des. 2010;16(16):1843–62. doi: 10.2174/138161210791209027. PMID:20337577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furcht CM, Muñoz Rojas AR, Nihalani D, Lazzara MJ. Diminished functional role and altered localization of SHP2 in non-small cell lung cancer cells with EGFR-activating mutations. Oncogene. 2013;32(18):2346–55, 2355.e1. doi: 10.1038/onc.2012.240. PMID:22777356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen G, Kim YH, Li H, Luo H, Liu D-L, Zhang Z-J, Lay M, Chang W, Zhang Y-Q, Ji R-R. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci. 2017;20(7):917–26. doi: 10.1038/nn.4571. PMID:28530662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C, Guan Q, Wang Y, Zhao ZJ, Zhou GW. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J Cell Biochem. 2003;90(5):1026–37. doi: 10.1002/jcb.10727. PMID:14624462. [DOI] [PubMed] [Google Scholar]
- 34.Huang T-T, Su J-C, Liu C-Y, Shiau C-W, Chen K-F. Alteration of SHP-1/p-STAT3 Signaling: A Potential Target for Anticancer Therapy. Int J Mol Sci. 2017;18(6). doi: 10.3390/ijms18061234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Nowak KA, Zaorsky NG, Winchester C-L, Dalal K, Giacalone NJ, Liu N, Werner-Wasik M, Wasik MA, Dicker AP, et al.. ALK inhibitor PF02341066 (crizotinib) increases sensitivity to radiation in non-small cell lung cancer expressing EML4-ALK. Mol Cancer Ther. 2013;12(5):696–704. doi: 10.1158/1535-7163.MCT-12-0868. PMID:23443800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang H, Wang P, Yuan Z, Lu B. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin Cancer Res. 2016;22(1):122–33. doi: 10.1158/1078-0432.CCR-15-0589. PMID:26728409. [DOI] [PubMed] [Google Scholar]
- 37.Du S, Lockamy V, Zhou L, Xue C, LeBlanc J, Glenn S, Shukla G, Yu Y, Dicker AP, Leeper DB, et al.. Stereotactic Body Radiation Therapy Delivery in a Genetically Engineered Mouse Model of Lung Cancer. Int J Radiat Oncol Biol Phys. 2016;96(3):529–37. doi: 10.1016/j.ijrobp.2016.07.008. PMID:27681749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shlyakhtina Y, Pavet V, Gronemeyer H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017;8(8):e3025. doi: 10.1038/cddis.2017.423. PMID:29048428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.