

Abstract
The advent of high resolution and frequency mass spectrometry has ushered in an era of data independent acquisition (DIA). This approach affords enormous multiplexing capacity and is particularly suitable for clinical biomarker studies. However, DIA-based quantification of clinical plasma samples is a daunting task due to the high complexity of clinical plasma samples, the diversity of peptides within the samples, and the high biologic dynamic range of plasma proteins. Here, we applied DIA methodology, including a highly reproducible sample preparation and LC-MS/MS analysis, and assessed its utility for clinical plasma biomarker detection. A pancreatic cancer relevant plasma spectral library was constructed consisting of over 14000 confidently identified peptides derived from over 2300 plasma proteins. Using a non-human protein as the internal standard, various empirical parameters were explored to maximize the reliability and reproducibility of the DIA quantification. The DIA parameters were optimized based on the quantification cycle times and profile complexity. Higher analytical and biological reproducibility was recorded for the tryptic peptides without labile residues and missed cleavages. Quantification reliability was developed for the peptides identified within a consistent retention time and signal intensity. Linear analytical dynamic range and the lower limit of quantification were assessed, suggesting the critical role of sample complexity in optimizing DIA settings. Technical validation of the assay using a cohort of clinical plasma indicated the robustness and unique advantage for targeted analysis of clinical plasma samples in the context of biomarker development.
Keywords: proteomics, mass spectrometry, data independent acquisition (DIA), plasma, pancreatic cancer
TOC Graphic
DIA based targeted proteomics for protein quantification in plasma
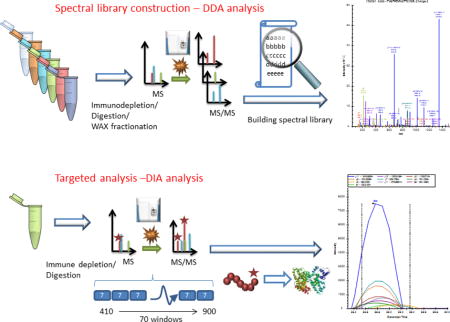
Introduction
The emerging application of data independent acquisition (DIA) has enabled the possibility of concomitant scanning of multiple co-eluted peptide fragments isolated within small consecutive isolation windows1;2. In DIA acquisition, MS/MS fragmentation is performed with a predefined m/z range. Hence, each recorded MS/MS spectrum is a multiplexed recording of the fragment ions derived from all peptides eluting in real time within the predefined m/z range of the precursor window. In a way, DIA-based targeted proteomics approach is equated with the selective reaction monitoring (SRM) of all peptides with any transition to the fingerprint fragments3–8. Due to its global nature and high reproducibility, this technique has a great potential for the large scale targeted quantification of key proteins in translational studies1;9. Different data independent acquisition methods have been developed based on a plethora of available high-frequency mass spectrometers, such as triple TOF based SWATH (sequential window acquisition of all theoretical spectra)5;8;10–12, Q-TOF based MSE 13 and Orbitrap based multiplexing strategy (MSX)3. The DIA technique thus should be assessed in the context of multiplexing biomarker detection from clinical specimens, such as plasma or serum, which are important biospecimens in clinical analysis14;15.
Plasma is a great source of clinical information underlying disease diagnosis, prognosis, and monitoring the response for a treatment16. Cellular proteins from the infected cells or tissue can potentially end up in blood and reveal the status of the infected cells or tissue17;18. Plasma, however, is a very complex fluid that almost every cell type contributes to its complexity, and it has a wide dynamic range in protein concentration exceeding 12 orders of magnitude16;17;19;20. An approach for the quantification of plasma proteins, especially the low abundant proteins, hence, requires a highly reproducible reduction of complexity and measurement within a wide dynamic range.
The present study aims at development of a DIA based assay and to study its utility in the translational purposes for clinical plasma proteome quantification. Due to the global nature of this technique, DIA acquisition contains the fragments from multiple co-eluted peptides in the mixture, so that consistent and reliable peak-picking and quantification becomes a daunting task. This challenge of analyzing clinical plasma samples is further complicated by the significant heterogeneity of plasma proteins among patients21. In this study, we developed a pancreatic cancer relevant spectral library, and established empirical parameters for the selection of quantifiable peptides for DIA analysis. The reproducibility of quantification from analytical and biological replicates is examined and the extent of linear dynamic range for the label-free quantification and the lower limit of quantification in crude plasma were evaluated. The optimized conditions were utilized in the analysis of a set of clinical plasma samples. The reliability and reproducibility of the technique were evaluated in the context of clinical analysis.
Experimental procedure
Plasma Samples
This study was approved by the Institutional Review Board at the University of Washington (Seattle, WA), University of Pittsburgh (Pittsburgh, PA), and Mayo Clinic (Rochester, MN). Plasma samples were collected from healthy subjects (NL), and patients with chronic pancreatitis (CP) and early stage pancreatic ductal adenocarcinoma (PDAC). The diagnosis of disease was made histologically in the case of pancreatic cancer patients. CP was diagnosed based on computed tomography (CT) scan showing calcifications, ductal dilation and atrophy, or by the presence of structural and functional abnormalities detected by combined endoscopic ultrasound (EUS) and/or secretin pancreatic function testing22. The PDAC patients with early stage disease were operable, representing a mixture of localized pancreatic cancer (stages 1 and 2). The cancer patients involved in this study did not receive any treatment prior to blood draw. The blood samples were processed using similar protocols within 4 hours after specimen collection. The plasma samples were collected into purple top tubes (Becton Dickinson, Franklin Lakes, NJ, USA) with EDTA, the potassium salt, as an anticoagulant. The blood was centrifuged at 330 × g for 20 minutes. The resultant plasma samples were aliquoted and stored in −80 °C until used.
Sample preparation
Sample preparation for profiling analysis
Equal volumes of the plasma samples of 5 healthy subjects, 5 chronic pancreatitis patients, and 5 pancreatic cancer patients were pooled to generate the pooled sample for profiling analysis for the development of a spectral library (Supplemental Table S1). Denatured invertase 2, (Sigma Aldrich, St. Louis, MO, USA) was added to the plasma samples as an internal standard at a final concentration of 25 μg/ml plasma. A total of 60 μL of the pooled sample (6 × 10 μL) was then depleted using Multiple Affinity Removal Column Human 6 (MARS6) column (Agilent technologies, Santa Clara, CA, USA) to remove the top 6 most abundant plasma proteins according to the manufacturer’s instruction. Using a 5 kDa filter, VIVASPIN 500 (Sigma Aldrich), the depletion buffer was exchanged to PBS by washing it 3 times with 500 μL PBS. The final volume was adjusted to 200 μL with PBS. The samples were then de-glycosylated by adding 0.5 μL of PNGase F (New England Biolabs, Ipswich, MA, USA), and incubated for 6 hours at 37 °C. The samples were reduced by adding 10 mM TCEP and incubation at 50 °C for 1 hour, and alkylated by adding 25 mM iodoacetamide (IDA) and incubation for 30 minutes at room temperature in the dark. After adjusting the pH to 7.5–8.5 with sodium hydroxide solution, the depleted plasma was digested with MS grade trypsin (Thermo Fisher Scientific, Waltham, MA, USA) at 1:30 enzyme to protein ratio, in a two-step fashion to improve digestion efficiency. In the first step, half of the trypsin was added and the mixture was incubated for 2 hours at 37 °C with vortexing every 30 minutes, then the remaining trypsin was added and the mixture was incubated for additional 16 hours at 37 °C. The digestion was terminated by adding 0.1% formic acid (V/V). Tryptic digested peptides were then fractionated using weak anionic exchange (WAX) spin column (The Nest Group, Southborough, MA, USA). After equilibration of the column, the dried sample was re-suspended with 100% Buffer A (acetonitrile:H2O:90:10 with 0.1% formic acid and 10 mM ammonium formate) and loaded onto the column, then eluted with Buffer B at 5, 7.5, 10, 12.5, 15, 17.5, 20, 30, 50, and 70% of buffer B (H2O with 0.1% formic acid). Fractionated samples were dried completely and re-suspended in 0.1% formic acid. One μg of each of the fractions was loaded for LC MS/MS analysis using data dependent acquisition (DDA) method.
Sample preparation for DIA analysis
Multiple depletion columns were used to evaluate the depletion effects on DIA analysis, including a re-usable MARS6 spin column, and two types of disposable spin columns with depletion of top 2 or top 12 most highly abundant proteins. Invertase 2 standard was added to crude plasma upfront to an initial plasma concentration of 25 μg/mL. Sample preparation using MARS6 was identical to the procedure used in the profiling analysis for the spectral library construction, except 8 μL of plasma was depleted every time to avoid the risk of column overloading. For top-12 depletion, a top 12 depletion spin columns (Thermo Fisher Scientific) was used. Six μL of plasma sample was directly loaded onto the buffer at the top of slurry, and the column was incubated with an end-over-end incubator for 1 hour at room temperature. The end of the column was removed and the sample was eluted with a centrifuge at 100 ×g for 2 minutes. The depleted mixture was collected and buffer exchanged to 50 mM ammonium bicarbonate using the 5 kDa spin filter, VIVASPIN 500 (Sigma Aldrich), and the final volume was adjusted to 100 μL. The top-2 depletion was performed using a ProteoPrep Immunoaffinity Albumin and IgG depletion kit (Sigma-Aldrich). The column was first conditioned with 0.4 mL of equilibration buffer, then the plug was removed and the buffer spun down at 5000 ×g for 30 seconds. Then 0.4 mL of PBS was added to condition the column and spun down. 25 μL of plasma sample was diluted 4 times in PBS and added to the top of column and incubated at room temperature for 10 minutes. The column was spun down at 8000 ×g for 10 seconds. The eluate was re-applied to the depletion column and the elution process was repeated once. After the depletion steps, the reduction, alkylation, and digestion procedures were performed similarly as previously described for the profiling analysis. The disposable 12 depletion column was chosen for DIA analysis of individual plasma samples to avoid inter-sample cross contaminations and achieve greater coverage of low abundant proteins.
LC-MS/MS set-up for data-dependent and data-independent analyses
The LC separation set up consisted of a trapping column and an analytical column connected back-to-back to increase the loading speed. The trapping column was a 360 μm and 100 μm outer and inner diameters self-packed integraFit column (Scientific Instrument Services, Ringoes, NJ, USA). The trapping column was packed to a length of about 3 cm with ProntoSIL 200Å (pore size) 5 μm (particle size)-C18 AQ (Mac-Mod, Chadds Ford, PA, USA). The analytical column was 25 cm long with 360 μm and 75 μm outer and inner diameters fused silica packed with ProntoSIL 120 Å -5μm-C18 AQ (Mac-Mod). A column tip was prepared by pulling the column with Laser Fiber Puller P-2000 (Sutter Instruments, Novato, CA, USA). Separation was done with a nanoACQUITY UPLC system from Waters. Buffer A and buffer B were water and acetonitrile with 0.1 % formic acid respectively. 1 μg of sample was loaded on the trapping column with 2% B at 2 μL/min flow rate for 10 minutes. Peptides were then resolved with a 90-minute gradient of 5 to 30% B followed by flushing at 80% B for 10 minutes and column equilibration with 2% B for 20 minutes. The analytical flow rate was 0.3 μL/min and entire data acquisition lasted for 120 minutes. For both DDA and DIA analysis, the same LC settings were used for retention time stability. DDA was performed on a Fusion mass spectrometer (ThermoFisher Scientific). The survey scan was done with 120K resolution at 400 m/z from 400 to 1600 m/z with AGC target of 4e5 and max injection time of 50 msec. Monoisotopic masses were then selected for further fragmentation for ions with 2 to 4 plus charge within a dynamic exclusion range of 30 seconds and a minimum intensity threshold of 5e3 ions. Fragmentation priority was given to the most intense ions. Precursor ions were isolated using the quadrupole with an isolation window of 1.6 m/z. Rapid scan speed in the ion trap was selected after HCD fragmentation (NCE 28%) and the AGC target of 1e4 and maximum injection time of 50 msec. The DDA cycle was limited to 3 seconds. DIA quantification was performed on a QE+ mass spectrometer (ThermoFisher Scientific) with the resolution of 17,500 at 200 m/z. AGC target was set at 1e6 with 55 msec max injection time. Optimal isolation windows were 7 m/z, and NCE was 28, the acquisition window covered a mass range from 410 to 900 m/z through 70 consecutive isolation windows.
Data analysis
DDA data for library construction was processed using Trans-Proteomic Pipeline (TPP)23;24. The MS raw files were converted to mzML open format and searched against the UniProt human protein database (2015-07-23) with the Comet algorithm25. The search parameters were set as follows: cysteine alkylation defined as static modification (+57.021464), and methionine oxidation (+15.9949) and asparagine-deamidation (+0.9840) as dynamic modifications. The search was limited to maximum five dynamic modifications and two miss-cleavages with a mass tolerance of 20 ppm. The peptide identification was statistically validated with PeptideProphet26 and only the peptides with a probability score ≥ 0.95 were retained for spectral library building.
The DIA data was analyzed using the Skyline software4. The Skyline library was devised by importing the search results generated from the plasma DDA data. Additional peptides belonging to the plasma protein categories were added from the tissue databases as well to enrich the identification and quantification. The transitions were limited to the peptides with 410 to 900 m/z, and only top 5 b or y ions with m/z value greater than 200 were selected for the data mining. The peptide profiles were explored through the highest library dot products in the 10 minutes of retention time from the spectral library. The signals from the transitions were summed to provide the quantification for the corresponding peptide. Similarly, the signals from peptides for each protein were summed to give the quantification at protein level.
Results and Discussion
Plasma peptide library building
The DIA based targeted proteomics analysis relies on the establishment of a comprehensive spectral library. The pancreatic cancer relevant plasma spectral library was constructed from the profiling analysis of plasma samples from healthy subjects and patients with resectable pancreatic cancer and patients with chronic pancreatitis, a disease which shares many physiological and molecular features with pancreatic cancer27;28. These plasma samples were pooled to represent the relevant proteomes that may present in various clinical samples. As more clinical plasma samples are analyzed, newly identified data will be introduced to this dynamic spectral library, expanding its capacity for a comprehensive analysis of DIA data. The depleted plasma samples were fractionated into 10 fractions using weak anionic exchange columns. Each of the fractions was then analyzed with LC-MS/MS using DDA approach. Due to the stochastic nature of the DDA analysis29, each sample was analyzed twice. The data were then combined together and only peptides with probability scores higher than 0.95 were selected for constructing the spectral library, which stores the identification information of each individual peptide, including retention time and fragmentation pattern. In addition to the plasma profiling experiment (peptide and protein identification results are provided in the Supplementary Information), additional peptides derived from the plasma proteins identified from pancreatic tissues were also added to enrich the library. The resulting library included 14320 unique peptides derived from 2317 proteins. The inclusion of deglycosylated N-glycopeptides during construction of the spectral library resulted in further improvement in identification of plasma proteins. 327 peptides with the consensus sequence for N-glycosylation (Asn-X-Ser/Thr), derived from 180 annotated glycoproteins, were identified from the fractionated human plasma samples. The identified glycoproteins, by large, belong to extracellular regions, plasma membrane, and blood microparticles according to gene-ontology annotations. Peptides derived from Invertase 2 - a yeast protein, which was spiked to the crude plasma samples and used as the internal standard for DIA analysis, were included in the spectral library. Tryptic digested peptides from Invertase 2 were verified to be unique to yeast and do not interfere with the quantification of human endogenous peptides based on the BLAST analysis30 and transition selection. Due to the wide Q1 window in DIA, dilution experiments were performed to optimize the spiking amount of Invertase 2 to subdue its impact on endogenous human peptides. Supplemental Table S2 shows the 22 peptides from Invertase 2 that were identified and included in the spectral library. The comparison of 2-depletion, 6-depletion, and 12-depletion columns, as well as non-depleted plasma for identification of peptides and proteins is provided in Supplemental Table S3. The depletion of abundant proteins appears to significantly increase the number of peptides and proteins identified in the plasma samples, especially the low abundant proteins. The performances of 6-depletion column and 12-depletion column appear to be similar and outperform the 2-depletion column. These observations are consistent with reported studies in evaluation of different depletion strategies and columns31–34. MS1 ion-current-based quantification was used to evaluate the abundant proteins in the non-depleted plasma samples and depleted plasma samples. All three depletion methods used showed an effective removal of more than 99% of plasma albumin and IgGs from the plasma samples. While similar performances were achieved for 6-depletion and 12-depletion, the disposable 12-depletion column was chosen for DIA analysis of individual samples, to avoid the risk of cross contamination between clinical samples (the 6-depeltion column is not disposable). Notably, while an optimal depletion strategy can significantly reduce the protein concentration range in a complex plasma sample, the combination of WAX fractionation aids in achieving a more comprehensive identification of peptides and proteins for building the spectral library.
The reproducibility of the disposable 12-depletion column was evaluated in the replicate experiments (see below). Only peptides identified with a PeptideProphet score ≥ 0.95 are included for library construction. A detailed analysis shows that 82% of the identified peptides from plasma don’t contain a miss-cleavage. Majority of the peptides (>95%) identified have a mass difference less than 5 ppm from the theoretical values, and this mass difference is independent from the m/z value of the peptides (Supplemental Figure S1). The quality of the fragmentation for the identified peptides was assessed based on the cumulative frequency distribution of the peptide fragmentation Xcorr values. Majority of the double-charged peptides shows Xcorr values greater than 2.45.
More than half of the quantifiable plasma proteins were quantified with either two or more peptides (Figure 1A). 580 proteins included in the library have known blood concentrations according to the plasma proteome database35. These proteins cover a dynamic range of more than 10 orders of magnitude. Most of these proteins are in the range of 1 to 106 ng/mL, and around 10% of the proteins have known concentrations less than 10 ng/mL (Figure 1B). This data shows the range of quantification can potentially reach the concentrations of invaluable key plasma protein biomarkers. Gene Ontology-based analysis36 of the identified proteins showed that from the library proteins, 680 proteins are intrinsic to membrane (including 472 plasma membrane proteins), 481 belong to the extracellular region, 221 are from cytoskeleton, and 186 are from various cellular fractions. The biological processes that they represent include 372 cell surface receptor linked signal transduction, 258 cell adhesion proteins, 187 immune response, 186 homeostasis process, and 162 regulations of apoptosis. These results suggest that a large number of proteins identified in plasma originated from the cellular fractions that are playing important roles in cell cycle, growth, immune response and apoptosis (Supplemental Figure S2).
Figure 1.
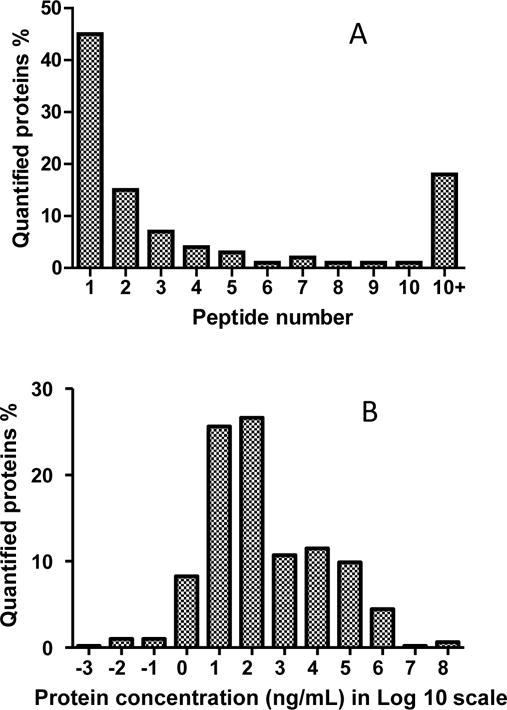
A) Percent distribution of the quantified proteins (2317) based on the number of quantified peptides used for protein quantification. More than half of the quantified plasma proteins were quantified with two or more peptides. B) Distribution of the 580 quantified proteins with known concentrations in plasma proteome database. Most of the quantified plasma proteins have a concentration in the range from 1 to 106 ng/mL. Around 10% of the plasma proteins have known blood concentrations less than 10 ng/mL.
DIA isolation windows
For the quantification purposes, DIA windows, with 5, 7, 10, and 20 m/z sizes were evaluated for depleted plasma detection. With a mass range from 410 to 900 m/z, it resulted in 100, 70, 50, and 25 consecutive DIA isolation windows, respectively. At a theoretical max scan speed of 13 Hz for the QEplus, this would result in cycle times of 7.7, 5.4, 3.8, and 1.9 sec. Using SENDLSYYK++ peptide from Invertase 2 as an example, the complexity factors associated with the width of DIA window were assessed. Five top b and y ions from the fragmentation pattern were selected for the quantification. Given the fact that the scan time (transient time) of the Orbitrap mass analyzer of QEplus at resolution of 17,500 at 200 m/z is 64 msec, a maximum fill time of 55 msec was chosen to assure that the fastest scan speed of about 13 Hz could be achieved. The actual cycle times estimated from the experiments and the number of quantification points as a criterion for the quality of quantification is presented in the Figure 2A. A wider DIA window corresponds to higher number of quantification points, providing a more precise monitoring on the elution profile of a peptide, thus, yields a better quantification. On the other hand, however, an increase of the DIA window size could compromise the identification (library matching) and quantification of a peptide by generating more complex MS/MS spectra due to inclusion of more peptides for fragmentation. This is particularly the case in a complex sample, such as plasma. As shown in Figures 2B to 2E, as the isolation window size increased, the DIA MS/MS spectra and the elution profiles became more complicated. Based on those initial experiments, the 7 m/z isolation window size was determined to be the optimal condition for peptide identification and quantification in our plasma analysis. This set up provided a cycle time of 5.7 seconds, with a justified tradeoff between isolation window size and cycle time due to the constant mass spectrometer acquisition frequency (13 Hz with 17,500 resolutions at 200 m/z). Narrower isolation windows though were necessary to obtain a less complex fragmentation patterns benefiting library matching, it comes with a cost on increasing cycle time. Using the peptides derived from Invertase 2 as examples, among the 4 different isolation windows sizes, the 7 m/z isolation window showed reasonable profile quality as represented by the library dot products value (Supplemental Figure S3). With a scan range of 410 to 900 m/z, our approach included 70 consecutive 7 m/z DIA isolation windows that resulted in 5.7 seconds of cycle time, balancing both factors for plasma analysis. The number of peptides that relies in this interval is c.a. 76% (Supplemental Figure S4), representing nearly 82% of proteins included in the library. Hence, confinement of quantification to the peptides with m/z between 410 and 900 results in augmented quality of quantification for a large set of peptides and proteins.
Figure 2.
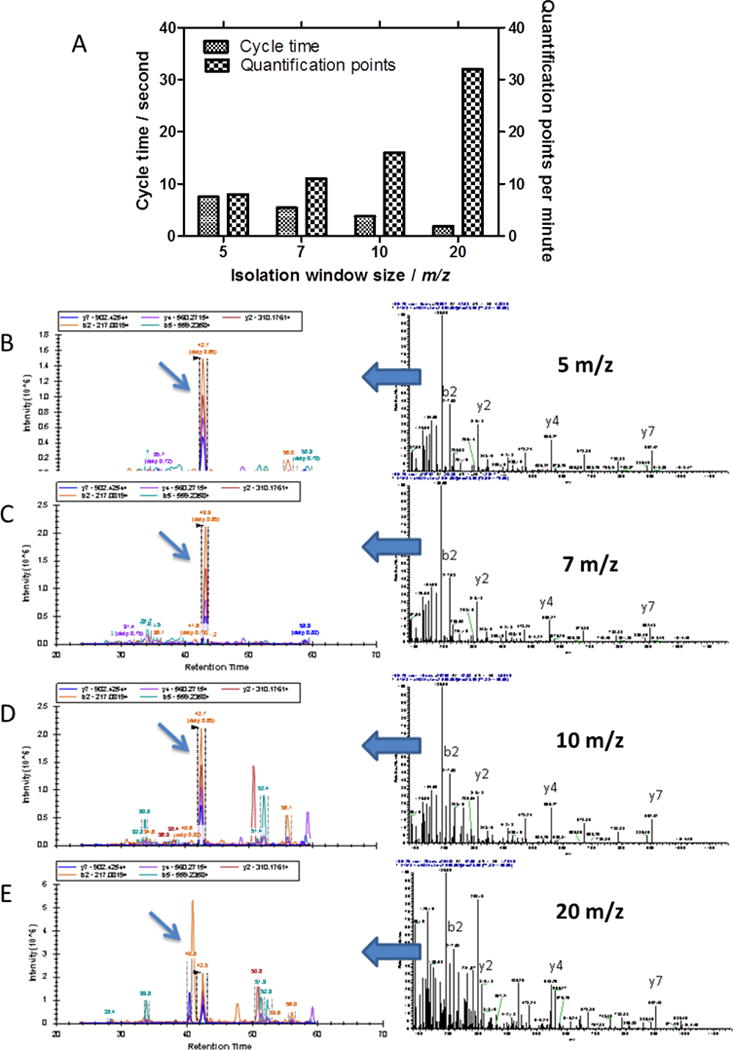
Optimization of the isolation windows size based on the profile quality and complexity for SENDLSYYK++ peptide from Invertase 2 standard protein. A) The observed cycle times for the different isolation windows of 5, 7, 10, and 20 m/z corresponding with the theoretical acquisition frequency of 13 Hz. B) to E) The MS/MS spectrum and extracted profile for the target peptide with different isolation windows of 5, 7, 10, and 20 m/z. The complexity of the MS/MS spectra and elution profile for the target peptides increase with the size of isolation windows. Blue arrow indicates the target peaks from SENDLSYYK++ peptide.
During the quantitative analysis of DIA data, the elution profiles of the peptides were extracted using a 10 minutes window of their corresponding identification retention time in the spectral library. This is justified based on the evaluation of Invertase 2 peptides and their retention time deviations between library and their DIA elution profiles. The library retention time of these peptides were compared with their retention times measured from DIA replicates. For these peptides, the average difference between the library retention time and the DIA measurements is 1.34 minute with a 3.11 minute standard deviation (Supplemental Table S4). Using the following formula, we approximate the 10 minute retention time window for library matching, assuming the retention time can be drifted in either direction: 2 × (average retention time + standard deviation + typical peak width (0.5 min)). While a shorter retention time window may affect the library matching, longer retention time windows may increase the risk of false discoveries. As shown in Supplemental Figure S5, while the retention time of a peptide may drift throughout the analysis of a large queue of clinical samples, the setting of 10 minute window appeared to be reasonable to contain the retention drift in the sequential analysis.
Technical and biological replicates
Absence of isotope-labeled internal standard necessitates a high level of reproducibility during sample preparation, LC-MS/MS analysis, and data mining37. The reproducibility and consistency of the DIA method was evaluated with the aid of a spiked-in standard protein, which was added to the crude plasma as an internal reference. The reproducibility assessment included 1) biological replicates, which evaluate the overall variations caused throughout sample preparation, LC-MS/MS analysis, and data mining; and 2) analytical replicates, which evaluated the variation that occurred during LC-MS/MS acquisition and data mining of the same tryptic plasma mixture. While biological replicates also cover the variations from analytical replicates, the analytical replicates provide insightful information regarding how intrinsic physicochemical properties of a peptide influence its MS sensitivity and quantification.
Four biological replicates were prepared under identical condition with the spike-in of 25 μg/mL of Invertase 2 standard protein and depletion of the top 12 abundant proteins. One biological replicate sample was analyzed by four analytical replicates. The correlations for the analytical replicates and the biological replicates were presented in Figure 3A & B, respectively. The regression curves for the 22 peptides (red dots) from Invertase 2 show a high linearity and reproducibility for both analytical and biological replicates, indicating the reliability of the quantification. Supplemental Figure S6 sorts the variation of Invertase 2 peptides based on their amino acid sequence characteristics. Evidently, peptides containing labile residues, in particular methionine and those containing miss-cleavages show higher variation. In addition, peptide IEIYSSDDLK++, which has flanking residues due to the close arginine and lysine residues at both N and C terminals, is also subjected to high variation. Therefore, six peptides from Invertase 2 that do not contain methionine or miss-cleavages were selected for Invertase 2 quantification to ensure the least variations. Supplemental Table S5 summarizes the analytical and biological variations for these 6 quantifiable peptides along with their mass spectrometry visibility, retention time variation, and library dot product values. Generally, biological variation feasibly is higher than analytical variations. Peptides that are difficult to digest or prone to degradation may show higher variations during sample preparation. The peptides were identified in consistent, narrow retention time windows, typically indicating the high reproducibility of LC separations regardless of their hydrophobicity. Using the intensity sum of these 6 quantifiable peptides to represent Invertase 2 quantification, the analytical and biological variation of Invertase 2 measurement was 4% and 14%, respectively. Quantifiable peptides with higher mass spectrometry visibility have a larger impact on the protein quantification as exemplified in Supplemental Table S5.
Figure 3.
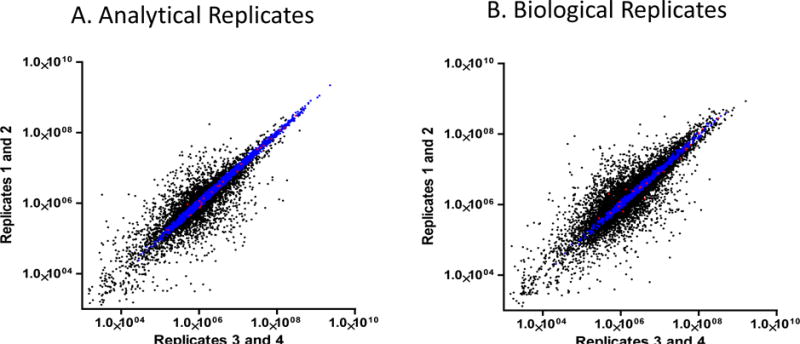
A). Analytical replicate measurements for the plasma peptides. Coefficient of variation for the analytical replicates is 0.9962. B) Biological replicate measurements for the plasma peptides. Coefficient of variation for the biological replicates is 0.9864. Red: peptides derived from Invertase 2 standard protein, Blue: endogenous plasma peptides with a CV less than 20%, Black: endogenous plasma peptides with a CV greater than 20%.
Quantifiable peptides
In DIA targeted quantitative analysis, it is critically important to identify robust “quantitative” peptides, which are not only “proteotypic” peptides38 that can be repeatedly identified in a complex sample, but also possess desired chromatographic and mass spectrometric characteristics allowing consistent DIA quantification. The analysis of the reproducibility of over 14,000 endogenous peptides identified in the analytical replicates indicated that the nature of a peptide (sequence, length and other physicochemical properties, etc.) plays a pivotal role in peptide quantification in addition to matrix associated factors such as protein abundance and sample complexity. Figure 4 A to D illustrate the CV distribution of the selected peptides based on their retention time deviation among the analytical replicate runs. Using small retention time variations (σ), we observed a symmetric frequency histograms distribution with a median peak at ~10% of variation for the endogenous peptides (Figure 4A & B). However, peptides with retention time variations more than 1 minute gradually pour into a second distribution curve as retention time variation increases, forming a bimodal peptide CV distribution (Figure 4C & D). This bimodal distribution may imply a general difference in peptide characteristics which influences peptide quantification robustness. For those peptides derived from the proteins with known plasma concentration, Figure 4E & F shows the correlation of peptide intensity and their corresponding protein concentration. For peptides with a retention time σ ≤ 0.2 minute, which mostly have a lower CV and distributed in the first modal, a large number of them were from extracellular proteins with higher concentration (Figure 4E). The peptides with a retention time σ > 0.2 minute appeared to include more intracellular proteins with a lower concentration (Figure 4F). Notably, 40% of the total proteins identified have peptides distributed in both modal, suggesting the role of peptide physicochemical properties in determining their quantification robustness. Further systematic investigations are needed to better address the correlation of such an observation with peptide physicochemical properties. In some cases, the higher retention time variations may indicate the incorrect and inconsistent identification of these peptides. They also have average lower library dot product scores that may have resulted from their inconsistent peak picking and discovery. These peptides may include peptides with reactive or variant amino acid residues or PTMs, peptides with low abundance, or peptides with poor chromatographic characteristics or low mass spectrometric sensitivity. Hence, we introduced the parameters derived from analytical replicates as empirical indicators for the reliable and consistent detection of peptides throughout multiple runs, namely the retention time sigma and peptide intensity CV. Using a sigma of 0.25 minute and a 20% CV, we were able to retain more than 4000 peptides derived from nearly 900 proteins for plasma detection. While determination of the stringency of such parameters depends on specific study design, in general, a small sigma and CV based on the replicate analysis warranted the selection of more consistent peptides for a more robust DIA quantification in multiple sample analysis.
Figure 4.
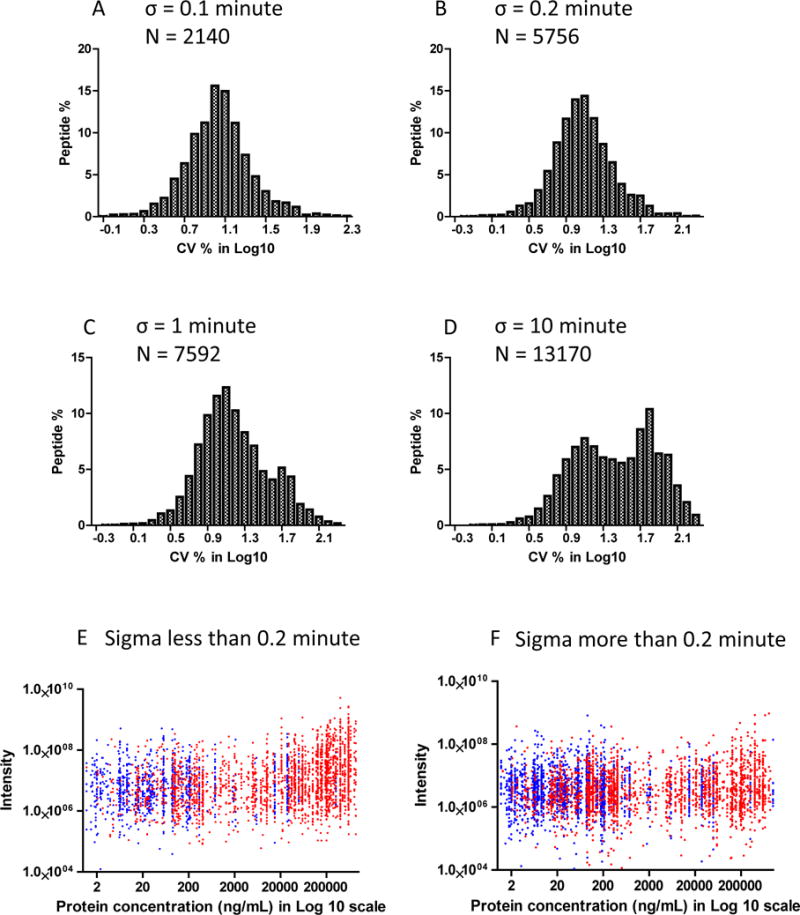
A) to D) Frequency distribution histogram for the variation of plasma peptides quantified within 4 analytical replicates with different retention time variations (sigma value): A) less than 0.1 minute, B) less than 0.2 minute, C) less than 1 minute, and D) less than 10 minutes. A singular distribution was observed for quantification of peptides with small retention time variations. A bimodal distribution started to emerge when retention time variations were increased to greater than 1 minute. N indicates the number of quantified peptides with the corresponding criterion. For the proteins with known plasma concentration, E) and F) illustrate the correlation of the peptide intensity and protein concentration: E) peptides with σ ≤ 0.2 min, F) peptides with σ > 0.2 min. Red dots represent the peptides from extracellular proteins with known concentrations. Blue dots represent the rest of the peptides.
Comparison of DIA-based quantification with DDA-based quantification in pooled plasma sample and HeLa cell line digest
DIA-based analysis is compared with the DDA-based analysis in profiling the same pooled plasma sample. DDA-based analysis doesn’t have the confinement of peptide m/z values and can be applied to the entire identified peptides in the library. The DDA-based analysis also can be performed with a high frequency as a single survey scan is needed to extract the MS1 profiles. In addition, the DDA-based quantification relies on the isotope distribution of MS1 ions rather than their fragmentation patterns. Considering that the fragmentation pattern depends on the instrument type and data acquisition condition, DDA can provide a global quantification. However, the main challenge for the conventional DDA-based quantification is the dynamic range of quantification, as a single survey of entire co-eluting ions is being used for the quantification. Supplemental Figure S7 compares the DDA and DIA profiles from two peptides with different visibility from the same standard protein.
DIA-based analysis can be applied to biological matrices other than clinical plasma samples. As a comparison, DIA-based method was performed to analyze HeLa cell line digest. A spectral library was constructed for HeLa cell digest, including information of 3525 proteins and 14204 peptides. Evidently, spectral library can be constructed easier for cell lines due to the lower dynamic range of proteins in a cell digest. In addition, the DIA-based profiling quality was compared for the plasma and cell line digests based on the library dot product values to represent the quality of peak picking using Skyline software (Supplemental Figure S8). While there are 11573 peptides from cell lysates have a library dot product value more than 0.8, there are only 1489 peptides with the same quality in plasma samples. Although cell lysates may present a more complex proteome than plasma, the enormous protein concentration difference inherent in plasma creates a significant technical hurdle preventing reliable identification and quantification of low-abundant peptides, underscoring the challenges in plasma analysis.
Quantification sensitivity
The detection sensitivity, lower limit of quantification, and analytical dynamic range were assessed with the spiked-in Invertase 2 standard protein using different concentrations in the crude plasma, including: 50, 25, 10, 1, 0.5, and 0.1 μg/mL. Figure 5A–F shows the extracted profiles for the top 5 b and y fragments from Invertase 2 peptide GLEDPEEYLR++ at different concentrations from 0.1 to 50 μg/mL. Evidently, the signal for this peptide (at retention time ~70 min) is detectable at 0.1 μg/mL concentrations. The dilution curves constructed for this peptide and the Invertase 2 protein based on the total of six quantifiable peptides are shown in Figure 5G & H, respectively. While the linear range for peptide GLEDPEEYLR++ is obvious from 0.1 to 50 μg/mL, the lower limit of the linear range for Invertase 2 protein appears to be higher than 0.1 μg/mL. This is because other quantifiable peptides, which also contribute to the Invertase 2 protein level quantification, have lower detection sensitivity, highlighting the implication of signature peptide selection in protein quantification. The library dot product score and retention times for the peptide GLEDPEEYLR++ from different concentrations is shown in Supplemental Figure S9. The data indicates a consistent detection of this peptide even at low concentrations, which is relevant to many quantifiable peptides in our analysis. In addition, the confinement of data-mining to the top five b and y ions (generally the most dominant fragments) with m/z values more than 200 aided in the exclusion of non-targeted fragments. However, the quality of profiles can be compromised at lower concentrations due to the loss of fragments – affecting peptide quantification. In addition, ion suppression effects and incorrect peak picking could be an issue for peptides with lower concentration in a complexed spectrum. For a quantifiable peptide within a given concentration, critical parameters that may influence the quantification sensitivity may include physicochemical properties of the peptide, as well as the interference from the complex nature of plasma. The complexity of a DIA spectrum should be minimized through selection of smaller size isolation windows, without compromising the quantification of the elution profile. In addition, retention time can be applied as a complementary parameter to optimize peak picking of the targeted peptide as adapted in mProphet39.
Figure 5.
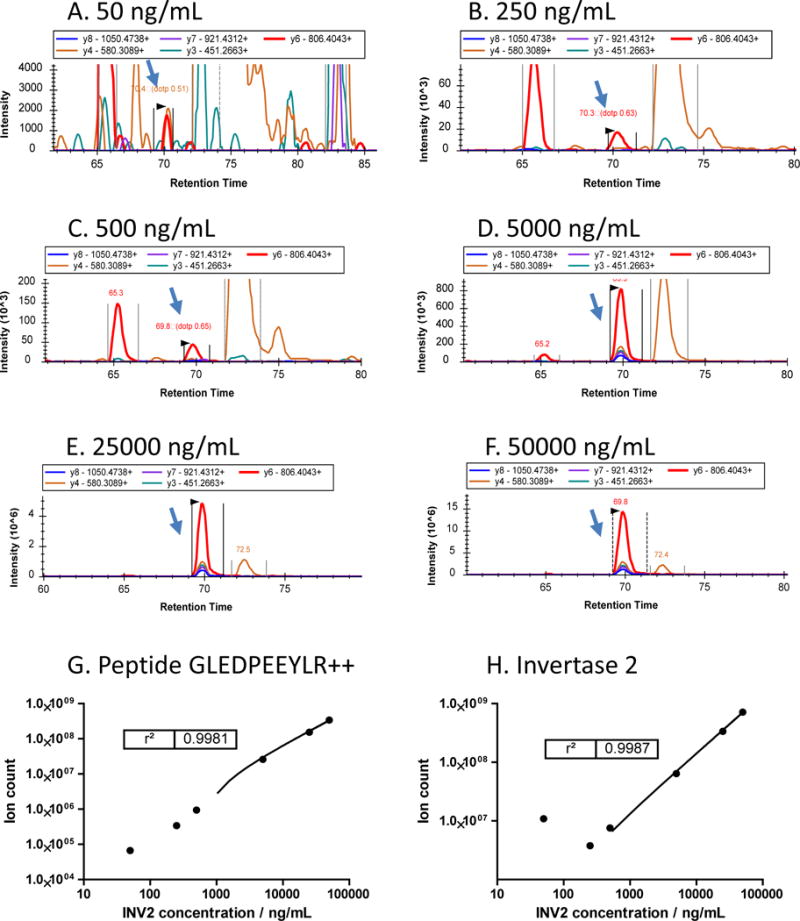
The profiles of the top 5 fragments (b and y ions with m/z more than 200) from peptide GLEDPEEYLR++ (Invertase 2) at different spiked-ion concentrations: A) 0.05 μg/mL, B) 0.25 μg/mL, C) 0.5 μg/mL, D) 5 μg/mL, E) 25 μg/mL, F) 50 μg/mL. G) Dynamic range for peptide GLEDPEEYLR++, and H) dynamic range for Invertase 2 summed from the 6 quantifiable peptides. Blue arrow indicates the target peaks from peptide GLEDPEEYLR++.
Technical validation on clinical samples
Clinical analysis of human samples introduces another dimension of complexity due to the innate differences among individual clinical samples. To test the robustness of the DIA assay, we evaluated the assay across 38 clinical plasma samples from diseased and non-diseased subjects, including 19 healthy subjects and 19 PDAC patients with stage 1 or 2 diseases. The samples were individually spiked with the Invertase 2 internal standard and were prepared in random order. Samples were analyzed in a blind fashion in a single queue with a 1 hour washing gradient between each analysis to avoid cross contamination or carry over from one sample to another. All 6 quantifiable peptides from the Invertase 2 were quantified in the 38 clinical samples resulted in a technical variation of 14.5% for Invertase 2 protein across the cohort (Figure 6). As shown in Supplemental Figure S10, the Invertase 2 quantifiable peptides were located within the volcano graphs of the total peptide variations. The sum of Invertase 2 quantifiable peptides showed a fold change of 0.99 and a p-value (based on t-test) of 0.88 when comparing Invertase 2 levels from cancer samples to healthy subjects. This high reproducibility in the ability to measure the internal standard will insure that the DIA assay is robust and that proteins of interest can be reliably measured across a variety of biosamples. Invertase 2 level can be further used for as an internal standard for normalization among inter-laboratory analysis results.
Figure 6.
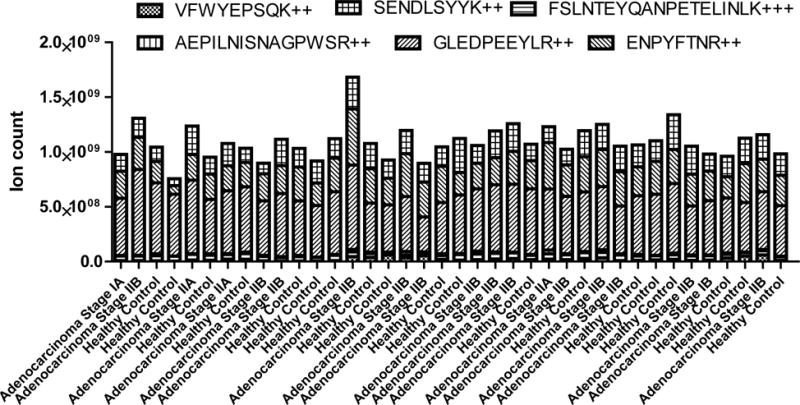
The measurement of Invertase 2 from the 38 clinical plasma samples that were prepared and analyzed in a blind fashion and a randomized order. Six quantifiable peptides were used for Invertase 2 quantification. The variation of total Invertase 2 level within the set of clinical samples is approximately 14.5%.
Conclusions
A DIA based targeted proteomics assay was devised and assessed from sample preparation to MS analysis for protein quantification in plasma, in the context of blood biomarker development. A pancreatic cancer relevant plasma spectral library was established by the extensive profiling of clinical samples from patients with PDAC, chronic pancreatitis and healthy controls. A reproducible sample preparation method was developed and verified in separate analytical and biological replicates. A set of criteria based on the characteristics of the peptides were introduced for the minimal variations. Empirical parameters, including retention time deviation and intensity CV based on replicate analysis, provided reasonable guidance in selecting quantifiable peptide for DIA analysis. The sensitivity of quantification was evaluated with different levels of spiked-in standard, suggesting that sample complexity is a significant factor in determining the limits of quantification, justifying the optimal selection of DIA isolation windows. The use of defined retention time windows can help in correct pin-pointing of the target profiles, and the advent of faster mass spectrometers with smaller isolation windows would minimize the complexity issue. The technical validation of the assay using clinical plasma samples confirmed its robustness for study of large scale clinical samples. The empirical data presented in this study also suggests an intrinsic link between peptide physicochemical characteristics and their robustness in DIA quantification, which warrants further investigation.
Supplementary Material
Supplemental Table S1. List of the plasma samples that are used to build the spectral library and to develop the analytical method.
Supplemental Table S2. The identified peptides and sequence coverage of Invertase 2 protein from yeast.
Supplemental Table S3. Comparison of different depletion columns for plasma depletion.
Supplemental Table S4. The comparison of the retention times from the spectral library with the observed retention times from the DIA analysis for the Invertase 2 peptides.
Supplemental Table S5. Selected six quantifiable peptides from Invertase 2 are presented with their corresponding retention time, retention time variation, analytical and biological variations, average library dot product, and mass spectrometry visibility.
Supplemental Figure S1. Characterization of peptides included in the spectral library.
Supplemental Figure S2. Gene Ontology analysis of the plasma proteins identified from the spectral library.
Supplemental Figure S3. Influence of isolation window size on library dot product value.
Supplemental Figure S4. Cumulative distribution of the library peptides based on their m/z value.
Supplemental Figure S5. Examples of retention time drift during sequential analysis of a large queue of clinical samples.
Supplemental Figure S6. The analytical and biological variations of the 22 peptides derived from Invertase 2.
Supplemental Figure S7. Comparison of DIA- and DDA-based quantification of peptides derived from Invertase 2 in the same plasma sample.
Supplemental Figure S8. The library dot product value for the plasma peptides in comparison with the library dot product values for the HELA cell line peptides.
Supplemental Figure S9. Characters of quantifiable peptide GLEDPEEYLR++ at different concentrations.
Supplemental Figure S10. The volcano plot of peptides in comparison of cancer vs normal control.
Summary of peptide and protein identification from the plasma profiling experiments for library construction.
Acknowledgments
This work was supported in part with federal funds from the National Institutes of Health under grants R01CA180949 and K25CA137222, and Donald E. Bocek Endowed Research Development Award in Pancreatic Cancer, and funds from the Canary Foundation and Swim Across America. This work is supported in part by the University of Washington’s Proteomics Resource (UWPR95794).
Reference List
- 1.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. MolCell Proteomics. 2012;11:O111. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 3.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS, Canterbury JD, Marsh DM, Kellmann M, Zabrouskov V, Wu CC, Maccoss MJ. Multiplexed MS/MS for improved data-independent acquisition. NatMethods. 2013;10:744–46. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egertson JD, Maclean B, Johnson R, Xuan Y, Maccoss MJ. Multiplexed peptide analysis using data-independent acquisition and Skyline. NatProtoc. 2015;10:887–903. doi: 10.1038/nprot.2015.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Buil A, Collins BC, Gillet LC, Blum LC, Cheng LY, Vitek O, Mouritsen J, Lachance G, Spector TD, Dermitzakis ET, Aebersold R. Quantitative variability of 342 plasma proteins in a human twin population. MolSystBiol. 2015;11:786. doi: 10.15252/msb.20145728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberger G, Koh CC, Guo T, Rost HL, Kouvonen P, Collins BC, Heusel M, Liu Y, Caron E, Vichalkovski A, Faini M, Schubert OT, Faridi P, Ebhardt HA, Matondo M, Lam H, Bader SL, Campbell DS, Deutsch EW, Moritz RL, Tate S, Aebersold R. A repository of assays to quantify 10,000 human proteins by SWATH-MS. SciData. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Searle BC, Egertson JD, Bollinger JG, Stergachis AB, Maccoss MJ. Using Data Independent Acquisition (DIA) to Model High-responding Peptides for Targeted Proteomics Experiments. MolCell Proteomics. 2015;14:2331–40. doi: 10.1074/mcp.M115.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selevsek N, Chang CY, Gillet LC, Navarro P, Bernhardt OM, Reiter L, Cheng LY, Vitek O, Aebersold R. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. MolCell Proteomics. 2015;14:739–49. doi: 10.1074/mcp.M113.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mischak H, Allmaier G, Apweiler R, Attwood T, Baumann M, Benigni A, Bennett SE, Bischoff R, Bongcam-Rudloff E, Capasso G, Coon JJ, D’Haese P, Dominiczak AF, Dakna M, Dihazi H, Ehrich JH, Fernandez-Llama P, Fliser D, Frokiaer J, Garin J, Girolami M, Hancock WS, Haubitz M, Hochstrasser D, Holman RR, Ioannidis JP, Jankowski J, Julian BA, Klein JB, Kolch W, Luider T, Massy Z, Mattes WB, Molina F, Monsarrat B, Novak J, Peter K, Rossing P, Sanchez-Carbayo M, Schanstra JP, Semmes OJ, Spasovski G, Theodorescu D, Thongboonkerd V, Vanholder R, Veenstra TD, Weissinger E, Yamamoto T, Vlahou A. Recommendations for biomarker identification and qualification in clinical proteomics. SciTranslMed. 2010;2:46ps42. doi: 10.1126/scitranslmed.3001249. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Huttenhain R, Surinova S, Gillet LC, Mouritsen J, Brunner R, Navarro P, Aebersold R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics. 2013;13:1247–56. doi: 10.1002/pmic.201200417. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Chen J, Sethi A, Li QK, Chen L, Collins B, Gillet LC, Wollscheid B, Zhang H, Aebersold R. Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. MolCell Proteomics. 2014;13:1753–68. doi: 10.1074/mcp.M114.038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, Maclean B, Aebersold R. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426–41. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 13.Chapman JD, Goodlett DR, Masselon CD. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass SpectromRev. 2013 doi: 10.1002/mas.21400. [DOI] [PubMed] [Google Scholar]
- 14.Sajic T, Liu Y, Aebersold R. Using data-independent, high resolution mass spectrometry in protein biomarker research: Perspectives and clinical applications. Proteomics Clin Appl. 2014 doi: 10.1002/prca.201400117. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., III Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113:2343–94. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. MolCell Proteomics. 2002;1:845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 17.Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. ClinChem. 2010;56:177–85. doi: 10.1373/clinchem.2009.126706. [DOI] [PubMed] [Google Scholar]
- 18.Surinova S, Schiess R, Huttenhain R, Cerciello F, Wollscheid B, Aebersold R. On the development of plasma protein biomarkers. J Proteome Res. 2011;10:5–16. doi: 10.1021/pr1008515. [DOI] [PubMed] [Google Scholar]
- 19.Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG, Fagan R, Lobley A. The human plasma proteome: a nonredundant list developed by combination of four separate sources. MolCell Proteomics. 2004;3:311–26. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Anderson NL, Anderson NG, Pearson TW, Borchers CH, Paulovich AG, Patterson SD, Gillette M, Aebersold R, Carr SA. A human proteome detection and quantitation project. MolCell Proteomics. 2009;8:883–86. doi: 10.1074/mcp.R800015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enroth S, Johansson A, Enroth SB, Gyllensten U. Strong effects of genetic and lifestyle factors on biomarker variation and use of personalized cutoffs. Nat Commun. 2014;5:4684. doi: 10.1038/ncomms5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majumder S, Chari ST. Chronic pancreatitis. Lancet. 2016;387:1957–66. doi: 10.1016/S0140-6736(16)00097-0. [DOI] [PubMed] [Google Scholar]
- 23.Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics ClinAppl. 2015 doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. MolSystBiol. 2005;1:2005. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 26.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 27.Chen R, Brentnall TA, Pan S, Cooke K, Moyes KW, Lane Z, Crispin DA, Goodlett DR, Aebersold R, Bronner MP. Quantitative proteomics analysis reveals that proteins differentially expressed in chronic pancreatitis are also frequently involved in pancreatic cancer. MolCell Proteomics. 2007;6:1331–42. doi: 10.1074/mcp.M700072-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Chen R, Yi EC, Donohoe D, Pan S, Eng J, Crispin DA, Lane Z, Goodlett DA, Bronner MP, Aebersold R, Brentnall TA. Pancreatic Cancer Proteome: the Proteins that Underlie Invasion, Metastasis, and Immunologic Escape. Gastroenterology. 2005;129:1187–97. doi: 10.1053/j.gastro.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Marcotte EM. How do shotgun proteomics algorithms identify proteins? Nat Biotechnol. 2007;25:755–57. doi: 10.1038/nbt0707-755. [DOI] [PubMed] [Google Scholar]
- 30.Mount DW. Using the Basic Local Alignment Search Tool (BLAST) CSHProtoc. 2007;2007 doi: 10.1101/pdb.top17. db. [DOI] [PubMed] [Google Scholar]
- 31.Bjorhall K, Miliotis T, Davidsson P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics. 2005;5:307–17. doi: 10.1002/pmic.200400900. [DOI] [PubMed] [Google Scholar]
- 32.Roche S, Tiers L, Provansal M, Seveno M, Piva MT, Jouin P, Lehmann S. Depletion of one, six, twelve or twenty major blood proteins before proteomic analysis: the more the better? J Proteomics. 2009;72:945–51. doi: 10.1016/j.jprot.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Tu C, Rudnick PA, Martinez MY, Cheek KL, Stein SE, Slebos RJ, Liebler DC. Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res. 2010;9:4982–91. doi: 10.1021/pr100646w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zolotarjova N, Martosella J, Nicol G, Bailey J, Boyes BE, Barrett WC. Differences among techniques for high-abundant protein depletion. Proteomics. 2005;5:3304–13. doi: 10.1002/pmic.200402021. [DOI] [PubMed] [Google Scholar]
- 35.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. MolCell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. NatProtoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 37.Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Nimeus-Malmstrom E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. SciTranslMed. 2012;4:142ra94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25:125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 39.Reiter L, Rinner O, Picotti P, Huttenhain R, Beck M, Brusniak MY, Hengartner MO, Aebersold R. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8:430–35. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. List of the plasma samples that are used to build the spectral library and to develop the analytical method.
Supplemental Table S2. The identified peptides and sequence coverage of Invertase 2 protein from yeast.
Supplemental Table S3. Comparison of different depletion columns for plasma depletion.
Supplemental Table S4. The comparison of the retention times from the spectral library with the observed retention times from the DIA analysis for the Invertase 2 peptides.
Supplemental Table S5. Selected six quantifiable peptides from Invertase 2 are presented with their corresponding retention time, retention time variation, analytical and biological variations, average library dot product, and mass spectrometry visibility.
Supplemental Figure S1. Characterization of peptides included in the spectral library.
Supplemental Figure S2. Gene Ontology analysis of the plasma proteins identified from the spectral library.
Supplemental Figure S3. Influence of isolation window size on library dot product value.
Supplemental Figure S4. Cumulative distribution of the library peptides based on their m/z value.
Supplemental Figure S5. Examples of retention time drift during sequential analysis of a large queue of clinical samples.
Supplemental Figure S6. The analytical and biological variations of the 22 peptides derived from Invertase 2.
Supplemental Figure S7. Comparison of DIA- and DDA-based quantification of peptides derived from Invertase 2 in the same plasma sample.
Supplemental Figure S8. The library dot product value for the plasma peptides in comparison with the library dot product values for the HELA cell line peptides.
Supplemental Figure S9. Characters of quantifiable peptide GLEDPEEYLR++ at different concentrations.
Supplemental Figure S10. The volcano plot of peptides in comparison of cancer vs normal control.
Summary of peptide and protein identification from the plasma profiling experiments for library construction.