

Abstract
A quantitative trait locus (QTL) on proximal chromosome (Chr) 10 accounts for >50% of the genetic variance in methamphetamine (MA) intake in mice selectively bred for high (MAHDR) and low (MALDR) voluntary MA drinking. The μ-opioid receptor (MOP-r) gene, Oprm1, resides at the proximal end of Chr 10, and buprenorphine reduces MA intake in MAHDR mice. However, this drug has only partial agonist effects at MOP-r. We investigated the impact of a full MOP-r agonist, morphine, on MA intake and saccharin intake, measured MOP-r density and affinity in several brain regions of the MA drinking lines and their C57BL/6J (B6) and DBA/2J (D2) progenitor strains, and measured MA intake in two congenic strains of mice to verify the QTL and reduce the QTL interval. Morphine reduced MA intake in the MAHDR line, but also reduced saccharin and total fluid intake. MOP-r density was lower in the medial prefrontal cortex of MAHDR, compared to MALDR, mice, but not in the nucleus accumbens or ventral midbrain; there were no MOP-r affinity differences. No significant differences in MOP-r density or affinity were found between the progenitor strains. Finally, Chr 10 congenic results were consistent with previous data suggesting that Oprm1 is not a quantitative trait gene, but is impacted by the gene network underlying MA intake. Stimulation of opioid pathways by a full agonist can reduce MA intake, but may also non-specifically affect consummatory behavior; thus, a partial agonist may be a better pharmacotherapeutic.
Keywords: addiction, selected lines, congenic, morphine, QTL, amphetamine
INTRODUCTION
Excessive, chronic methamphetamine (MA) use has debilitating consequences, and existing findings support a genetic component to risk for chronic use (Aoyama et al. 2006; Iamjan et al. 2015). The MA high drinking (MAHDR) and MA low drinking (MALDR) lines of mice were selectively bred based on level of voluntary MA intake and provide a relevant animal model of differential genetic risk for MA use, as recently reviewed (Phillips and Shabani 2015; Shabani et al. 2016). Critical features of the model include binge-level MA intake in the MAHDR line (Shabani et al. 2016); no difference between the lines in quinine, saccharin (SAC), potassium chloride or sodium chloride preference or consumption (Shabani et al. 2011; Wheeler et al. 2009); equivalent patterns of learning to operantly self-administer natural rewards (Shabani et al. 2012a); operant intracranial and oral MA self-administration in the MAHDR line that is absent in the MALDR line (Shabani et al. 2012a); evidence of MA-conditioned reward in MAHDR mice that is absent in the MALDR line (Shabani et al. 2011; Wheeler et al. 2009); high sensitivity to aversive effects of MA in MALDR mice that is drastically reduced in the MAHDR line (Shabani et al. 2011; 2012b; Wheeler et al. 2009); and equivalent sensitivity to cocaine effects (Gubner et al. 2013). Results for male and female mice have been largely comparable across all of these traits, and when found have never been genotype-dependent. Gene mapping using genetic samples from the MAHDR and MALDR lines, identified a quantitative trait locus (QTL) on proximal chromosome (Chr) 10 that accounts for >50% of the genetic variance in MA intake (Belknap et al. 2013), and existing evidence indicates that the trace amine-associated receptor 1 (Taar1) gene at 23.9 Mb on mouse Chr 10 influences MA intake (Harkness et al. 2015). However, there could be more than one influential gene on Chr 10 and Oprm1, the mu-opioid receptor (MOP-r) gene at 6.75 Mb, attracted our attention as a potential candidate for several reasons. Human OPRM1 is associated with MA-induced psychosis and dependence (Ide et al. 2004), and MOP-r drugs attenuate MA intake and other MA-related traits in humans and rodents (Dlugos et al. 2011; Eastwood and Phillips 2014b; Ide et al. 2004; Jayaram-Lindstrom et al. 2008a; 2008b; 2017), although there are mixed results for agonists vs. antagonists. Microarray gene expression analysis in MA-naïve MA drinking (MADR) mice, using tissue from several reward pathway brain regions, including the medial prefrontal cortex (mPFC), nucleus accumbens (NAc), and ventral midbrain (Vmb), identified Oprm1 as a differentially expressed gene, with MAHDR mice having lower Oprm1 expression only in the mPFC, compared to MALDR mice (Belknap et al. 2013). This suggested that reduced MOP-r-regulated function in the mPFC might lead to greater risk for MA intake.
MALDR mice exhibit greater MOP-r agonist-stimulated locomotor activity and voluntary morphine (MOR) intake, compared to MAHDR mice (Eastwood and Phillips 2014a). The MOP-r antagonist, naltrexone, had no effect on MA intake in MAHDR mice, and slightly enhanced MA intake in MALDR mice, whereas low doses of buprenorphine (BUP), a MOP-r partial agonist, reduced MA consumption in MAHDR mice (Eastwood and Phillips 2014b), without effects on general fluid intake. BUP binds to receptors other than the MOP-r (Wang et al. 2015) and the effects of BUP on MA intake were characterized by a U-shaped dose-response curve, possibly due to MOP-r agonist effects at lower doses and MOP-r antagonist effects at higher doses (Sadée et al. 1982; Pick et al. 1997). In the current studies, MOR, which has full agonist and no antagonist effects on the MOP-r, was examined for its effects on MA intake. In addition, MOR effects on SAC and total fluid intake were assessed to address whether effects of MOR were specific to MA intake, as opposed to other consummatory behaviors. The founding population for the MADR lines was the F2 cross of the C57BL/6J (B6) and DBA/2J (D2) inbred mouse strains. To determine whether selective breeding had effects on MOP-r that were different than those pre-existing in the progenitor strains, and confirm that gene expression differences in the MADR mice are accompanied by differences in protein level, MOP-r density was assessed in the MADR lines and B6 and D2 strains; we also examined affinity. Finally, MA intake was examined in two Chr 10 congenic strains to confirm the MA drinking QTL on an isogenic background, and potentially reduce the size of the QTL interval. Each congenic possessed a different length Chr 10 segment from the B6 strain introgressed onto the D2 strain background.
MATERIALS AND METHODS
Animals and Husbandry
The MADR mouse lines were created in replicate, using short-term, mass selection (Belknap et al. 1997; Shabani et al. 2011; Wheeler et al. 2009). MADR mice used here were second or later litter MA-naïve offspring of the fifth selection generation of replicate 2. Congenic strain breeders were received from the University of Pennsylvania and have been previously described (Doyle et al. 2014). The congenic strains possess a Chr 10 B6 segment of 7.72 (D2.B6 0–7.72 Mb) or 20.4 (Chr 10 D2.B6 0–20.4 Mb) Mb in size. Within each congenic, mice heterozygous for the Chr 10 introgressed B6 segment were mated to produce the homozygous D2 and homozygous D2.B6 littermate mice used here. PCR genotyping of polymorphic markers was used to determine where the transition from B6 to D2 markers occurred. Mice were weaned at 21–23 days of age and same-sex housed (2–5 mice per shoebox cage; 28.5 × 17.5 × 12 cm with wire cagetops) on Bed-O-Cob™ bedding (The Anderson Inc., Maumee, OH). Room temperature was 21±1°C and mice were maintained on a 12:12 h light:dark cycle. B6 and D2 mice were obtained from The Jackson Laboratory (Bar Harbor, ME) at 10 weeks of age and housed for 2 weeks in our animal facility before use, under the same conditions as all others. All procedures were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the VA Portland Health Care System. Measures were taken to minimize pain and discomfort.
Drugs
(+) MA hydrochloride (HCl), SAC sodium salt, and [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO), were purchased from Sigma (St. Louis, MO); morphine sulfate and [3H]DAMGO (46 ci/mmol) were obtained from the NIDA drug supply program (Bethesda, MD). For drinking, MA and SAC were dissolved in tap water. For injections, MOR was dissolved in saline (0.9% NaCl, Baxter Healthcare Corporation, Deerfield, IL). All injections were intraperitoneal (IP; 10 ml/kg).
Effect of MOR on MA two-bottle choice drinking
Male and female mice were placed on a reverse 12:12h light:dark cycle (lights off at 0700h and on at 1900 h) at least 2 weeks before the study began, to allow drinking behavior to be monitored during the dark phase, the natural period of consummatory behavior in mice. A 6-h monitoring period was chosen to allow for detection of potentially short-lasting MOR effects, since MOR has an approximately 75-minute half-life in brain after reaching systemic circulation (Kalvass et al. 2007). The procedure matched that used to examine the effect of BUP on the acquisition of MA drinking (Eastwood and Phillips 2014b). Mice were isolate housed on day 1 and offered two, 25-ml, water-filled graduated cylinders with stoppers and ball-bearing sipper tubes for 2 days. One tube was removed at 1300 h on day 2. On days 3 and 4, mice were injected with saline 30 min prior to dark-phase onset, the water tube was replaced at 0700h, and drinking tube volumes were measured every 2h for 6h; these procedures familiarized the mice with handling, injection and the tube-reading process, which occurred under red light conditions. On days 5–12, the same procedures were followed, except that saline or MOR (5, 10, or 15 mg/kg) was administered 30 min before dark onset and an MA tube was placed on the cage at dark onset for 6 h. The MA concentration was 20 mg/l on days 5–8 and 40 mg/l on days 9–12. The doses of MOR were based on previous experiments in which 20 and 30 mg/kg MOR significantly increased locomotor activity in MALDR, but had no effect in MAHDR mice (Eastwood and Phillips 2014a); thus, an impact of these doses on MA intake in MAHDR mice would not be associated with effects on general locomotor behavior. The positions of the water and MA tubes were alternated every 2 days, consistent with selection trait testing (Shabani et al. 2011). At the end of each 6-h period, the MA tube was removed and the water tube was left in place for the remaining 18-h. Mice were 71–97 days old at study initiation, and data were collected in 4 cohorts (N=2–3/sex/line/dose/cohort; for MAHDR: 9 female and 7 male, 9 female and 7 male, 7 female and 8 male, and 9 female and 7 male for MOR doses 0, 5, 10 and 15 mg/kg, respectively; for MALDR: 8 female and 8 male, 9 female and 9 male, 9 female and 8 male, and 7 female and 8 male for MOR doses 0, 5, 10 and 15 mg/kg, respectively).
Effect of MOR on SAC two-bottle choice drinking
The effect of MOR on SAC drinking in MADR mice was examined using the same procedures and 0.033 and 0.066% SAC solutions that are equivalently preferred by the MADR lines (Shabani et al. 2011). Due to limited availability of the mice, only two doses of MOR (7.5 and 15 mg/kg) were used, which were chosen based on the results of the MOR-MA study. Data were collected in 2 cohorts (N=4–7/line/dose/cohort; for MAHDR: 8, 11 and 11 female mice for MOR doses 0, 7.5 and 15 mg/kg, respectively; for MALDR: 11, 10 and 10 female mice for MOR doses 0, 7.5 and 15 mg/kg, respectively) and mice were 73–115 days old at study initiation. Only female mice were used for this study, as no significant sex × line interaction effects have been found for the MADR mice in previous studies of MA or SAC intake (Shabani et al. 2011; Wheeler et al. 2009), and the sexes did not differ with regard to MOR dose effects in the current MOR-MA study or our previous BUP-MA study (Eastwood and Phillips 2014b).
Brain tissue preparation
The mPFC and NAc were collected from a 1–1.3 mm slice, 0.5 mm anterior to the anterior commissure, using a micropunch (16 gauge blunt cut needle). The Vmb included the ventral tegmental area and was collected from the distal 1.0 mm slice of the hypothalamic region. Because regions and protein content were small, tissue from 5 animals was pooled to comprise a sample for each region. Samples were placed in assay buffer (50 mM ice-cold Tris buffer, pH 7.5) and homogenized for 30 seconds. Homogenates were microfuged at 14,000 rpm for 20 min at 4° C. The resulting pellet was washed with assay buffer and microfuged at 14,000 rpm for 20 min at 4° C. The pellet was re-suspended in ice-cold assay buffer, and homogenized. Protein concentration was assayed using a BCA protein assay kit (Thermo Scientific, Rockford, IL) with bovine serum albumin as the standard. Thirty drug-naïve female mice per line or strain were dissected for a final N=6 samples/line or strain. Male mice were in short supply and we have not found sex × genotype interaction effects for the MA drinking trait (Shabani et al. 2011; Wheeler et al. 2009). MADR mice were 95–121 days old at the time of dissection; B6 and D2 mice were 84–94 days old.
Ligand binding assay
For saturation binding, membranes were incubated (2–20 μg protein) in assay buffer in a water bath at 25°C for 60 min with 5 concentrations of [3H]DAMGO (0.145–4.85 nM), a MOP-r agonist. Non-specific binding was measured in the presence of 1 μM unlabeled DAMGO. The incubation was terminated by rapid filtration through PerkinElmer (Waltham, MA) Filtermat A filters presoaked in 0.05% polyethylenimine on a Tomtec (Hamden, CT) cell harvester. Filters were dried and spotted with scintillation cocktail and the amount of radioactivity retained on the filters was determined using a PerkinElmer microBeta plate 1405.
Two-bottle choice MA drinking in congenics
D2.B6 congenic strain mice were tested for MA intake using the 2-bottle choice procedure utilized to create the MADR lines. Thus, 20 and then 40 mg/l MA was offered vs. water for 18 h each day on 8 consecutive days (Shabani et al. 2011). The 18-h period extended from 3 h prior to the beginning of the dark phase until 3 h after the dark phased ended. Mice were 80–140 days of age and data were collected in 7 cohorts (N=13–48/cohort). The number of mice tested was 78 Chr 10 D2.B6 0–7.72 Mb congenic and background strain mice (N=16–25/sex/strain; 19 female and 25 male Chr 10 D2.B6 0–7.72 Mb controls, and 16 female and 18 male Chr 10 D2.B6 0–7.72 Mb congenics) and 45 Chr 10 D2.B6 0–20.4 Mb congenic and background strain mice (N=2–18/sex/strain; 14 female and 18 male Chr 10 D2.B6 0–20.4 Mb controls, and 2 female and 11 male Chr 10 D2.B6 0–20.4 Mb congenics). Fewer animals were produced by the longer segment het × het breeders and only 2 congenic females out of 8 litters were produced. Therefore, the effect of sex was not examined within this congenic, but it was for their background strain littermates.
Statistical analysis
Behavioral data were analyzed by repeated measures Analysis of Variance (ANOVA; Statsoft Version 9, Tulsa, OK) with MA concentration and time as possible repeated measures. Independent variables, depending on the study, were line or strain, sex, MOR dose, MA concentration and time. Dependent variables are described with experimental results. Significant multifactor interactions were deconstructed by identifying significant two-way interactions within particular levels of a factor and then further resolved by simple main effects analysis and the Newman Keuls post-hoc test for mean comparisons. Binding data were analyzed using Prism GraphPad software (version 6.04; San Diego, CA, USA). For saturation isotherms, the total binding capacity (Bmax) and equilibrium constant (KD) were determined using a one-site model for MOP-r binding sites and Scatchard analyses were performed. Specific binding was determined by subtracting nonspecific binding from total binding. Genotype effects for Bmax and KD were determined by Student’s t-test. The criterion for significance was p<0.05. Illustrations were created with Powerpoint version 14.0.7184.5000 (2010 Microsoft Corporation, Redmond, WA) or SigmaPlot version 12.5 (Systat Software, Inc., San Jose, CA).
RESULTS
Effects of MOR on MA two-bottle choice drinking
MOR had dose-, line- and time-dependent effects on MA intake, measured in mg/kg using body weight data collected every 2 days. Initial analysis identified significant line × MOR dose × MA concentration [F(3,121)=2.7; p<0.05], time × line × MOR dose [F(6,242)=2.7; p<0.05], and time × line × MA concentration [F(2,242)= 14.0; p<0.001] interactions. There were no significant sex effects. For 20 mg/l MA, there was a significant time × line × MOR dose interaction [F(6,242)=3.5; p<0.01]. MOR dose effects were line-dependent during the first 2h [F(3,121)=3.9; p<0.05], with the 10 and 15 mg/kg doses eliminating the selected line difference in MA consumption (Fig. 1a). There was a significant effect of MOR dose within the MAHDR, but not MALDR, mice [F(3,121)=5.5; p<.01). During the second 2h, there was a trend for a line × MOR dose interaction (p=0.06) and a significant main effect of line [F(1,121)=9.6; p<0.01], reflecting greater MA intake in MAHDR mice (Fig. 1b). During the final 2h (Fig. 1c), only a significant line difference in MA intake was found [F(1,121)=18.3; p<0.001]. For the 40 mg/l MA concentration, there was a significant time × line interaction [F(2,242)=16.0; p<0.001], but no significant effects of MOR (Fig. 1d–f). For each 2-h period, MAHDR mice consumed more MA than MALDR mice [F(1,121)=6.6; p<0.05; F(1,121)=113.8; p<0.001; F(1,121)=29.7; p<0.001, for each 2h, respectively].
Fig. 1. Effect of MOR treatment on MA intake.
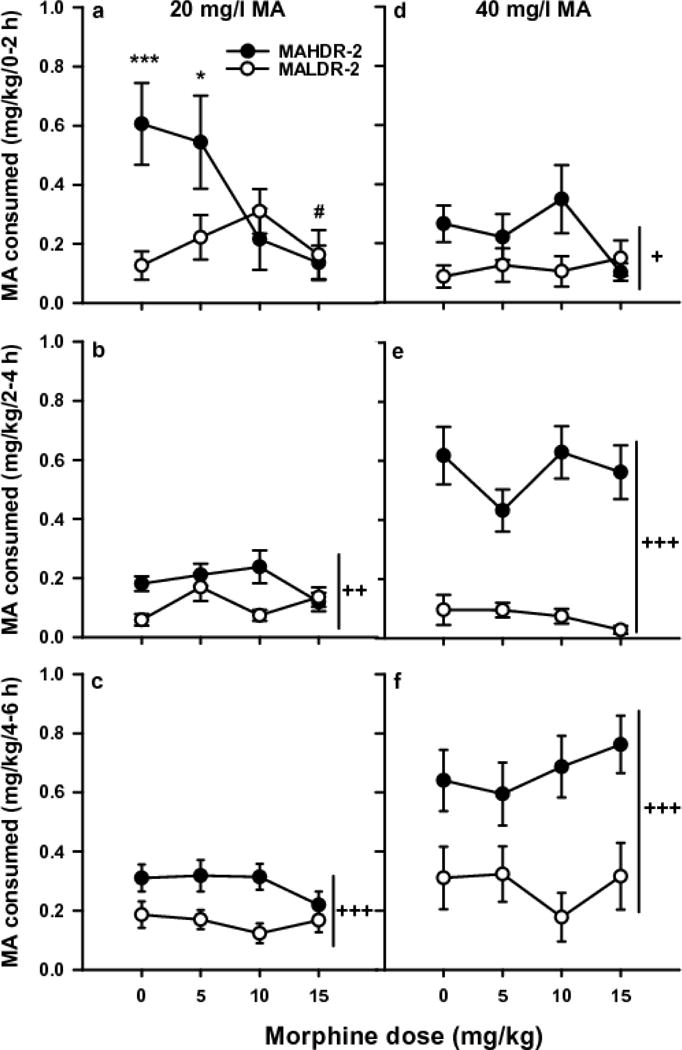
Means ± SEM for MA consumed from the 20 mg MA/l (a–c) and 40 mg MA/l (d–f) solutions for the three, 2-h periods of the 6-h MA vs. water limited access study. Each data point is a 2-day average for days 2 and 4 of the 4-day period for each MA concentration. *p<0.05, ***p<0.001 for the line difference at the indicated MOR dose. #p<0.01 for the main effect of MOR dose within the MAHDR line. +p<0.05, ++p<0.01, +++p<0.001 for the main effect of line. N=15–18/line/dose
Total volume consumed from the water and MA tubes during the 6-h access periods, corrected for body weight (ml/kg), are shown in Table 1. There were significant MOR dose × MA concentration × sex [(F(3,113)=2.9; p<0.05], time × MOR dose × sex [F(6,226)=2.2; p<0.05], and line × MA concentration [F(1,113)= 12.3; p<0.001] interactions, but no significant interactions for MOR dose that involved line. For the 20 mg/l MA access period, significant time × MOR dose, and time × sex [F(6,226)= 2.2; p<0.05 and F(2,226)=3.2; p<0.05] interactions were detected. There was a significant effect of MOR dose during the first 2h [F(3,113)=7.4; p<0.001]; 10 and 15 mg/kg MOR reduced total volume consumed compared to saline. For the second 2-h period, there was a significant effect of MOR dose [F(3,113)=4.5; p<.01], as well as a significant effect of sex [F(1,113)=13.7; p<.001]. Total volume was lower for the 15 mg/kg MOR-treated mice compared only to the 5 mg/kg MOR-treated mice, and females consumed more total volume per body weight than males. For the third 2-h period, there was only a significant main effect of sex [F(1,113)= 10.4; p<0.01]; female mice consumed more total volume per body weight, compared to male mice. For the 40 mg/l MA access period, significant time × MOR dose [F(6,226)=2.5; p<0.05] and time × sex [F(2,226= 7.7; p<0.001] interactions were found. Again, there were no significant interactions with MOR dose that involved line. There was a significant effect of MOR dose during the first 2h [F(3,113)=5.1; p<0.01]; the 15 mg/kg dose reduced total volume intake compared to all other dose groups. During the next two, 2-h periods there was a significant effect of sex [F(1,113)=9.4; p<0.01 and F(1,113)=6.2; p<0.05, respectively]; female mice consumed more in total volume per body weight, compared to male mice.
Table 1.
Total volume consumed during the MOR treatment, MA vs water two-bottle choice drinking study.
| 20 mg MA/l | 40 mg MA/l | ||||||
|---|---|---|---|---|---|---|---|
| Line | MOR dose | 0-2 h (ml/kg) | 2-4 h (ml/kg) | 4-6 h (ml/kg) | 0-2 h (ml/kg) | 2-4 h (ml/kg) | 4-6 h (ml/kg) |
| MAHDR | 0 mg/kg | 17.5 ± 2.0 | 27.2 ± 3.7 | 36.9 ± 3.2 | 24.7 ± 4.1 | 41.0 ± 4.4 | 44.8 ± 4.2 |
| MALDR | 0 mg/kg | 21.8 ± 8.4 | 28.1 ± 4.3 | 41.1 ± 5.4 | 20.3 ± 4.0 | 32.5 ± 5.1 | 39.7 ± 5.9 |
| MAHDR | 5 mg/kg | 12.2 ± 2.4 | 29.3 ± 2.3 | 35.1 ± 4.2 | 20.1 ± 3.9 | 38.6 ± 4.1 | 49.4 ± 3.4 |
| MALDR | 5 mg/kg | 14.9 ± 2.5 | 38.8 ± 4.4 | 40.3 ± 3.5 | 17.3 ± 2.8 | 38.5 ± 4.8 | 45.0 ± 4.4 |
| MAHDR | 10 mg/kg | 7.8 ± 1.4* | 29.3 ± 4.2 | 38.6 ± 4.1 | 22.5 ± 5.0 | 46.0 ± 4.2 | 46.2 ± 5.7 |
| MALDR | 10 mg/kg | 11.9 ± 2.6* | 28.3 ± 3.6 | 36.9 ± 6.2 | 15.5 ± 3.5 | 40.6 ± 5.5 | 36.3 ± 5.4 |
| MAHDR | 15 mg/kg | 3.4 ± 1.0++ | 16.3 ± 1.7 | 27.5 ± 3.3 | 5.9 ± 1.4+ | 31.2 ± 3.6 | 39.4 ± 3.3 |
| MALDR | 15 mg/kg | 3.7 ± 1.3++ | 27.1 ± 2.7 | 32.1 ± 4.4 | 12.5 ± 2.8+ | 33.3 ± 5.0 | 42.6 ± 5.3 |
Shown are means ± SEM total volume consumed in ml/kg for each 2-h period of MA vs water access. MOR pretreatments were given 30 min before the onset of the MA vs water access period. Listed is the MA concentration that was offered vs. water at the time that total volume was assessed. Significant results for MOR in comparison to saline are indicated;
p<.05 for the effect of the 10 mg/kg MOR dose (collapsed on line), compared to the 0 mg/kg MOR dose (collapsed on line);
p<.01 and
p<.001 for the effect of the 15 mg/kg MOR dose (collapsed on line), compared to the 0 mg/kg MOR dose (collapsed on line).
Effects of MOR on SAC two-bottle choice drinking
MOR dose-dependently reduced intake (mg/kg) of the lower SAC concentration, an effect that was not dependent upon time of measurement. Initial analyses identified a significant time × line × MOR dose × SAC concentration interaction [F(4,110)=2.9; p<0.05]; however, subsequent analyses did not detect any significant effects of line. For 0.033% SAC (Fig. 2a–c), there was a significant main effect of MOR dose [F(2,55)=4.3; p<.05] and of time [F(2,110)=10.0; p<0.001]. Less SAC was consumed after treatment with the 15 mg/kg MOR dose, compared to saline (ps<.05), and more SAC was consumed during the final 2h, compared to the first and second 2h periods (p<0.01). For 0.066% SAC (Fig. 2d–f), there was a significant main effect of time [F(2,110)=4.4; p<0.05], with more SAC consumed during the second 2h period than during the third. There was no significant effect of MOR dose.
Fig. 2. Effect of MOR treatment on SAC intake.
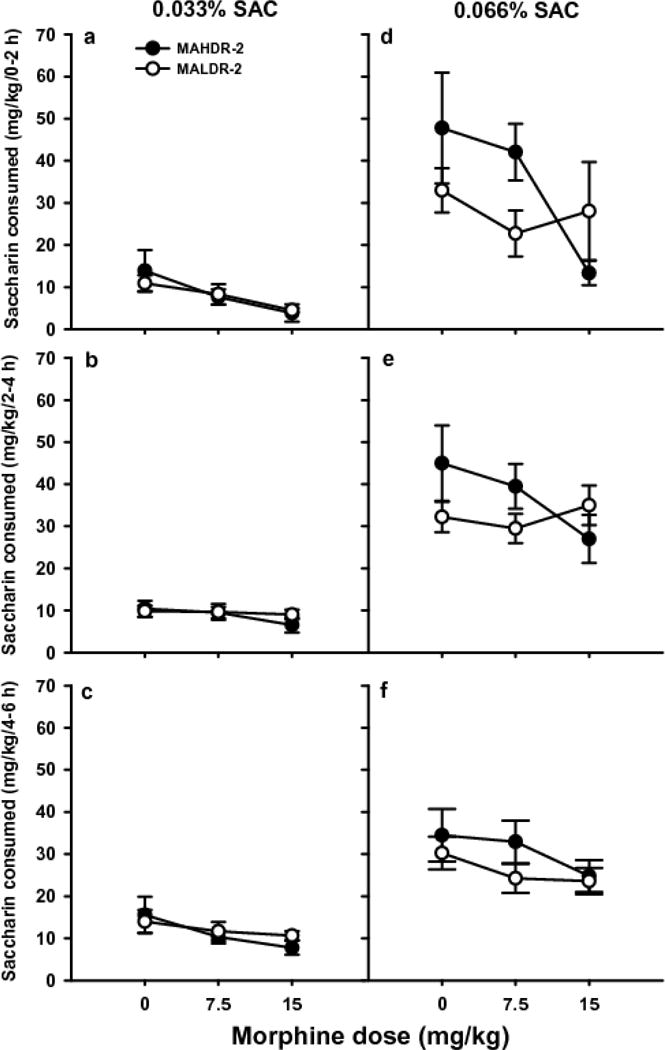
Means ± SEM for SAC consumed from the 0.033% (a–c) and 0.066% (d–f) solutions for the three, 2-h periods of the 6-h SAC vs. water limited access study. Each data point is a 2-day average for days 2 and 4 of the 4-day period for each SAC concentration. N=8–11/line/dose
Data for total volume consumed (ml/kg) during the SAC study are summarized in Table 2. Initial analyses identified significant line × SAC concentration [F(1,55)=6.7; p<0.05], time × SAC concentration [F(2,110)=42.4; p<0.001], and time × MOR dose [F(4,110)=4.4; p<0.01] interactions. Subsequent analyses did not detect any significant effects of line. For total volume during the 0.033% SAC access period, a significant time × MOR dose interaction was detected [F(4,110)=4.2; p<0.01]. For the first 2-h period, there was a significant effect of MOR dose [F(2,55)=5.8; p<0.01]; the 15 mg/kg dose reduced total volume consumed compared to saline. There was no significant effect of MOR dose for the other 2-h periods. For total volume during the 0.066% SAC access period, a significant effect of MOR dose [F(2,55)= 3.2; p<0.05] was detected; post-hoc tests indicated a strong statistical trend (p=0.06) toward lower total volume in the 15 mg/kg MOR dose group, compared to the saline group. There was a significant effect of time [F(2,110)= 6.0; p<0.01] associated with more total volume consumed during the second 2h, compared to the first and last 2h periods.
Table 2.
Total volume consumed during the MOR treatment, SAC vs water, two-bottle choice drinking study.
| 0.33% SAC | 0.66% SAC | ||||||
|---|---|---|---|---|---|---|---|
| Line | MOR dose | 0–2 h (ml/kg) | 2–4 h (ml/kg) | 4–6 h (ml/kg) | 0–2 h (ml/kg) | 2–4 h (ml/kg) | 4–6 h (ml/kg) |
| MAHDR | 0 mg/kg | 33.0 ± 6.6 | 41.6 ± 4.5 | 61.5 ± 13.2 | 76.2 ± 19.1 | 76.4 ± 12.2 | 60.6 ± 7.2 |
| MALDR | 0 mg/kg | 25.3 ± 2.8 | 40.4 ± 4.1 | 65.0 ± 6.8 | 53.7 ± 7.5 | 56.1 ± 5.4 | 55.7 ± 4.8 |
| MAHDR | 7.5 mg/kg | 20.5 ± 8.0 | 42.3 ± 4.0 | 44.6 ± 3.8 | 70.5 ± 10.2 | 69.3 ± 7.7 | 58.0 ± 7.4 |
| MALDR | 7.5 mg/kg | 23.5 ± 3.7 | 44.3 ± 4.8 | 48.3 ± 5.7 | 42.8 ± 7.6 | 58.6 ± 4.2 | 45.2 ± 4.3 |
| MAHDR | 15 mg/kg | 5.1 ± 1.1+ | 27.0 ± 5.6 | 50.2 ± 7.4 | 22.1 ± 4.7* | 46.8 ± 8.5 | 45.8 ± 4.4 |
| MALDR | 15 mg/kg | 15.2 ± 5.2+ | 43.8 ± 4.6 | 58.2 ± 4.8 | 46.0 ±17.3* | 64.1 ± 7.9 | 45.9 ± 5.7 |
Shown are means ± SEM total volume consumed in ml/kg for each 2-h period of SAC vs water access. MOR pretreatments were given 30 min before the onset of the SAC vs water access period. Listed is the SAC concentration that was offered vs. water at the time that total volume was assessed.
p=.06 for a trend toward an effect of the 15 mg/kg MOR dose (collapsed on line), compared to the 0 mg/kg MOR dose (collapsed on line) at the same time period;
p<.01 for the effect of the 15 mg/kg MOR dose (collapsed on line), compared to the 0 mg/kg MOR dose (collapsed on line) at the same time period.
[3H]DAMGO binding
Binding data are summarized in Table 3. MOP-r density (Bmax) did not significantly differ between the MADR lines for the NAc or Vmb, but was more than 2-fold higher in mPFC tissue from MALDR compared to MAHDR mice [t(12)=3.98; p<0.001]. There were no differences between the MADR lines in affinity (KD) for [3H]DAMGO in any of these brain regions, with affinity ranging from 1.5–4.7 nM. There were no differences in receptor density or affinity for [3H]DAMGO between the B6 and D2 mice.
Table 3.
MOP-r density and affinity in 3 brain regions for MADR, B6 and D2 mice.
| Region | Line/Strain | Bmax (fmol bound/mg protein) | KD (nM) |
|---|---|---|---|
| mPFC | MALDR | 152.7 ± 54.8 | 2.8 ± 2.0 |
| mPFC | MAHDR | 65.7 ± 18.3*** | 1.5 ± 1.0 |
| mPFC | B6 | 48.6 ± 11.1 | 1.3 ± 0.8 |
| mPFC | D2 | 53.1 ± 9.5 | 1.6 ± 0.7 |
| NAc | MALDR | 366.0 ± 108.5 | 2.6 ± 1.6 |
| NAc | MAHDR | 337.5 ± 119.0 | 3.0 ± 2.1 |
| NAc | B6 | 176.2 ± 29.8 | 1.2 ± 0.5 |
| NAc | D2 | 254.4 ± 52.3 | 4.1 ± 1.5 |
| Vmb | MALDR | 345.0 ± 109.4 | 4.7 ± 2.5 |
| Vmb | MAHDR | 262.9 ± 51.1 | 2.5 ± 1.0 |
| Vmb | B6 | 374.4 ± 133.9 | 3.1 ± 2.1 |
| Vmb | D2 | 541.3 ± 107.4 | 3.8 ± 1.4 |
Values were determined using concentration of [3H]DAMGO from 0.145–4.85 nM as described in the methods section. Each value represents the mean ± SEM of tissues from 6 samples (5 animals pooled for each), all performed in duplicate.
p<0.001, for the comparison between MAHDR and MALDR for the mPFC. All saturation curves were best fit using a one-site model.
MA Drinking in Chr 10 D2.B6 (0–7.72) and Chr 10 D2.B6 (0–20.4) congenic and background strain mice
Markers used for genotyping of the congenic mice are listed in Fig. 3a, along with genotype information. For MA intake (mg/kg) in the Chr 10 D2.B6 (0–7.72) congenic and background strain littermates, there was a significant main effect of sex [F(1,74)=4.3, p<0.05], due to higher intake in females (3.5 ± 0.2 mg/kg vs. 3.0± 0.2 mg/kg in females vs. males, respectively), regardless of genotype. More MA was consumed when the 40 mg/l concentration was available, compared to the 20 mg/l concentration [F(1,74)=83.3, p<0.001], but there were no significant effects of genotype on MA intake (Fig. 3b). For total volume consumed (ml/kg) during the MA access period, the only significant result was a significant main effect of MA concentration [F(1,74)=31.1; p<0.001], with more total volume consumed when the higher MA concentration was offered (Fig. 3c). For the Chr 10 0–20.4 Mb congenic and background strain littermates, there was a significant strain × MA concentration interaction [F(1,43)=7.5; p<0.01] for MA consumption (Fig. 3d). The congenic strain with the introgressed B6 segment consumed significantly less MA from both concentrations, compared to their D2 background strain littermates, confirming the MA intake QTL on Chr 10 and reducing the interval to a 12.82 Mb segment. Sex could only be included as a factor in the analysis of the background strain data, due to an inadequate number of female congenic mice to analyze this factor. There were no significant effects of sex. For total volume consumed, there was only a main effect of concentration [F(1,43)=7.6; p<0.01], with greater total volume consumed when the higher MA concentration was offered (Fig.3e). Female non-congenic mice consumed more total volume than males (267.5 ± 11.6 vs. 233 ± 10.3 ml/kg in females vs. males, respectively).
Fig. 3. Marker and genotype information and MA drinking data for the congenic and D2 background strain mice.
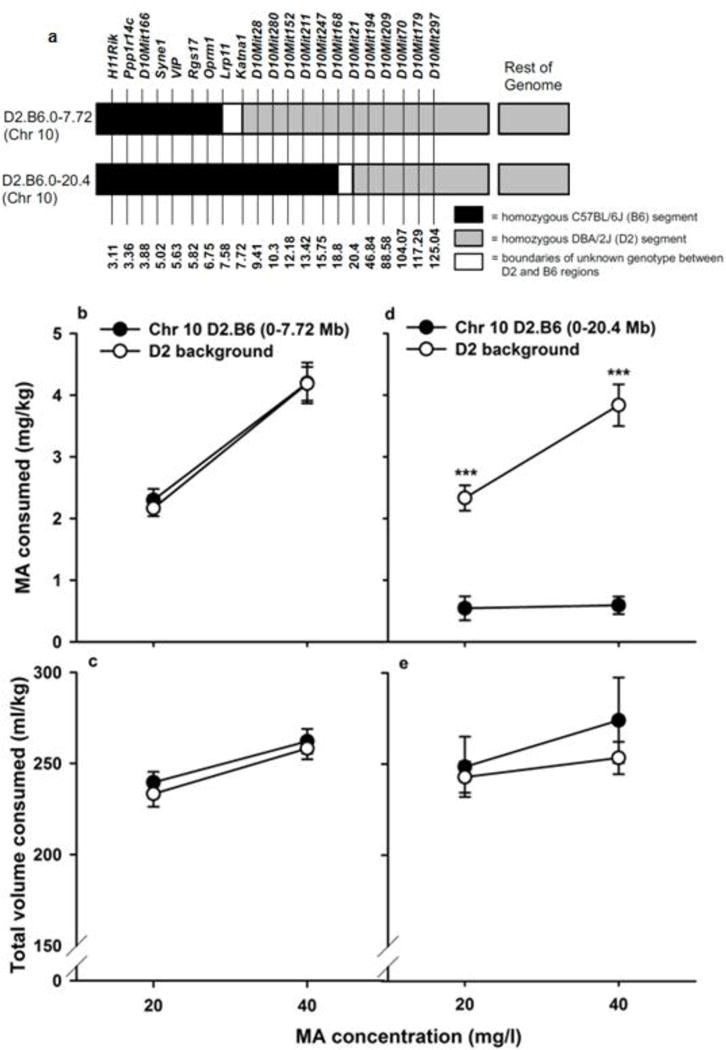
(a) Each congenic strain had a DBA2/J (D2) background genotype (grey) on all chromosomes, with a C57BL/6J (B6) segment (black) inserted on mouse Chr 10. White segments represent boundaries between confirmed B6 and D2 genome and are of unknown genotype. Above the diagram are the names of DNA microsatellite markers used to identify genotype, and the numbers correspond to genetic map positions in Megabases (Mb). (b and c) Mean ± SEM for MA consumed in mg/kg for the 20 and 40 mg MA/l solutions. Each data point is a 2-day average for days 2 and 4 of the 4-day period for each MA concentration. (d and e) Mean ± SEM total volume consumed in ml/kg during the same MA access periods shown in b and c. ***p<0.001 for the effect of strain at the indicated concentration. N=34–44/strain for the D2:B6 Chr 10 0–7.72 Mb and D2 mice, and N=13–32/strain for the Chr 10 D2.B6 0–20.4 Mb and D2 mice
DISCUSSION
As hypothesized, MOR reduced MA consumption, specifically in MAHDR mice. Although doses were chosen that did not have locomotor depressant effects, because MOR also reduced total volume consumed and SAC intake, non-specific effects that impacted consummatory behavior cannot be ruled out. A drug with partial agonist effects at MOR-r, such as buprenorphine that did not have non-specific effects (Eastwood and Phillips, 2014b), remains a better option for treatment. Consistent with previously published Oprm1 expression data indicating higher expression specifically in the mPFC of MALDR compared to MAHDR mice (Belknap et al., 2013), MOP-r density was also significantly higher specifically in mPFC tissue from MALDR than from MAHDR mice. Differences were not found in the NAc or Vmb, and there were no differences in MOP-r density between B6 and D2 mice, indicating that MOP-r density in the mPFC is a trait genetically correlated with MA intake in the selected lines. However, congenic strain analysis confirmed a QTL on Chr 10 for MA intake and reduced the relevant interval to a 12.82 Mb region that excluded Oprm1. These findings are consistent with the results of expression analyses indicating that Oprm1 is regulated by a significant MA intake risk network (Belknap et al., 2013), but that polymorphisms in Oprm1 do not play a role in risk for MA intake in the MADR lines.
There were significant effects of MOR on MA intake in MAHDR mice that were dose-dependent and also dependent on the concentration of the MA drinking solution. Thus, MOR effects on MA intake were larger for the 20 than 40 mg/l MA solution. This may have been due to tolerance development to MOR effects on MA intake or competing behaviors with repeated exposure, since a consecutive testing procedure was used. For example, in a study of antinociceptive potency in mice, 3 MOR treatments were sufficient for tolerance to develop (Duttaroy and Yoburn 1995). Results for the current SAC study are also consistent with the tolerance hypothesis, since MOR only had an impact on consumption of the lower SAC concentration,. An alternative explanation is that the mice have greater drive to consume the higher MA and SAC concentrations.
Although it is possible that MOR had an impact on the desire to consume both MA and SAC via interactions with reward mechanisms, because total volume intake was also reduced, it seems more likely that general behavioral effects of MOR interfered with drinking behavior. However, we chose doses of MOR that do not impact locomotor behavior in MAHDR mice (Eastwood and Phillips 2014a); thus, it is unlikely that an effect on general mobility explains these results. We previously examined the effects of BUP on MA intake, based on treatment efficacy in human psychostimulant users (Foltin and Fischman 1996; Salehi et al. 2015; Schottenfeld et al. 1993), and as a potentially more acceptable treatment drug than MOR. MA intake was reduced by BUP in the absence of significant reductions in total fluid consumption (Eastwood and Phillips 2014b).
We focused on MOP-r in the current work, based on evidence for regulation of Oprm1 by a gene network associated with differential risk for MA intake (Belknap et al. 2013). MOR has high affinity for both the μ- and к-opioid receptor subtypes (Raynor et al. 1994); however, the ĸ- and μ-opioid receptors have opposing actions on mesolimbic dopamine release, decreasing and increasing dopamine in the NAc, respectively (Di Chiara and Imperato 1988). Following acute subcutaneous MOR, drug levels were detectable in mouse brain within 5 min and had a 75 min half-life (Kalvass et al. 2007). MOR effects during the first 2 h of our 6-h limited access sessions are consistent with a relatively short half-life. A potential advantage of BUP as a treatment for MA addiction, in addition to potentially fewer non-specific behavioral effects, is that BUP takes longer to disassociate from the MOP-r (166 min for BUP vs 7 min for MOR) (Boas and Villiger 1985). BUP also binds to δ- and ĸ-opioid receptors, but has 10-fold lower affinity for δ compared to μ and ĸ (Lutfy and Cowan 2004). In addition, BUP activates the opioid receptor like 1 (ORL-1), which compromised the MOP-r-mediated antinociceptive effect of BUP (Lutfy et al. 2003). Activation of ORL-1 receptors by BUP could at least partially counteract some of its MOP-r-mediated effects on MA intake as well.
Amphetamines promote the release of endogenous opioids, and intra-mPFC DAMGO attenuated glutamate-induced mPFC neuron firing, an effect reversed by naloxone (Schad et al. 2002). The PFC is a major source of glutamate projections to the NAc and Vmb (Carr and Sesack 2000; Taber and Fibiger 1995). In the presence of MA, more mPFC MOP-r in MALDR mice could lead to a reduction in the activity of mPFC glutamate projections onto GABA-containing neurons in the NAc, and ultimately reduce dopamine levels. This would be predicted to reduce sensitivity to rewarding and reinforcing effects of MA in the MALDR line, as has been reported (Shabani et al. 2011; 2012a; Wheeler et al. 2009). However, in a microdialysis study, the dopamine response to MA was larger in the mPFC in MAHDR mice, but equivalent in the NAc in the 2 lines (Lominac et al. 2014). For extracellular glutamate, MAHDR mice exhibited an MA-induced reduction in in the mPFC and an increase in the NAc that was not seen in MALDR mice (Lominac et al. 2016; Szumlinski et al. 2017). The possibility that MA-stimulated dopamine in the mPFC results in reduced mPFC glutamate in MAHDR mice is consistent with other reports demonstrating antagonistic dopamine-glutamate interactions within the PFC (Abekawa et al. 2000). Such an inverse relationship has also been observed with excessive cocaine-taking (Ben-Shahar et al. 2012) and repeated MA exposure (Althobaiti et al. 2016). Determination of how the difference in mPFC MOP-r density interacts with the existing neurochemical findings in the MADR lines will require more direct investigation.
B6 and MALDR mice have similar low MA intake profiles, whereas D2 and MAHDR mice have higher MA intake profiles (Eastwood and Phillips 2014a). We predicted that the inbred strain results would mirror those for the selected lines, so that the B6 strain would have greater MOP-r density in the mPFC, but not NAc or Vmb, compared to D2 mice. In fact, we examined the fourth selection generation breeders of 3 replicate sets of the MADR lines and found that a large percentage of the MAHDR line breeders were homozygous for the D2-Oprm1 allele (72%), whereas a large percentage of the MALDR line breeders were homozygous for the B6-Oprm1 allele (68%). However, no difference in mPFC MOP-r density between D2 and B6 mice was found. Thus, the differences in Oprm1 expression and MOP-r density in the mPFC of the MADR lines are genetically correlated traits with selective breeding for MA intake. The current results confirm a brain region-specific pattern for MOP-r protein levels in the MADR lines that is similar to their Oprm1 gene expression profile (Belknap et al. 2013). The expression difference was originally identified in the first replicate set of MADR lines and the protein difference was confirmed here in the second replicate set of lines, indicating consistency across replicate selections. Our studies using systemically administered opioid-receptor agonists indicate that increased opioid activity results in reduced MA intake (data herein and Eastwood and Phillips 2014b). However, to identify a brain region-specific role of MOP-r on MA intake, studies are needed in which a MOP-r-specific agonist is delivered directly into the mPFC and other regions in which MOP-r are found.
Previous expression network analyses and current congenic data are consistent with the conclusion that Oprm1 is not a QTG contributing to genetic risk for MA intake. Within the reduced 12.82 Mb region on Chr 10 associated with MA intake, there are 60 protein coding genes. Finer mapping of the QTL would make investigation of specific genes more feasible. We have considerable evidence that the Taar1 gene, at 23.9 Mb, just outside of the congenic interval, impacts MA intake (Harkness et al. 2015). The location of Taar1 outside of the congenic interval would appear to indicate that more than one gene on mouse Chr 10 influences MA intake. However, recent sequencing of the Chr 10 D2.B6 0-20.4 Mb congenic has revealed that some of those mice possess the B6-Taar1 allele that is associated with reduced MA intake, when it was expected that all would possess the D2-Taar1 allele. Genotyping of the 45 mice used in the current study, revealed a frequency of 0.94 for the D2-Taar1 allele, which is associated with higher MA intake, in the non-congenic mice and a frequency of only 0.15 for the D2-Taar1 allele in the congenic mice (those with an introgressed B6 segment). There is dominance of the B6-Taar1 allele for low MA intake (Harkness et al. 2015), and the only congenic mice possessing a D2-Taar1 allele were 3 Taar1 heterozygotes. The correlation between Taar1 allele frequency and MA intake using data from both strains, was r=0.77, p<.00001; when dominance was taken into consideration by grouping the heterozygous mice with the homozygous B6-Taar1 mice, the correlation was 0.80, p<.0001. All of the shorter segment congenic and D2 background strain mice possessed only the D2-Taar1 allele, as expected, and both exhibited high MA intake, consistent with previous data for the impact of Taar1 (Harkness et al. 2015). There was no error made in the way in which the congenics were created. Standard methods were applied; however, the single nucleotide polymorphism that differentiates the B6 and D2 alleles is not detectable with the standard PCR marker genotyping methods that are used to develop congenic strains or to verify their genotype. The problem that relevant genetic information may be missed using marker-based PCR genotyping has been previously discussed (Shi et al. 2016; Shifman et al. 2006). It is possible, and in fact likely, that the B6-Taar1 allele is responsible for the low MA intake of the Chr 10 D2.B6 0–20.4 Mb congenic, as it is in the MALDR mice. This relationship has been more intensively investigated in multiple models and is the topic of another paper, which provides strong evidence supporting the conclusion that Taar1 plays a significant role in level of MA intake.
In summary, opioid systems appear to have a role in MA consumption; however, partial MOP-r agonists may have greater promise than full agonists as pharmacotherapeutics for MA use disorders. MOP-r activity in the mPFC may be particularly important, as greater MOP-r density in this region is found in mice that exhibit low MA intake, reduced MA reward, increased MA aversion and increased sensitivity to some physiological effects of MA, such as MA-induced hypothermia. Additional study is needed to determine which of these traits are impacted by opioid pathway activity in the mPFC. Data in congenics eliminated a segment of Chr 10 from further consideration and also supported previous data indicating that Taar1 impacts MA consumption.
Acknowledgments
The research presented here was supported by Department of Veterans Affairs grants I01BX002106 and I01BX002758, National Institutes of Health grants T32DA07262, P50DA018165, U01DA041579 and R24AA020245, and a National Institutes of Health-Veterans Affairs interagency agreement. The authors declare no conflicts of interest. The views and opinions expressed are those of the authors and should not be construed to represent the views of the affiliated institutions or the funding agencies. We thank Harue Baba for her help with genotyping and data collection and Robert Johnson for his technical support with the receptor binding assays. We also thank Drs. Glenn Doyle and Thomas Ferraro for provision of the congenic mice from which we established our breeding stock for the current study.
Footnotes
Ethical Approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
References
- Abekawa T, Ohmori T, Ito K, Koyama T. D1 dopamine receptor activation reduces extracellular glutamate and GABA concentrations in the medial prefrontal cortex. Brain Res. 2000;867:250–254. doi: 10.1016/s0006-8993(00)02298-8. [DOI] [PubMed] [Google Scholar]
- Althobaiti YS, Almalki AH, Das SC, Alshehri FS, Sari Y. Effects of repeated high-dose methamphetamine and ceftriaxone post-treatments on tissue content of dopamine and serotonin as well as glutamate and glutamine. Neurosci Lett. 2016;634:25–31. doi: 10.1016/j.neulet.2016.09.058. [DOI] [PubMed] [Google Scholar]
- Aoyama N, Takahashi N, Kitaichi K, Ishihara R, Saito S, Maeno N, Ji X, Takagi K, Sekine Y, Iyo M, Harano M, Komiyama T, Yamada M, Sora I, Ujike H, Iwata N, Inada T, Ozaki N. Association between gene polymorphisms of SLC22A3 and methamphetamine use disorder. Alcohol Clin Exp Res. 2006;30:1644–1649. doi: 10.1111/j.1530-0277.2006.00215.x. [DOI] [PubMed] [Google Scholar]
- Belknap JK, McWeeney S, Reed C, Burkhart-Kasch S, McKinnon CS, Li N, Baba H, Scibelli AC, Hitzemann R, Phillips TJ. Genetic factors involved in risk for methamphetamine intake and sensitization. Mamm Genome. 2013;24:446–458. doi: 10.1007/s00335-013-9484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belknap JK, Richards SP, O’Toole LA, Helms ML, Phillips TJ. Short-term selective breeding as a tool for QTL mapping: ethanol preference drinking in mice. Behav Genet. 1997;27:55–66. doi: 10.1023/a:1025615409383. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar OM, Szumlinski KK, Lominac KD, Cohen A, Gordon E, Ploense KL, DeMartini J, Bernstein N, Rudy NM, Nabhan AN, Sacramento A, Pagano K, Carosso GA, Woodward N. Extended access to cocaine self-administration results in reduced glutamate function within the medial prefrontal cortex. Addict Biol. 2012;17:746–757. doi: 10.1111/j.1369-1600.2011.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boas RA, Villiger JW. Clinical actions of fentanyl and buprenorphine. The significance of receptor binding. Br J Anaesth. 1985;57:192–196. doi: 10.1093/bja/57.2.192. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther. 1988;244:1067–1080. [PubMed] [Google Scholar]
- Dlugos AM, Hamidovic A, Hodgkinson C, Shen PH, Goldman D, Palmer AA, de Wit H. OPRM1 gene variants modulate amphetamine-induced euphoria in humans. Genes Brain Behav. 2011;10:199–209. doi: 10.1111/j.1601-183X.2010.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle GA, Schwebel CL, Ruiz SE, Chou AD, Lai AT, Wang MJ, Smith GG, Buono RJ, Berrettini WH, Ferraro TN. Analysis of candidate genes for morphine preference quantitative trait locus Mop2. Neuroscience. 2014;277:403–416. doi: 10.1016/j.neuroscience.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- Eastwood EC, Phillips TJ. Opioid sensitivity in mice selectively bred to consume or not consume methamphetamine. Addict Biol. 2014a;19:370–379. doi: 10.1111/adb.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood EC, Phillips TJ. Morphine intake and the effects of naltrexone and buprenorphine on the acquisition of methamphetamine intake. Genes Brain Behav. 2014b;13:226–235. doi: 10.1111/gbb.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltin RW, Fischman MW. Effects of methadone or buprenorphine maintenance on the subjective and reinforcing effects of intravenous cocaine in humans. J Pharmacol Exp Ther. 1996;278:1153–1164. [PubMed] [Google Scholar]
- Gubner NR, Reed C, McKinnon CS, Phillips TJ. Unique genetic factors influence sensitivity to the rewarding and aversive effects of methamphetamine versus cocaine. Behav Brain Res. 2013;256:420–427. doi: 10.1016/j.bbr.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkness JH, Shi X, Janowsky A, Phillips TJ. Trace amine-associated receptor 1 regulation of methamphetamine intake and related traits. Neuropsychopharmacology. 2015;40:2175–2184. doi: 10.1038/npp.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iamjan SA, Thanoi S, Watiktinkorn P, Nudmamud-Thanoi S, Reynolds GP. BDNF (Val66Met) genetic polymorphism is associated with vulnerability for methamphetamine dependence. Pharmacogenomics. 2015;16:1541–1545. doi: 10.2217/pgs.15.96. [DOI] [PubMed] [Google Scholar]
- Ide S, Kobayashi H, Tanaka K, Ujike H, Sekine Y, Ozaki N, Inada T, Harano M, Komiyama T, Yamada M, Iyo M, Ikeda K, Sora I. Gene polymorphisms of the mu opioid receptor in methamphetamine abusers. Ann N Y Acad Sci. 2004;1025:316–324. doi: 10.1196/annals.1316.039. [DOI] [PubMed] [Google Scholar]
- Jayaram-Lindström N, Guterstam J, Häggkvist J, Ericson M, Malmlöf T, Schilström B, Halldin C, Cervenka S, Saijo T, Nordström AL, Franck J. Naltrexone modulates dopamine release following chronic, but not acute amphetamine administration: a translational study. Transl Psychiatry. 2017;7:e1104. doi: 10.1038/tp.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram-Lindström N, Hammarberg A, Beck O, Franck J. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry. 2008a;165:1442–1448. doi: 10.1176/appi.ajp.2008.08020304. [DOI] [PubMed] [Google Scholar]
- Jayaram-Lindström N, Konstenius M, Eksborg S, Beck O, Hammarberg A, Franck J. Naltrexone attenuates the subjective effects of amphetamine in patients with amphetamine dependence. Neuropsychopharmacology. 2008b;33:1856–1863. doi: 10.1038/sj.npp.1301572. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Olson ER, Cassidy MP, Selley DE, Pollack GM. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50,u and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther. 2007;323:346–355. doi: 10.1124/jpet.107.119560. [DOI] [PubMed] [Google Scholar]
- Lominac KD, McKenna CL, Schwartz LM, Ruiz PN, Wroten MG, Miller BW, Holloway JJ, Travis KO, Rajasekar G, Maliniak D, Thompson AB, Urman LE, Phillips TJ, Szumlinski KK. Mesocorticolimbic monoamine correlates of methamphetamine sensitization and motivation. Front Syst Neurosci. 2014;8:70. doi: 10.3389/fnsys.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominac KD, Quadir SG, Barrett HM, McKenna CL, Schwartz LM, Ruiz PN, Wroten MG, Campbell RR, Miller BW, Holloway JJ, Travis KO, Rajasekar G, Maliniak D, Thompson AB, Urman LE, Kippin TE, Phillips TJ, Szumlinski KK. Prefrontal glutamate correlates of methamphetamine sensitization and preference. Eur J Neurosci. 2016;43:689–702. doi: 10.1111/ejn.13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Cowan A. Buprenorphine: a unique drug with complex pharmacology. Curr Neuropharmacol. 2004;2:395–402. doi: 10.2174/1570159043359477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, Kieffer BL, Takeshima H, Carroll FI, Maidment NT, Evans CJ. Buprenorphine-induced antinociception is mediated by μ-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci. 2003;23:10331–10337. doi: 10.1523/JNEUROSCI.23-32-10331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips TJ, Shabani S. An animal model of differential genetic risk for methamphetamine intake. Front Neurosci. 2015;9:327. doi: 10.3389/fnins.2015.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick CG, Peter Y, Schreiber S, Weizman R. Pharmacological characterization of buprenorphine, a mixed agonist-antagonist with kappa 3 analgesia. Brain Res. 1997;744:41–46. doi: 10.1016/s0006-8993(96)01069-4. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned ĸ-, δ-, and μ-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- Sadée W, Rosenbaum J, Herz A. Buprenorphine: differential interaction with opiate receptor subtypes in vivo. J Pharmacol Exp Ther. 1982;223(1):157–162. [PubMed] [Google Scholar]
- Salehi M, Emadossadat A, Kheirabadi GR, Maracy MR, Sharbafchi MR. The effect of buprenorphine on methamphetamine cravings. J Clin Psychopharmacol. 2015;35:724–727. doi: 10.1097/JCP.0000000000000408. [DOI] [PubMed] [Google Scholar]
- Schad CA, Justice JB, Jr, Holtzman SG. Endogenous opioids in dopaminergic cell body regions modulate amphetamine-induced increases in extracellular dopamine levels in the terminal regions. J Pharmacol Exp Ther. 2002;300:932–938. doi: 10.1124/jpet.300.3.932. [DOI] [PubMed] [Google Scholar]
- Schottenfeld RS, Pakes J, Ziedonis D, Kosten TR. Buprenorphine: dose-related effects on cocaine and opioid use in cocaine-abusing opioid-dependent humans. Biol Psychiatry. 1993;34:66–74. doi: 10.1016/0006-3223(93)90258-f. [DOI] [PubMed] [Google Scholar]
- Shabani S, Dobbs LK, Ford MM, Mark GP, Finn DA, Phillips TJ. A genetic animal model of differential sensitivity to methamphetamine reinforcement. Neuropharmacology. 2012a;62:2169–2177. doi: 10.1016/j.neuropharm.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabani S, Houlton SK, Hellmuth L, Mojica E, Mootz JR, Zhu Z, Reed C, Phillips TJ. A mouse model for binge-level methamphetamine use. Front Neurosci. 2016;10:493. doi: 10.3389/fnins.2016.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabani S, McKinnon CS, Cunningham CL, Phillips TJ. Profound reduction in sensitivity to the aversive effects of methamphetamine in mice bred for high methamphetamine intake. Neuropharmacol. 2012b;62:1134–1141. doi: 10.1016/j.neuropharm.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabani S, McKinnon CS, Reed C, Cunningham CL, Phillips TJ. Sensitivity to rewarding or aversive effects of methamphetamine determines methamphetamine intake. Genes Brain Behav. 2011;10:625–636. doi: 10.1111/j.1601-183X.2011.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Walter NA, Harkness JH, Neve KA, Williams RW, Lu L, Belknap JK, Eshleman AJ, Phillips TJ, Janowsky A. Genetic polymorphisms affect mouse and human trace amine-associated receptor 1 function. PLoS One. 2016;11:e0152581. doi: 10.1371/journal.pone.0152581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman S, Bell JT, Copley RR, Taylor MS, Williams RW, Mott R, Flint J. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumlinski KK, Lominac KD, Campbell RR, Cohen M, Fultz EK, Brown CN, Miller BW, Quadir SG, Martin D, Thompson AB, von Jonquieres G, Klugmann M, Phillips TJ, Kippin TE. Methamphetamine addiction vulnerability: The Glutamate, the Bad, and the Ugly. Biol Psychiatry. 2017;81:959–970. doi: 10.1016/j.biopsych.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber MT, Fibiger HC. Electrical stimulation of the prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. J Neurosci. 1995;15:3896–3904. doi: 10.1523/JNEUROSCI.15-05-03896.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PC, Ho IK, Lee CW. Buprenorphine-elicited alteration of adenylate cyclase activity in human embryonic kidney 293 cells coexpressing ĸ-, μ-opioid and nociceptin receptors. J Cell Mol Med. 2015;19:2587–2596. doi: 10.1111/jcmm.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, Reed C, Burkhart-Kasch S, Li N, Cunningham CL, Janowsky A, Franken FH, Wiren KM, Hashimoto JG, Scibelli AC, Phillips TJ. Genetically correlated effects of selective breeding for high and low methamphetamine consumption. Genes Brain Behav. 2009;8:758–771. doi: 10.1111/j.1601-183X.2009.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]