

Abstract
Cytosine methylation is widespread among organisms and essential for mammalian development. In line with early postulations of an epigenetic role in gene regulation, symmetric CpG methylation can be mitotically propagated over many generations with extraordinarily high fidelity. Here, we combine BrdU labeling and immunoprecipitation with genome-wide bisulfite sequencing to explore the inheritance of cytosine methylation onto newly replicated DNA in human cells. Globally, we observe a pronounced lag between the copying of genetic and epigenetic information that is reconsolidated within hours to accomplish faithful mitotic transmission. Populations of arrested cells show a global reduction of lag induced intermediate CpG methylation when compared to proliferating cells, while sites of transcription factor engagement appear cell-cycle invariant. Alternatively, the cancer cell line HCT116 preserves global epigenetic heterogeneity independently of cell-cycle arrest. Taken together, our data suggest that heterogeneous methylation largely reflects asynchronous proliferation, but is intrinsic to actively engaged cis-regulatory elements and cancer.
Introduction
Cytosine methylation represents a classic epigenetic modification that is faithfully transmitted over DNA replication by recognition of information retained on the parental strand. In mammals, its prevalence within the CpG dinucleotide context provides a symmetrical substrate to restore transiently hemi-methylated states, an elegant mechanism that resembles the Watson-Crick model of genetic inheritance1,2. Three enzymes are generally responsible for establishing and maintaining this modification: DNA methyltransferases 1 (DNMT1), 3A (DNMT3A), and 3B (DNMT3B), all of which are essential for normal mammalian development3. Maintenance appears to be predominantly accomplished by DNMT1, which localizes to replication foci4 and exhibits 10-40 fold higher binding affinity and catalytic activity towards hemi-methylated DNA substrates5–7. DNMT1 is also recruited to nascent DNA by the essential cofactor UHRF1 (ubiquitin-like, with PHD and RING finger domains 1), which exhibits a high affinity for hemi-methylated DNA through its SRA domain8,9 and ubiquitinates the histone H3 tail to facilitate DNMT1 recruitment10. DNMT1 activity is further directed to the replication fork through its interaction with the proliferating cell nuclear antigen (PCNA) DNA clamp11, and deletion of DNMT1s PCNA-binding domain has been reported to delay post replication remethylation12. More conceptually, accurate reestablishment of the human methylome requires catalytic activity at ~45 million heterogeneously distributed CpGs (roughly 80% of CpG sites within the diploid genome) that must be completed within a single cell cycle13. Given this scale, it may not be surprising that some earlier studies have observed a lag in nascent strand methylation in somatic and transformed cells14–18, which presumably reflects the kinetic discrepancy between rapid polymer extension from the 3′-OH of the previously incorporated base versus the multistep transfer of a methyl-group to hemi-methylated CpG dyads19,20. However, the global scale, kinetics and possible implications of this disconnect between copying genetic versus epigenetic information remain to be determined.
Results
Repli-BS identifies a global delay in methylating nascent DNA
To investigate the acquisition of CpG methylation on nascent DNA, we combined Repli-seq21 (immunoprecipitation of bromodeoxyuridine (BrdU) labeled nascent strands followed by sequencing) with bisulfite treatment to measure post-replication cytosine methylation at base pair resolution (Repli-bisulfite seq: Repli-BS, Fig. 1a, Supplementary Fig. 1a, Methods). Human embryonic stem cells (ESCs; male HUES64) were treated for one hour with BrdU and sorted into six S-phase fractions (S1-6) before BrdU-immunoprecipitation, followed by bisulfite sequencing (Fig. 1a,b, Supplementary Data Set 1, Supplementary Fig. 1b). We initially pooled data from the six fractions and compared the methylation level of around 24.5 million newly replicated (nascent) CpGs to bulk (non-sorted, no BrdU-immunoprecipitation) whole genome bisulfite sequencing (WGBS) data. While our bulk reference population exhibited a canonical methylation landscape with high CpG methylation (mean 0.83), the average for DNA synthesized within our 1 hour BrdU pulse was globally reduced (mean 0.64; Fig. 1c, Supplementary Fig. 1c). This discrepancy was consistent across early (S1 + S2; mean 0.63), mid (S3 + S4; mean 0.63) and late (S5 + S6; mean 0.66) stages of S-phase (Supplementary Fig. 1d). Moreover, we found that all measured genomic features appeared equally affected by this delay including promoters, enhancers and gene bodies of genes with a range of different expression levels (Supplementary Fig. 1e,f). CpG density as well as enrichment for the polycomb repressive complex 2 (PRC2) subunit EZH2 appeared to have some influence on a very small subset of CpGs (Supplementary Fig. 1g–j). We also observed a global delay for non-CpG methylation, which was more apparent for gene bodies, repetitive elements and other known DNMT3A and 3B targets (Supplementary Fig. 1k,l). Notably, the emergence of non-symmetric methylation on the nascent strand requires de novo activity as the parental strand cannot serve as a template and hence follows an alternative mechanism. Finally, we used single cell RNA-sequencing to determine expression of key epigenetic regulators throughout the cell cycle (Supplementary Data Set 2, Supplementary Fig. 2a) and in parallel applied a new multiplexed single cell reduced representation bisulfite sequencing (RRBS) approach to measure cytosine methylation across individual cells sorted by DNA content. Cells in S-phase showed lower mean methylation values compared to cells in G1 or G2-M, which independently points towards a replication-associated reduction in methylation (Fig. 1d, Supplementary Fig. 2b,c).
Figure 1. Repli-BS identifies a global delay in the acquisition of CpG methylation on nascent DNA.
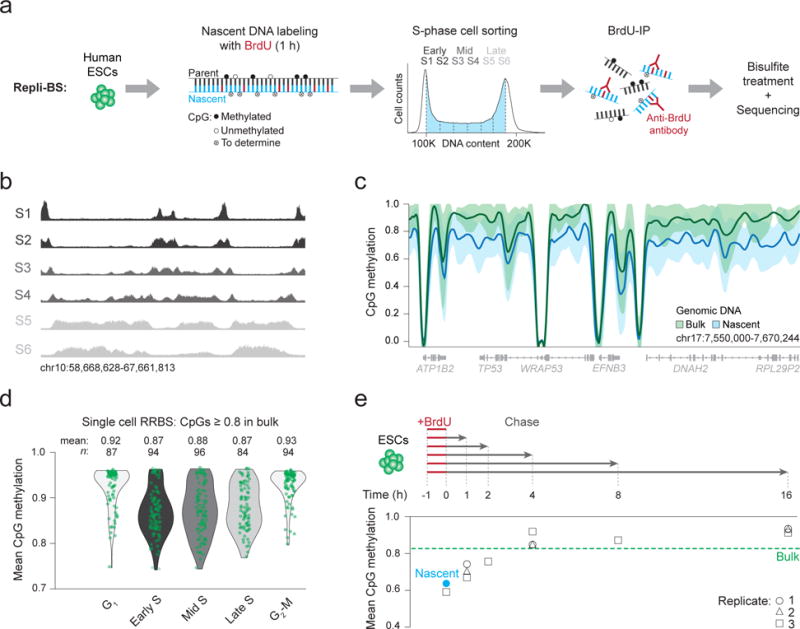
(a) Schematic representation of our experimental design. Asynchronously growing human embryonic stem cells (ESCs) are labeled with bromodeoxyuridine (BrdU) for 1 hour (h). During the pulse, BrdU (red) is incorporated into newly synthesized nascent DNA (blue) as it is copied from the parental strand (black). Filled circles = methylated CpGs, empty circles = unmethylated CpGs. Empty circles marked with an x, (‘To determine’) = CpGs investigated by Repli-BS for methylation status. Six gates of equal proportion were used to sort S-phase cells according to DNA content. BrdU immunoprecipitation (IP) was then performed to capture newly replicated DNA strands, which were treated with bisulfite prior to library generation and sequencing. (b) Genome browser tracks show the read density and corresponding replication pattern across each of the six collected S-phase fractions. (c) A local regression analysis was performed using methylation values for CpGs within nascent (S1-6) and bulk genomic DNA. Loess smoothed mean methylation levels are displayed as a bold line. Shaded areas: one standard deviation. (d) Single cell reduced representation bisulfite sequencing (RRBS) data for cells sorted by cell cycle stage. Violin plots with overlaid strip plots display mean methylation per cell for all CpGs with methylation ≥ 0.8 in bulk cells. Each green dot represents one cell. Despite not isolating nascent DNA, 85% S-phase cells (n = 236/274) display lower mean methylation levels than bulk ESCs. (e) ESCs were pulsed with BrdU for 1 h, chased in normal growth medium for 1 to 16 h and analyzed by Repli-BS. Mean CpG methylation levels for nascent DNA samples are shown on the bottom. Dashed green line: Genomic average for bulk DNA. Nascent mean methylation after the initial labeling (0 h) from a is indicated with the blue dot and the black circles, triangles and squares show mean methylation for three replicates (replicates 1 and 2: same cellular material but different IPs and replicate 3: biological replicate).
Biphasic remethylation of nascent DNA
To better understand the kinetics with which nascent DNA restores methylation over time, we chased BrdU-pulsed cells for 1-16 hours prior to performing Repli-BS (Fig. 1e; Supplementary Data Set 1). The most notable methylation increase occurred within the first hour following replication, which could be associated with active recruitment of DNMT1 to the replication fork, followed by a slower, incremental period that stabilized to bulk reference levels after around 4 hours (Fig. 1e, Supplementary Fig. 2d). The delayed kinetics of the second phase could reflect a replication-uncoupled search for unevenly dispersed hemi-methylated targets. These dynamics affected the entire genome equally (Supplementary Fig. 2e) however, while the mean methylation and rate of increase were remarkably similar between replicates (Fig. 1e), methylation typically emerged on distinct CpGs (Supplementary Fig. 2f). This suggests that the fidelity of this modification is not genetically-encoded and may instead be determined by the molecular limits of DNMT1 activity near the replication fork, where additional processes may act as obstructions. The gain of methylation over time can also be measured at single molecule resolution by determining the read-level progression from partial to fully methylated states (Supplementary Fig. 2g,h), which confirmed a high correlation between neighboring CpGs that would be consistent with in vitro measurements reporting a processive activity for DNMT1 (Refs6,22) (Supplementary Fig. 2i).
DNMT3A and 3B have a limited effect on post-replication methylation rate
As previously observed, genetic deletion of DNMT1 in mouse and human ESCs results in rapid, global loss of CpG methylation confirming the central role of DNMT1 in methylation maintenance23,24. Long term passaging of DNMT3A and DNMT3B double knockout (DKO) ESCs results in a gradual loss of methylation showing that the de novo methyltransferases are required to compensate for the incomplete maintenance fidelity of DNMT1 (Refs24,25). After approximately 20 passages, our human DKO cells show a global decrease in CpG methylation of ~10% (Supplementary Figure 3a). To assess the contribution of the de novo DNMTs to post-replication nascent strand methylation, we repeated the 1 hour BrdU pulse-chase Repli-BS experiment in the DKO cells and observed that DNMT1 is able to methylate nascent DNA to bulk levels in the absence of any de novo activity (Supplementary Figure 3b–d). In fact, the nascent strand methylation rate appeared slightly faster than in WT cells, which may be due to the reduced number of targets (hemi-methylated CpGs) in the comparatively hypomethylated DKO line.
Distinct origins of global intermediate methylation
In both cell lines, our data show that the delay in methylation of nascent DNA reduces global methylation levels. Given that ~35% cells within an unsynchronized ESC population are undergoing replication at any given time, we hypothesized that the intrinsically lower global methylation of S-phase cells may contribute to the intermediate methylation (values other than 0% or 100%) observed in bulk measurements, which are often attributed to intercellular heterogeneity. To investigate this, we arrested ESCs in prometaphase and performed WGBS to determine whether absence of cells in S-phase would reduce the fraction of intermediately methylated CpGs (Fig. 2a, Supplementary Fig. 4a,b, Supplementary Data Set 1) which comprise the majority (58%) of CpGs in proliferating ESCs (Fig. 2b). In line with our hypothesis, the number of unmethylated CpGs (with 0% methylation) remained roughly constant between proliferating and arrested cells (5.6 and 6.9%). In contrast, intermediately methylated CpG sites were reduced from 58% in proliferating cells to 25% in arrested cells with a concomitant increase in the number of fully methylated CpGs from 35.5% to 68.9% (Fig. 2b, Supplementary Fig 4c). This shift was independent of CpG coverage (Supplementary Fig. 4d) and was further validated by differentiating ESCs into post-mitotic spinal motor neurons (MNs)26, which reduces the proportion of proliferating cells without artificial arrest (Fig. 2c). After two weeks, the majority (~90%) of cells on the plate were Ki67 negative and the proportion of intermediately methylated CpGs indeed decreased as predicted (Fig. 2c), which suggests that global methylation heterogeneity in ESCs arises largely as a byproduct of time-dependent nascent strand methylation (Fig. 2d). Of interest, intercellular DNA methylation heterogeneity has been frequently noted in cancer cells in association with reduced global methylation levels27,28 (Fig. 3a). Considering our results, one may speculate that the elevated fraction of intermediate methylation levels in cancer cells could be associated with the highly proliferative nature of these transformed cells. To explore this, we arrested the colon cancer cell line HCT116 and found an expected increase in mean methylation levels (from eliminating contribution of nascent strands with reduced methylation) but not a shift towards full methylation as observed in ESCs (Fig. 3b, Supplementary Fig. 4e,f). Although we only investigate HCT116 cells here, it suggests that the epigenetic heterogeneity in cancer cells is a combination of both replication-associated and elevated cell-to-cell variation.
Figure 2. Mitotically arrested cells show reduced intermediate CpG methylation suggesting time-dependent methylation heterogeneity.
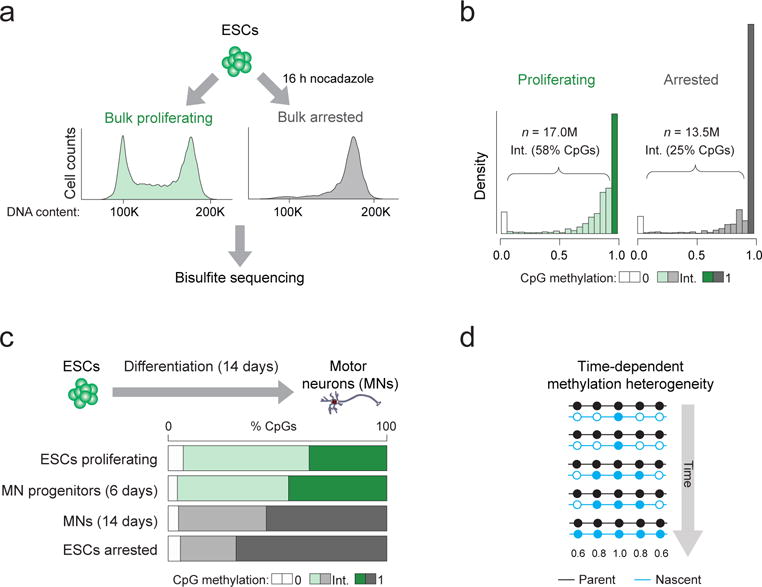
(a) ESCs were grown in either normal (“proliferating”) culture conditions (green) or in the presence of the mitotic inhibitor nocodazole for 16 h (“arrested”; grey). The fluorescent activated cell sorting (FACS) plots show representative histograms according to DNA content (propidium iodide intensity) for cells from each experimental condition confirming the shift to G2-M phase (4N) for nocodazole treated cells (right). Genomic DNA was extracted from both conditions and bisulfite sequencing was performed. (b) Histograms displaying the distribution of CpG methylation in cells grown in proliferating and arrested conditions. Intermediate (Int.) methylation (shown in intermediate shading) refers to CpGs with methylation levels other than 0 (light shading) or 1 (dark shading). (c) ESCs were differentiated towards post-mitotic motor neurons (MNs) and samples on day 6 and 14 were analyzed by WGBS to determine the proportion of CpGs with intermediate methylation levels. (d) Simplified schematic illustrating a hypothetical scenario of methylation heterogeneity arising from a lag in maintenance methylation following DNA synthesis. The grey arrow indicates time during S-phase. In an unsynchronized population, cells exist in all stages of S-phase and therefore each display different methylation levels with respect to ‘time’. Nascent DNA is colored blue and is gradually methylated over time. Filled circle = methylated CpG, empty circle = unmethylated CpG. Numbers beneath indicate the expected mean methylation for the CpGs at that position based on the illustrated pattern.
Figure 3. Cancer cells and active TF binding sites display inherent cell-to-cell methylation heterogeneity.
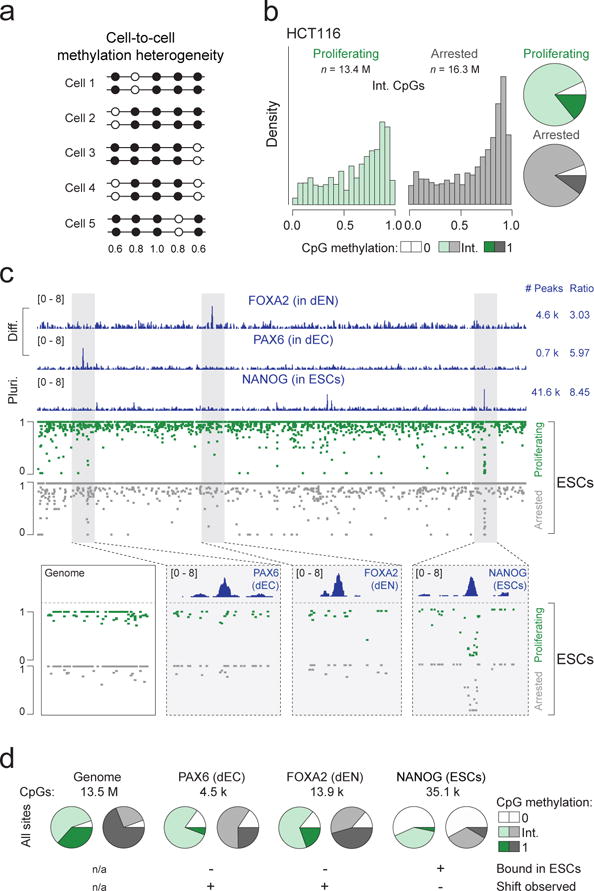
(a) Simplified schematic analogous to the one in Fig. 2d, showing an alternative hypothetical scenario that represents CpG methylation heterogeneity arising from cell-to-cell differences. Filled circle = methylated CpG, empty circle = unmethylated CpG. Numbers beneath indicate the expected mean methylation for the CpGs at that position based on the illustrated pattern. (b) Histograms displaying methylation levels for bulk and arrested CpGs with intermediate methylation in HCT116 cells (left). Pie charts summarizing the distribution of unmethylated, intermediate and fully methylated CpGs in proliferating and arrested HCT116 cells for CpGs covered at ≥ 15X (right). (c) ChIP-seq tracks (chr5:163,394,518-163,756,719) for FOXA2 in ESCs differentiated for 5 days into endoderm (dEN), PAX6 in ESCs differentiated for 5 days into ectoderm (dEC) and NANOG in undifferentiated pluripotent ESCs. ‘Diff.’ = Differentiated, ‘Pluri.’ = Pluripotent. Peaks = total number of peaks called by MACS2, ratio = average number of CpGs per peak (top). WGBS tracks for proliferating and arrested ESCs (middle). For each TF, methylation levels in proliferating and arrested ESCs are displayed that show a shift to high methylation levels (as in the ‘genome’ example) for non-bound sites but no shift in the engaged NANOG binding site. Genome: chr5:163,477,070-163,491,961, PAX6: chr5:163,434,313-163,439,131, FOXA2: chr5:163,527,525-163,531,337, NANOG: chr5:163,721,080-163,726,485. (d) The analysis displayed in c is expanded to the whole genome. When considering all CpGs or the ones found at not actively bound (but experimentally confirmed) TF binding sites (FOXA2 and PAX6) we observe a similar decrease in the percentage of CpGs with intermediate methylation levels (decrease of 33%, 22% and 20% respectively; “+ shift observed”) while sites that are bound by NANOG in ESCs do not show this behavior and remain largely unchanged despite the arrest (decrease of only 4%; “- Shift observed”).
Intermediate methylation at bound transcription factor targets points to intercellular heterogeneity
Prior studies have also suggested that intermediate levels of methylation are frequently found around transcription factor (TF) binding sites13,29,30. To compare these local events with the global patterns described above, we re-examined our bulk and arrested ESC datasets and utilized previously determined TF binding sites based on chromatin immunoprecipitation followed by sequencing (ChIP-seq) experiments in ESCs as well as their differentiated derivatives. We first identified TF targets that are engaged and hence actively regulated in pluripotent cells (OCT4 or NANOG in ESCs) as well as differentiation-specific target sites (PAX6 in ectoderm, FOXA2 in endoderm, GATA6 in mesoderm and EOMES in mesendoderm) that are not bound in undifferentiated ESCs and should therefore behave like the genomic average (Fig. 3c, Supplementary Fig. 4g,h). We then compared methylation levels for bulk and arrested ESCs at actively engaged vs. differentiation-specific regions and found that the latter show a similar decrease in the percentage of intermediately methylated CpGs upon arrest to that observed genome-wide, while actively bound sites remained largely unchanged. This suggests the presence of intercellular heterogeneity at engaged TF binding sites (Fig. 3c,d, Supplementary Fig. 4g,h).
Discussion
We applied a genome-wide approach to measure cytosine methylation levels of nascent DNA (Repli-BS) and found a persistent lag in the reestablishment of both CpG and non-CpG methylation following DNA synthesis. For CpG methylation, the rapid initial surge that we observe is consistent with the replication fork recruiting DNMT1 to newly synthesized DNA via its PCNA binding domain, which, when absent, reduces nascent strand methylation rate as previously noted31. It appears that the initial seeding of DNMT1 generally does not occur at fixed locations, as we were not able to identify commonly or disproportionately hypermethylated features in our pulse-chase experiment. Consecutive CpGs were methylated in the majority of nascent reads, supporting observations made in vitro that suggest DNMT1 acts in a processive manner on the same molecule. However, the possibility cannot be excluded that DNMT1 may also methylate larger chromatin domains more rapidly through alternative modes of recruitment. In our DKO experiments we made the intriguing observation that reduced global methylation levels appear to increase the nascent strand methylation rate. In the future, it will be interesting to further explore how substrate (hemi-methylated targets) and enzyme (DNMT1) levels affect the kinetics of post-replication methylation and whether this enzyme to substrate ratio can be mechanistically linked to the reduced, but generally stable, levels of global methylation across cancer types32–34. In this context, we show that intermediate methylation in cancer cells arises as a result of both inherent intercellular epigenetic heterogeneity and cell cycle linked heterogeneity. In contrast, the extensive methylation maintenance lag in human ESCs is largely explained by the fraction of cells in S-phase. We expect that the insights that emerge from our study may allow for a more precise analysis of the biological function of regions that display true intermediate methylation values in non-transformed cells. Finally, it remains to be seen whether the global lag of remethylation after replication has a biologically relevant role at some sites including, but not limited to, providing a window of opportunity for TFs to access their genomic targets35, as recently reported for H3K27me3 (Ref36). In conclusion, our description of the genome-wide lag between the copying of genetic and epigenetic information adds further insight to our understanding of how epigenetic information is mitotically inherited.
Online methods
Cell culture
Human embryonic stem cells (male HUES64; obtained from Harvard University) and the modified variant DNMT3A−/−DNMT3B−/− (HUES64 DKO) cells were grown in feeder-free conditions using Geltrex (Thermo Fisher Scientific) and mTeSR (STEMCELL Technologies). HCT116 cells (obtained from Dr. Baylin at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) were grown in McCoy’s 5A (modified) medium supplemented with 10% FBS and 1% penicillin and streptomycin. All cell lines tested negative for mycoplasma.
Bulk vs arrested population measurements
Cells were either grown with no treatment (bulk) or arrested using 2 μg/ml nocodazole for 16 h, then collected and washed twice with PBS. Arrested cells were stained using the live-dead Fixable Blue Dead Cell Stain kit (Thermo Fisher Scientific) according to manufacturer’s recommendations, then fixed by adding 100% ethanol dropwise to the cell suspension until a final ethanol concentration of 75% was achieved. Fixed arrested cells were then counter-stained with propidium iodide (PI) prior to FACS sorting to collect those in G2-M phase.
BrdU treatment
To label nascent strands of DNA, BrdU (5-bromo-2′-deoxyuridine; BD Biosciences, cat# 550891) was added to cell culture medium at a final concentration of 50 μM. Cells were incubated for 1 h at 37 °C to allow incorporation of BrdU nucleotides into newly synthesized DNA. Cells were washed three times with basal media (DMEM-F12), then either collected immediately or incubated at 37 °C for the duration of the chase before collection. To collect, cells were detached from tissue culture plates and disaggregated into single cells using StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific) for ESCs and 0.05% trypsin for HCT116. Cells were then pelleted, resuspended in PBS and either fixed in ethanol as described above or snap frozen.
Enrichment for BrdU labelled DNA (original)
Genomic DNA was isolated by incubating cells in SDS-PK buffer (0.5% (wt/vol) sodium dodecyl sulfate (SDS), 50 mM Tris-HCL, 0.01M EDTA, 1M NaCl and 0.2 mg/ml proteinase K) at 56 °C for 2 h. Phenol-chloroform extraction was performed and genomic DNA was precipitated in 1 volume of isopropanol at -20 °C. DNA was pelleted, washed with ethanol and resuspended in TE buffer. Genomic DNA was fragmented to an average size of 200 bp using a Branson Digital Sonifier (Model 250) and a Branson 101-148-063 microtip. DNA was sonicated using 40% amplitude for a total of 4 minutes. A double-antibody immunoprecipitation was performed to isolate nascent (BrdU labeled) strands of DNA. For each immunoprecipitation reaction, at least 120 ng of sonicated DNA was used. DNA was first heat denatured at 95 °C for 5 min and rapidly cooled on ice. 12.5 μg primary antibody (mouse anti-BrdU, BD Biosciences Pharmingen, cat. no. 555627) was added to the ssDNA suspension with constant rocking for 20 minutes followed by addition of 20 μg of secondary antibody (rabbit anti-mouse IgG, Sigma, cat. no. M-7023) for 20 min. Antibody-DNA complexes were then pelleted, resuspended and incubated in digestion buffer (50mM Tris-HCl, 0.01M EDTA, 0.5% SDS and 0.25 mg/ml proteinase K) at 37 °C overnight. Nascent ssDNA was subsequently purified by phenol-chloroform extraction and ethanol precipitation.
Enrichment for BrdU labelled DNA (optimized)
The above protocol was adapted to decrease the length of sample processing specifically for non-fixed, non-sorted cells. DNA was isolated using the Quick-DNA Universal kit (Zymo Research Cat# D4069). Sonication was performed at 40% amplitude for 2 min for samples resuspended in 200 μl, 4 min for samples resuspended in 500 μl or in a 96-well plate using a Covaris 96-well sonicator (E220) for 75 s/sample. BrdU-immunoprecipitation was performed for 60 min and 30 min for primary and secondary antibodies listed above, then nascent ssDNA was purified using the Agencourt AMPure XP system (Beckman Coulter Cat# A63881).
Fluorescent activated cell sorting (FACS) and cell cycle analysis
Fixed cells, (in some cases BrdU labeled and/or with live-dead stain) were pelleted and washed twice in FACS buffer (1% FBS in PBS) and then stained with PI for 20-30 min. Cells were also co-incubated with RNAse A (0.250 mg/ml). Prior to sorting, cells were passed through 40 μm filter FACS tubes. FACS was performed using a BD FACSAria Cell Sorter with a 100 μm nozzle under low pressure. To identify G1, S and G2-M cell populations, three sequential gating strategies were applied to the bulk sample population. Cell debris were removed using a forward versus side scatter comparison, dead cells were removed based on live/dead Fixable Blue stain intensity, and finally single cells (singlets) were enriched for using a PI signal width versus height comparison strategy.
Motor neuron differentiation
Human ESC colonies were dissociated using Accutase and plated at a density of 74,000 cells/cm2 with 10 μM ROCK inhibitor (Y-27632, DNSK International) in mTeSR1 for 48 h. Media was replaced on day 0 with N2B27 medium (50% DMEM:F12, 50% Neurobasal, supplemented with NEAA, Glutamax, N2 and B27; Gibco, Life Technologies) containing 10 μM SB431542 (DNSK International), 100 nM LDN-193189 (DNSK International), 1 μM Retinoic Acid (RA, Sigma) and 1 μM of Smoothened-Agonist (SAG, DNSK International). Culture medium was changed daily for 6 days and then was switched to N2B27 medium supplemented with 1 μM RA, 1 μM SAG, 5 μM DAPT (DNSK International) and 4 μM SU5402 (DNSK International). Cells were fed daily until day 14 of differentiation. At day 6 and 14, cells were collected for WGBS.
Multiplexed single cell RRBS
ESCs were collected, fixed in 75% ethanol and stained with PI. Cells were then sorted by cell cycle phase into G1, early S, mid S, late S and G2-M phase to obtain 1 cell per well in a 96-well plate containing 5 μl 0.1X CutSmart buffer (New England BioLabs Cat#B7204S) per well (480 cells total). Sorted cells were lysed at 50 °C for 2 h in a reaction containing 0.2 U proteinase K (New England Biolabs), 0.2% Triton X-100 (EMD Millipore) and 1x CutSmart buffer. After heat-inactivation of proteinase K at 75 °C for 30 min, we added 2 μl of digestion buffer consisting of 1xCutSmart buffer and 0.5 μl MspI (20 U/μl, New England Biolabs) directly to each cell lysis reaction. The digested DNA was end-repaired and adenylated by adding to each sample 2 μl of a mixture containing 2.5 U Klenow fragment (3′-5′exo-, NEB), 0.4 μl of dNTP mixture (10 mM dATP, 1 mM dCTP and 1 mM dGTP) and 1x CutSmart buffer and incubating at 30 °C for 25 min followed by 37 °C for another 30 min. After heat inactivation at 70°C for 10 min, adenylated DNA fragments were ligated with 5mC substituted indexed adapters overnight at 16 °C through distributing to each well 3 μl of reaction containing 800 U of T4 ligase (NEB), 0.1 μl of 100mM ATP (Roche), 1.5 μl of 0.1μM methylated indexed adapter and 1x CutSmart buffer. 24 indexed ligation reactions were pooled, purified on AmPure XP beads and subjected to sodium bisulfite treatment using an EpiTect Fast Bisulfite kit (QIAGEN) following the manufacturer’s recommendations with extended conversion time (20 min each cycle). The bisulfite converted multiplex RRBS library was amplified for a total of 18 cycles utilizing KAPA HiFi Uracil+ DNA Polymerase. The PCR program consisted of 98 °C denaturation for 45 s, 6 cycles of 98 °C for 20 s, 58 °C annealing for 30 s and 72 °C extension for 1 min, followed by 12 cycles of 98 °C for 20 s, 65 °C annealing for 30 s and 72 °C extension for 1 min and a final extension at 72 °C for 5 min. Pooled multiplex RRBS libraries with a size range of 150-700 bp were size-selected and gel-purified to remove adapter dimers before loading onto an Illumina HiSeq2500 sequencer. We performed paired-end sequencing runs for a total 200 cycles. In total, only 25/480 cells failed to generate sufficient reads.
Single cell RNA-seq
Two 96-well plates of human ES and mesoderm single cells were used to make libraries for single cell RNA-sequencing. Library generation and analysis was done as described in Ref37. Briefly, RNA-seq reads were first trimmed using Trimmomatic38. Trimmed reads were aligned to the RefSeq hg38 genome and transcriptome (GRCh38.2) using Bowtie2 (Ref39) and TopHat40, respectively. The resulting transcriptome alignments were processed by RSEM to estimate the abundance of RefSeq transcripts, in Transcripts Per kilobase Million (TPM). All cells with less than 4000 detectable annotated transcripts were removed from further analysis. Detectable transcript were defined as transcripts with (TPM > 1). After removing apoptotic cells and background differentiation, cells were ordered according progress in the cell cycle as done previously41. Expression values displayed in Supplementary Fig. 2a were averaged using a moving window of 20 cells.
Quantifying gene expression cycling in G1-S and S-phase
We ordered all G1-S and S-phase cells according to progress along the cell cycle, found the 20-cell window with highest and lowest expression for each gene and calculated a t-test P value. We then permuted the order of all G1-S and S-phase cells at random and calculated the highest and lowest expression 20-cell windows for each gene and corresponding control t-test P value. A false discovery rate (FDR) of finding a gene with a treatment P value was measured by counting the number of genes with a more significant P value in the control experiment with permuted cell order normalized to the total number of control gene experiments. Genes were ranked in their cycling of expression using the FDR measure, ordering from smallest to largest.
Bisulfite sequencing
Either total genomic DNA (for WGBS) or DNA fragments recovered from the BrdU antibody pull-down (for Repli-BS) were concentrated by ethanol precipitation (final volume 20 μl). For the motor neuron samples, genomic DNA was fragmented using a Covaris S2 for 6 min according to the following program: duty cycle 5%; intensity 10; cycle per burst 200. The sheared DNA was purified using the DNA Clean & Concentrator kit from Zymo Research per the manufacturer’s recommendations. For all samples, bisulfite conversion was carried out using the Zymo DNA Methylation-Gold™ Kit (Cat# D5005) according to the manufacturer’s instructions and the bisulfite converted DNA was recovered with 15 μl elution buffer. The eluted DNA was processed immediately utilizing the Accel-NGS Methyl-seq DNA library kit (Cat# DL-ILMMS-12, Swift Biosciences) following the manufacturer’s recommendations with slight modifications. Specifically, we optimized the PCR cycle number required for library DNA enrichments by performing a range of PCR cycles (10, 12, 14, and 16) using 10% of the post-ligation DNA. The minimal cycle number was then used for the sample PCR to generate the sequencing libraries. PCR products were cleaned up using the Agencourt AMPure XP system (Beckman Coulter Cat# A63881) and the final library DNA was eluted in 12 μl elution buffer. Using the Agilent Bioanalyzer we confirmed the absence of adapter-dimers. If residual traces were detected, we performed an agarose gel size-selection to further clean up the final library. Up to 12 indexed sequencing libraries were pooled together for sequencing using a 100-cycle paired-end (PE) run on an Illumina HiSeq 2500 or HiSeq 4000. For 6 nascent WT ESC time course samples (replicate 1, Figure 1e), 75 base paired-end sequencing was performed.
Data processing and analysis
Raw sequencing reads were aligned against human genome version hg19/GRCh37 using BSMAP42. Custom Perl scripts were used to remove read pairs that aligned to different chromosomes. Paired-end read alignments rates varied between 55-94%, mean = 83% (Supplementary Data Set 1). Methylation calls were made by comparing the sequenced reads to the reference genome using custom Perl scripts. Unless otherwise indicated, only CpGs with a minimum coverage of ≥ 5X were used for the downstream analysis.
For comparison between nascent and bulk methylation levels, methylation ratios were generated by dividing the mean level of methylation for all nascent CpGs by the mean level of methylation for matched bulk CpGs. For non-CpGs, cytosine methylation levels were generated using BSMAP. To visualize read enrichment, IGV was used and maximum coverage values were set to 20 for all tracks (scaled by 1,000,000/total number of mapped reads). CpG density was calculated as the percentage of CpG dinucleotides per 100 bp.
To identify replication-timing domains specific to each S-phase fraction (S1-S6), we utilized a 10-kb sliding window to identify regions in which the average read density was 1.5X greater than the genomic average within each fraction. For the methylation analysis in Supplementary Figure 1d and 2e, regions from S1 and S2, S3 and S4, and S5 and S6 fractions were merged and termed early, mid, and late fractions, respectively.
For genome methylation plots, loess smoothing and standard deviation calculations were derived using the msir package in R (V1.3.1) with span set to 0.04 for the 120 kb region on chromosome 17 and 0.1 for the POU5F1 (OCT4) locus. All genomic regions displayed are euchromatic. Violin plots were created using the R package ‘vioplot’ using standard parameters. Boxplots and heat maps were created using R. For boxplots: the median is shown in bold, the box displays interquartile range and whiskers extend to the most extreme data point that is no more than 1.5 times the interquartile range.
For analysis of single cell -RRBS data, only CpGs that had methylation ≥ 0.8 in the ESC bulk sample were used to study methylation levels in single cells. This was done because RRBS captures predominantly regions with high CpG density which are typically lowly methylated CpGs.
When comparing replicates, we confirmed the absence of coverage dependent effects by comparing methylation of matched CpGs with ≥ 10X coverage in both samples. For the time course heat maps, only CpGs with ≥ 5X coverage in the combined replicate dataset, and with ≥ 5X coverage in the bulk dataset were included.
For the genomic feature comparisons, CpGs from nascent or bulk DNA were intersected using BedTools v2.25.0 with: CpG islands, as previously defined in Ref43, H1-specific typical or super-enhancers, exons, introns, high CpG density or low CpG density promoters (Ref44), Long Interspersed Nuclear Elements (LINEs) and Short Interspersed Nuclear Elements (SINEs) downloaded from the UCSC Genome Browser. H3K27me3, H3K4me3, H3K27ac and EZH2 annotations were downloaded for H1 ESCs from UCSC. CpG density was calculated for 100 bp tiles as the total number of CpG dinucleotides/100 base pairs.
To compare methylation levels between gene bodies of expressed vs. non-expressed genes, bulk RNA-seq data was used from Gifford et al., Table S2 Ref45. Genes were binned into not expressed (FPKM < 1), expressed (FPKM > 10) and highly expressed (FPKM > 100). For all three categories, CpGs within gene bodies (using the UCSC RefSeq gene annotation) of the respective genes were taken and nascent and bulk DNA methylation levels were compared.
For individual read analyses, we first extracted individual methylation read patterns using custom scripts written in Perl and applied custom R scripts to determine the CpG methylation status of consecutive sites for each read. Only reads with a minimum of three CpGs were considered for this analysis.
To account for differential coverage among samples (e.g. arrested and bulk methylation levels; see Supplementary Data Set 1), we downsampled the higher coverage sample to match the number of aligned reads in the comparison set. Specifically, for the arrested to bulk comparison, 28.5% of reads within the ESC bulk DNA dataset were selected at random and methylation ratios were called as described above.
To investigate CpG methylation within transcription factor binding sites we used our previously described transcription factor ChIP peak sets in human ESCs and their derivative germ cell layers (GSE61475): NANOG (ESC; GSM1505698), OCT4 (ESC; GSM1505724), FOXA2 (endoderm; GSM1505639), EOMES (mesendoderm; GSM1505630), GATA6 (mesoderm; GSM1505661) and PAX6 (Ectoderm; GSM1505715). Bulk and arrested datasets were intersected with the peak sets to obtain only CpGs located within transcription factor binding sites. The proportion of CpGs that were equal to 0, 1 or intermediate were then calculated in bulk and arrested conditions. We repeated this analysis for only CpGs covered with at least 10 reads and confirmed no coverage-dependent effects.
Data availability
Data have been deposited in the Gene Expression Omnibus (GEO) under accession GSE82045. Other published data sets used in this study include: HUES64 (GSM1112841), HUES64 DNMT3A and DNMT3B DKO (GSM1545007), transcription factor binding data (GSE61475) and motor neuron WGBS data (GSM2406773, MN-d6 and GSM2406772, MN-d14). A Life Sciences Reporting Summary for this article is available.
Supplementary Material
Acknowledgments
We thank all members of the Meissner laboratory and in particular Rahul Karnik. We would also like to thank Albert Jeltsch for providing thoughtful feedback on the manuscript. TLD was supported in part by postdoctoral fellowships from the Ford Foundation, UNCF/Merck Science Initiative, Harvard Medical School, and the Broad Institute Diversity Initiative. The Kiskinis lab gratefully acknowledges financial support from the Les Turner ALS Foundation, Muscular Dystrophy Association and the Feinberg School of Medicine. AM is a New York Stem Cell Foundation Robertson Investigator. The Max Planck Society, the New York Stem Cell Foundation, the Broad Institute (SPARC funding to develop single cell RRBS) and NIH grants (1P50HG006193, P01GM099117, R01DA036898), supported this work.
Footnotes
Author Contributions
JC, TLD and AM designed the study with input from ZDS. TLD, JC, RP and VA performed the experiments. HG and AG developed the multiplexed single cell RRBS protocol, generated the sequencing libraries and helped with design of experiments and the analysis. KC, SK, BT and MJZ assisted in data processing. JC performed bioinformatics analyses. AT performed the single cell RNA-seq cell cycle analysis. DPS and EK performed the MN differentiation, characterization and sample collection. JC, TLD, ZDS, AG and AM interpreted the data. JC, TLD, ZDS and AM wrote the manuscript with assistance from the other authors.
The authors declare no competing financial interests.
References
- 1.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- 2.Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenetics and cell genetics. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 3.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 4.Prelich G, Stillman B. Coordinated leading and lagging strand synthesis during SV40 DNA replication in vitro requires PCNA. Cell. 1988;53:117–126. doi: 10.1016/0092-8674(88)90493-x. [DOI] [PubMed] [Google Scholar]
- 5.Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A. 1983;80:5559–5563. doi: 10.1073/pnas.80.18.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004 doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 7.Pradhan S, et al. Baculovirus-mediated expression and characterization of the full-length murine DNA methyltransferase. Nucleic Acids Res. 1997;25:4666–4673. doi: 10.1093/nar/25.22.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 9.Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 10.Qin W, et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell research. 2015;25:911–929. doi: 10.1038/cr.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang LS, et al. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 12.Schermelleh L, et al. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–4312. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziller MJ, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams RL. The relationship between synthesis and methylation of DNA in mouse fibroblasts. Biochim Biophys Acta. 1971;254:205–212. doi: 10.1016/0005-2787(71)90829-x. [DOI] [PubMed] [Google Scholar]
- 15.Liang G, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodcock DM, Adams JK, Cooper IA. Characteristics of enzymatic DNA methylation in cultured cells of human and hamster origin, and the effect of DNA replication inhibition. Biochim Biophys Acta. 1982;696:15–22. doi: 10.1016/0167-4781(82)90004-5. [DOI] [PubMed] [Google Scholar]
- 17.Woodcock DM, et al. Delayed DNA methylation is an integral feature of DNA replication in mammalian cells. Exp Cell Res. 1986;166:103–112. doi: 10.1016/0014-4827(86)90511-2. [DOI] [PubMed] [Google Scholar]
- 18.Shirodkar AV, et al. A mechanistic role for DNA methylation in endothelial cell (EC)-enriched gene expression: relationship with DNA replication timing. Blood. 2013;121:3531–3540. doi: 10.1182/blood-2013-01-479170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- 21.Hansen RS, et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci. 2014;39:310–318. doi: 10.1016/j.tibs.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Lei H, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 24.Liao J, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet. 2015;47:469–478. doi: 10.1038/ng.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson M, et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos DP, Kiskinis E. Generation of Spinal Motor Neurons from Human Pluripotent Stem Cells. Methods Mol Biol. 2017;1538:53–66. doi: 10.1007/978-1-4939-6688-2_5. [DOI] [PubMed] [Google Scholar]
- 27.Hansen KD, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landau DA, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 30.Elliott G, et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nat Commun. 2015;6:6363. doi: 10.1038/ncomms7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Oakeley EJ, Sun L, Jost JP. Multiple domains are involved in the targeting of the mouse DNA methyltransferase to the DNA replication foci. Nucleic Acids Res. 1998;26:1038–1045. doi: 10.1093/nar/26.4.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith ZD, et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017;549:543–547. doi: 10.1038/nature23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witte T, Plass C, Gerhauser C. Pan-cancer patterns of DNA methylation. Genome Med. 2014;6:66. doi: 10.1186/s13073-014-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donaghey J, et al. Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat Genet. 2018 doi: 10.1038/s41588-017-0034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petruk S, et al. Delayed Accumulation of H3K27me3 on Nascent DNA Is Essential for Recruitment of Transcription Factors at Early Stages of Stem Cell Differentiation. Molecular cell. 2017;66:247–257 e245. doi: 10.1016/j.molcel.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trombetta JJ, et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol. 2014;107(4):22, 21–17. doi: 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kowalczyk MS, et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. doi: 1471-2105-10-232 [pii] 10.1186/1471-2105-10-232 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Illingworth R, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gifford CA, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data have been deposited in the Gene Expression Omnibus (GEO) under accession GSE82045. Other published data sets used in this study include: HUES64 (GSM1112841), HUES64 DNMT3A and DNMT3B DKO (GSM1545007), transcription factor binding data (GSE61475) and motor neuron WGBS data (GSM2406773, MN-d6 and GSM2406772, MN-d14). A Life Sciences Reporting Summary for this article is available.