

Abstract
Excitation-contraction (EC) coupling denotes the conversion of electric stimulus in mechanic output in contractile cells. Several studies have demonstrated that calcium (Ca2+) plays a pivotal role in this process. Here we present a comprehensive and updated description of the main systems involved in cardiac Ca2+ handling that ensure a functional EC coupling and their pathological alterations, mainly related to heart failure.
Keywords: Calcium, RyR, Mitochondria, SerCa, Contraction, Heart Failure
1 Introduction
Each heartbeat is the result of calcium (Ca2+) release and reuptake. During this cycle, the presence of this cation is essential to convert electric stimulation (action potential) in mechanic output (contraction), in a process commonly termed excitation-contraction (EC) coupling (Santulli et al. 2017a; Fabiato and Fabiato 1975). The action potential is the electrical signal that depolarizes the plasma membrane of cardiac myocytes, allowing the entrance of a relatively low amount of extracellular Ca2+, which in turn induces high Ca2+ release from the sarcoplasmic reticulum (SR) (Lenzi and Caniggia 1953). Cytosolic Ca2+ binds to myofilaments, activating contractile machinery (Ebashi et al. 1967; Reddy and Honig 1972). This “Ca2+-induced Ca2+ release” (CICR), represents a positive feedback mechanism that allows the functional coupling between plasma membrane and SR (Katz 1967; Fabiato and Fabiato 1979; Fabiato 1983; Isenberg and Han 1994).
2 Microanatomy of EC Coupling
In cardiac cells, the structural units that mediate CICR are termed “diads” (Page et al. 1971; Soeller and Cannell 1997), consisting in specialized cellular micro-domains each composed by terminal cisternae of SR, also known as junctional SR, localized in close proximity to a tubular invagination of the plasma membrane, the transverse tubule (T-tubules) (Soeller and Cannell 1997; Shacklock et al. 1995; Clark et al. 2001). On the side of plasma membrane, along T-tubules, there are voltage-gated Ca2+ channels (T-Type and L-type, also known as dihydropyridine receptors, DHPRs), whereas on the side of SR cisternae there are intracellular Ca2+ release channels (ryanodine receptors, RyRs) (Pinali et al. 2017; Crocini et al. 2016; Li et al. 2015; Fameli et al. 2014).
Ca2+ channels on T-tubules are activated by depolarization and through them Ca2+ from the extracellular compartment goes in the cytosol, and activates RyR channels located on the SR of the same diad (Keizer and Levine 1996; Nakai et al. 1997). It has been estimated that for each Ca2+ channel opened on T-tubules, about 4–6 RyR channels are activated on SR, which in turn can activate neighboring RyR channels, resulting in massive release of Ca2+ from SR (Wang et al. 2001; Paolini et al. 2004).
3 Troponin, Tropomyosin, and Ca2+: How Force Is Produced
The troponin complex consists of three subunits: tropomyosin-binding (T), inhibitory (I), and Ca2+ sensor (C). Troponin-T anchors the other two subunits on tropomyosin. Troponin I, under resting intracellular levels of Ca2+, competes with tropomyosin for a common binding site on actin myofilaments, forcing tropomyosin into a position where it sterically blocks the binding of myosin heads (Lehman et al. 2013; Rao et al. 2014). The binding of cytosolic Ca2+ to Troponin-C induces conformational changes leading to the so-called activated state, in which myosin-binding sites on actin are exposed (Wang et al. 1999; Lehman et al. 2009). Then, the formation of cross-bridges between actin and myosin produces the sliding of myofilaments one over the other, eventually resulting in muscle contraction (Brunello et al. 2014; Kawai et al. 2006; Piazzesi and Lombardi 1996).
The inodilator levosimendan is a Ca2+ sensitizer that acts via binding the Ca2+-saturated Troponin-C (Gustafsson et al. 2017). In particular, hydrogen-bond donor and acceptor groups on the pyridazinone ring and on the mesoxalonitrile–hydrazone moieties of levosimendan bind to a hydrophobic pocket of the Ca2+-saturated amminoterminal domain of Troponin-C (Robertson et al. 2008). The main consequence of levosimendan binding is that the Ca2+-saturated Troponin-C is stabilized in the presence of the drug, thereby promoting contractile force without an increase in the amplitude of intracellular Ca2+ transient. Levosimendan also acts via activation of ATP-sensitive K+ channels (Grossini et al. 2009). In clinical trials levosimendan exhibited non-consistent results, especially in terms of mortality (Mebazaa et al. 2007; Polzl et al. 2017; Landoni et al. 2017).
Ca2+ handling within cardiac myocytes is in a delicate and dynamic equilibrium, which relies on several physiological modulators and subcellular systems, structurally and functionally related. Importantly, the amount of Ca2+ extruded by the cell during relaxation must be the same as the amount entering during the contraction (Karlstad et al. 2012). After the contraction phase, several mechanisms contribute to relaxation. Notably, Ca2+ is a fundamental determinant also during relaxation, since the correct functioning of channels responsible for cytosolic Ca2+ removal strictly depends on Ca2+ concentration (Allen et al. 1988; Plummer et al. 2011; Subramani et al. 2005).
4 Ca2+ Handling in the Pathogenesis of Heart Failure: New Insights
An accurate cardiac Ca2+ handling warrants the appropriate contraction that allows the heart to pump sufficient blood to meet the metabolic demand of the body. Impairment in cardiac pump function characterizes the pathological condition of heart failure (HF) (Karlstad et al. 2012; Kitsis and Narula 2008). Malfunction of the processes involved in Ca2+ handling plays an important role in the pathogenesis of HF, and this kind of alteration in myocytes is a primary defect causing contractile dysfunction in HF (Yano et al. 2005). Modifications in the expression and/or activity of Ca2+ channels, alongside with alterations of cardiomyocyte architecture (T-tubules), seem to have a determinant role in failing myocytes (Balke and Shorofsky 1998). The involvement of different Ca2+ transporters has been related to the stage and etiology of HF; indeed, studies in human failing hearts suggest that patients with ischemic cardiomyopathy are characterized by impaired Ca2+ uptake, while patients with idiopathic dilated cardiomyopathy exhibits mainly modifications in Ca2+ release (Sen et al. 2000).
Cardiac contraction is the result of isometric force (or ventricular pressure) and rapid shortening to allow ejection of the blood (Caremani et al. 2016; Colomo et al. 1994). Both of them rely on Ca2+, specifically on amplitude/duration of Ca2+ transients, and on sensitivity of myofilaments to Ca2+. To allow the generation of Ca2+ transients appropriate in terms of duration and amplitude, cardiac myocytes need a high – and at the same time flexible – capacity of Ca2+ buffering. Normally the amount of Ca2+ bound is in a 100:1 ratio compared to Ca2+ free in the cytosol, and during contraction such ratio immediately increases (about tenfold) in favor to free Ca2+ (MacGowan et al. 2006). Ca2+ buffering should ensure an optimal removal of free Ca2+ from cytosol in order to have an appropriate amount of Ca2+ available to the contraction in the next beat.
Each cycle of EC coupling needs a dynamic interplay between Ca2+ transient and myofilaments. The sensitivity of myofilaments to Ca2+ is high when the myofilaments are stretched, i.e. in the phase of cardiac blood filling (diastole) (Zhao et al. 2016). Therefore, right after relaxation the myofilaments are more responsive to Ca2+ binding. Such autoregulation is a key mechanism underlying the coordination in EC coupling. Hence, when a perfect overlap of high Ca2+ transient and high myofilaments sensitivity to Ca2+ is obtained, an appropriate force is generated, leading to an optimal contraction.
Although the exact mechanisms involved in cardiac Ca2+ handling are not entirely understood, it is known (Benitah et al. 2003) that EC coupling and cardiac performance are mainly based on two mechanisms: (1) Synchronized Ca2+ release during contraction; (2) Effective Ca2+ reuptake that ensure a good and robust termination of Ca2+ dependent contraction.
To satisfy the first point, there are systems that mediate Ca2+ flux towards the cytosol, from extracellular compartment or from intracellular Ca2+ stores, i.e. SR, during EC coupling (Fig. 1). For the second one, there are systems involved in retrograde Ca2+ flux, to terminate EC coupling (Fig. 2).
Fig. 1.
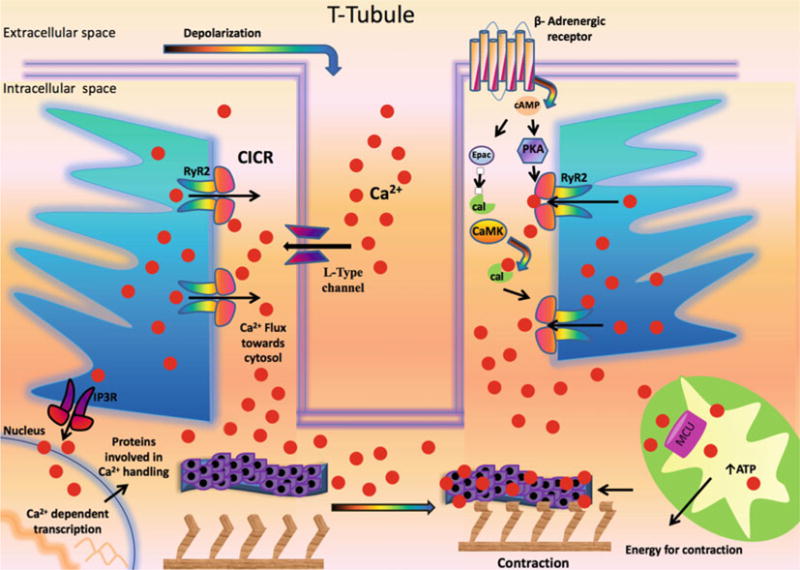
Role of Ca2+ in excitation-contraction coupling in cardiomyocyte. Ca2+: calcium; cal: calmodulin; CaMK: Ca2+/calmodulin-dependent protein kinase; CICR: Ca2+-induced Ca2+ release; Epac: exchange protein directly activated by cAMP; IP3R: inositol 1,4,5-trisphosphate receptor; MCU: mitochondrial Ca2+ uniporter; PKA: Protein Kinase A; RyR2: Type 2 ryanodine receptor Ca2+ release channel
Fig. 2.
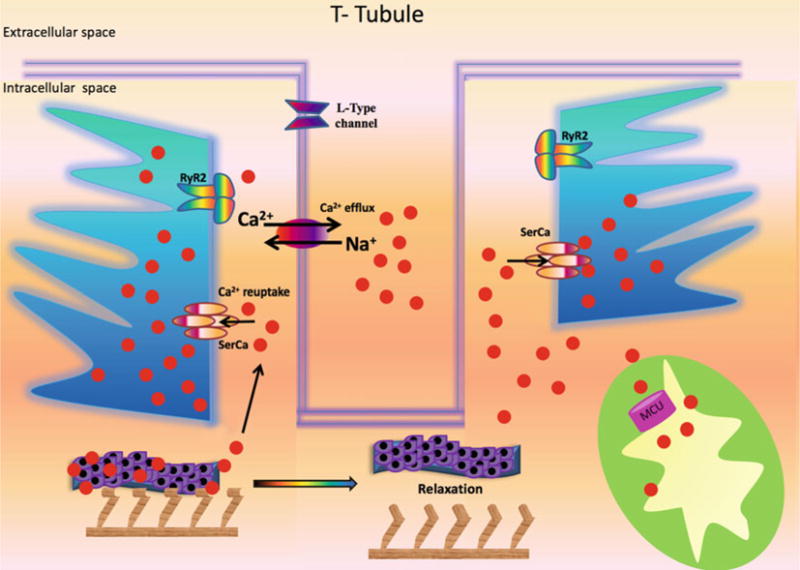
Mechanistic role of Ca2+ in the relaxation phase. Ca2+: calcium; MCU: mitochondrial Ca2+ uniporter; Na+: sodium; RyR2: Type 2 ryanodine receptor Ca2+ release channel; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase
5 Ca2+ Fluxes during Systole
Ca2+ flux towards the cytosol starts with the entrance of a small amount of Ca2+ from extracellular space, triggering EC coupling: the depolarization wave is “transformed” in high Ca2+ release from intracellular store for contraction (Nanasi et al. 2017). The individual events of local Ca2+ release from SR are named Ca2+ sparks and reflect the activation of a cluster of about 6–20 Ca2+ channels on SR (Xie et al. 2013). Single Ca2+ sparks occur also at rest, and a crucial difference from Ca2+ sparks events during EC coupling is the synchronization; indeed, the action potential is able to synchronize several thousand of Ca2+ sparks within the same cell (Louch et al. 2013). Thus, individual events of local Ca2+ release are overlapping in time and space during EC coupling.
The main cellular complexes mediating Ca2+ flux towards cytosol for EC coupling are: voltage sensitive Ca2+ channels (T- and L-type) on the plasma membrane and RyRs on the SR.
6 Voltage-Dependent Ca2+ Channels
In myocytes there are two main types of voltage-dependent Ca2+ channels: T-type and L-type Ca2+ channels (Gonzalez-Rodriguez et al. 2015; Shaw and Colecraft 2013). The T-type channels are not preferentially located in the junctional region inside the diads, and the Ca2+ current generated by this kind of channels is negligible, with a minimal contribute to EC coupling (Zhou and January 1998). The L-type channels, also known as dihydropyridine receptors (DHPRs), are the main voltage dependent Ca2+ channels involved in EC coupling; they mainly localize on the T-tubule, in close proximity of Ca2+ channels on SR terminal cisternae (Bodi et al. 2005). DHPRs are activated by depolarization, allowing the Ca2+ to enter inside the cytosol and trigger CICR. A strategic auto-regulation of the CICR mechanism occurs at this level: indeed, DHPRs are inhibited by Ca2+ itself at the cytosolic side, limiting the amount of Ca2+ entry after depolarization. This local inhibitory effect on DHPRs is mediated by calmodulin, which binds the C-terminal domain on the receptor (Pott et al. 2007). Ca2+ released from SR in the same sarcolemmal-SR junction has also a modulatory effect on L-type receptors: when SR Ca2+ release occurs, the Ca2+ flux through DHPRs is reduced (Shiferaw et al. 2003). These events confirm that the Ca2+ needed for the contraction primarily derives from SR, since Ca2+ channels on plasma membranes are quickly inhibited after the entrance of small amount of Ca2+.
As mentioned above, action potential is responsible for synchronization of Ca2+ release from SR. The specific organization of ion channels on the plasma membrane, and in particular on the specialized invaginations of the T-tubules, plays a pivotal role in EC coupling (Oyehaug et al. 2013; Galbiati et al. 2001). Indeed, the loss of these structures leads to aberrant Ca2+ handling with blunted contractility, and such alteration has been mechanistically associated to HF (Wei et al. 2010).
In humans, HF secondary to dilated, hypertrophic, and ischemic cardiomyopathy, is related to aberrant alterations of T-tubule structure and the subsequent not-appropriate organization of voltage-dependent Ca2+ channels (Lyon et al. 2009). These anomalies could also be related to impaired function of junctophilins, proteins that are responsible for appropriate positioning of voltage-gated Ca2+ channels on T- tubules (Pinali et al. 2017; Schobesberger et al. 2017).
7 Key Role of RyR2 in Cardiac EC Coupling
RyRs are intracellular release channels, deputed to Ca2+ release from intracellular stores; the RyR2 isoform represents the most abundant isoform of this family in cardiomyocytes (Tunwell et al. 1996). This name was attributed based on the molecule with a high binding affinity to these receptors: the ryanodine, a natural product, known to induce a paralytic effect on striated muscle (Sutko et al. 1997; Rumberger and Ahrens 1972).
These channels have a tetrameric structure, similar to a mushroom, with the stalk across the SR membrane and the cap towards the cytosol. In this large structure, it is possible to identify at least two functional domains: C-terminus, which represents the ion pore across the membrane, and N-terminus that contains numerous modulatory sites (Gao et al. 1997). Indeed, RyR activity is highly regulated by several molecules – including calmodulin, FK-506 binding protein (FKBP), sorcin, junctin, and triadin (Balshaw et al. 2001; Anthony et al. 2007) – linked to the receptor, which thus represents the scaffold of a large macromolecular complex (Santulli et al. 2017b). Calmodulin and sorcin, two small proteins with high affinity for Ca2+, are able to inhibit RyR when Ca2+ levels in the cytosol are higher than a threshold value, preventing further Ca2+ release from SR. This phenomenon is involved in EC coupling termination.
RyR has also several sites modulated by Ca2+, Mg2+, phosphorylation, and oxidation, making this receptor also a sensor of intracellular redox state (Hain et al. 1995; Chugun et al. 2007; Xie et al. 2015). Therefore, RyR activity is the result of a multifaceted interactome specialized in Ca2+ handling.
Inositol-1,4,5, trisphosphate (IP3) receptors (IP3Rs) represent another example of intracellular Ca2+ release channel. Located on the SR as RyR, they are activated by a second messenger, IP3, involved in several pathways. These receptors are closely related to the RyR family, sharing a high structural homology (Santulli et al. 2017a, b; Maltsev et al. 2017). Although there are several stimuli that induce IP3 accumulation, the physiological depolarization of cardiac myocyte, does not seem to activate IP3Rs and their involvement seem to be limited to the regulation of transcription of important modulator of Ca2+ handling machinery (e.g. CamKII, calcineurin), indirectly contributing to regulate the capacity of the cells to maintain Ca2+ homeostasis, indispensable for appropriate EC coupling and contractility (Santulli et al. 2017a; Santulli and Marks 2015). Moreover, IP3R has a different distribution in atrial and ventricular myocytes, compared to RyR2; in particular, IP3Rs are principally located in the atrial myocyte and also in Purkinje fibers (Luo et al. 2008; Mignery et al. 1990). Purkinje fibers are a specialized conduction system, and IP3Rs can modulate the transfer of depolarization wave through ventricular mass.
8 Ca2+ Fluxes During Diastole
To ensure muscle relaxation, mechanisms of Ca2+ reuptake from the cytosol actively mediate a retrograde Ca2+ flux. The main “cytosolic Ca2+ scavengers”, Sarco-Endoplasmic reticulum Ca2+ ATPase (SerCa) and Na+/Ca2+ exchanger, achieve this function (Fig. 2). A large portion of Ca2+ is pumped by SerCa from the cytosol back to the SR, but the relative contribute of different Ca2+ removal systems can depend on the species.
9 Functional Role of Na+/Ca2+ Exchanger in Ca2+ Reuptake
Na+/Ca2+ exchanger mediates reversibly influx or efflux of Ca2+, depending on concentration of free Ca2+ inside the cytosol, but normally it works in Ca2+ efflux mode (Fujioka et al. 2000; Luongo et al. 2017). In cardiomyocytes this channel represents the major Ca2+ efflux system, with a critical role in the regulation of Ca2+ cellular content (Zhu et al. 2015; Acsai et al. 2011). Indeed, some alterations of ionic equilibrium inside the cell, that induce increase in Na+ intracellular levels, can trigger changes in the activity of the channel, which starts to work in reverse mode, mediating Na+ efflux, with influx of Ca2+ ions (Kang and Hilgemann 2004).
10 SerCa-ATPase
SerCa ATPase represents a fundamental pump system mediating Ca2+ reuptake in the SR/ER (Zima et al. 2014). In cardiomyocytes, SerCa removes the Ca2+ from cytosol during relaxation with an active process (ATP hydrolysis), in order to terminate the EC coupling cycle and to restore Ca2+ levels inside SR, necessary for the next contraction.
The correct activity of SerCa, and the indirect interplay with RyR2 that promotes Ca2+ flux in the opposite direction, are essential to have an EC coupling cycle with appropriate amplitude and synchronization in space and time. Ca2+ itself plays a pivotal role in mediating such interplay (Collins and Thomas 2001). SerCa, as well as RyR2, is highly regulated not only directly by Ca2+, but also by other molecules that are sensitive (CaMKII) or not (PKA) to Ca2+. These two kinases, as seen for RyR2, are able to regulate SerCa, but not via direct phosphorylation, but through phosphorylation of the most important regulator of SerCa, phospholamban. Phospholamban (PLB), is an endogenous inhibitor of SerCa that limits the affinity of the pump for Ca2+. PKA can phosphorylate PLB at Ser16, inhibiting PLB and therefore activating SerCa, increasing its pumping rate (Simmerman et al. 1986). CaMKII, whose activation relies also on the exchange protein directly activated by cAMP (Epac) pathway (de Rooij et al. 1998; Lymperopoulos et al. 2014; Parnell et al. 2015), is able to phosphorylate PLB at Thr17, leading also in this case to an acceleration of SR Ca2+ uptake (Simmerman et al. 1986). SerCa is also regulated by the metabolic state of the cell in terms of ATP/ADP ratio, and redox-status (Zima et al. 2014). Alterations in expression or activity of SerCa, strongly contribute to HF development and progression, through dysfunctional Ca2+ handling. In particular, several studies show that both mRNA and protein levels of SerCa pump are downregulated in HF (Currie and Smith 1999; Armoundas et al. 2007). The decrease in SerCa/PLB ratio results in impaired SR Ca2+ uptake (Linck et al. 2000). Moreover, the regulation of SerCa by both PKA (Schwinger et al. 1998) and CaMKII (Schwinger et al. 1999) is significantly impaired in failing hearts.
11 Intracellular Ca2+ Leak: Definition, Pathophysiology, and Interventions
RyR2 interactome ensures the appropriate amplitude and kinetics of Ca2+ cycling, indispensable for cardiac contractility. Alterations in RyR2 and/or in RyR2 interactome, are among the main identified aspects of HF (Ono et al. 2000; Kohno et al. 2003). All the events that are able to affect the opening frequency of RyR2, determining changes in amplitude or duration of Ca2+ release, can predispose to contractility dysfunction (Patel et al. 2000). In particular, the phenomenon of Ca2+ leak, defined as inappropriate release of Ca2+ from the SR (e.g. during the diastolic phase), is determinant in HF progression (Hofer et al. 1996). In pathological conditions such as HF, there is a high release of Ca2+ from SR during diastole, reducing the availability of SR Ca2+ for the subsequent contraction, thereby impairing contractility (Santulli et al. 2017b).
FKBP12.6, also known as Calstabin2, plays an important pathophysiological role in the regulation of RyR2 function. Indeed, it stabilizes a conformation of the channel that prevents Ca2+ leak (Timerman et al. 1996; Lam et al. 1995; Xin et al. 1999; Yuan et al. 2014). There are numerous events, both acute and chronic, that trigger instability of RyR macromolecular complex. One typical example is represented by its phosphorylation, which induces the dissociation from the receptor of its stabilizing molecule, FKBP12.6 (Yuan et al. 2014). Chronic activation of beta-adrenergic system increases PKA activity (Santulli et al. 2013), with consequent phosphorylation of RyR (Takasago et al. 1991). Phosphorylation of RyR by CaMKII has also been reported in HF (Kushnir et al. 2010). Furthermore, ROS-linked oxidation of RyR affects the normal Ca2+ handling, compromising the correct contraction of myocytes during HF (Santulli et al. 2015).
Several benzothiazepine derivatives, including JTV519 (also known as K201, 4-[−3{1-(4-Benzyl) piperidinyl}propionyl]-7-methoxy-2,3,4,5-tetrahydro-1,4-benzothiazepine) (Kohno et al. 2003; Hachida et al. 1999a, b; Sacherer et al. 2012) and its substructure S107 (7-methoxy-4-methyl-2,3,4,5-tetrahydro-1,4-benzothiazepine) (Thevis et al. 2009; Matecki et al. 2016; Lukyanenko et al. 2017) have shown cardioprotective and antiarrhythmic properties by decreasing Ca2+ leak (Santulli et al. 2017a; Xie et al. 2013, 2015).
Henceforth, RyR2 represents the central target of many pathways dysregulated in cardiac pathological conditions, including adrenergic dysfunction, metabolic disorders, ROS production: all of these conditions are accompanied by alterations in Ca2+ handling and subsequent impairment in contractility.
12 Role of Mitochondrial Ca2+ in Metabolism-Contraction Coupling
Mitochondria play a strategic role in ensuring an adequate EC coupling (Miragoli and Cabassi 2017; Umanskaya et al. 2014; Gambardella et al. 2017; Torrealba et al. 2017). Indeed, cardiac myocytes are critically dependent on constant and appropriate energy supply, alongside with a finely tuned Ca2+ handling (Torrealba et al. 2017; Sorriento et al. 2017; Sheeran and Pepe 2017). Mitochondria and SR are functionally and structurally associated and, at the point of interaction (mitochondrial associated membranes) Ca2+ transits from SR to mitochondria (Min et al. 2012; Bononi et al. 2017). Ca2+ that enters in mitochondria is able to increase their bioenergetics, but at the same time, mitochondria, allowing Ca2+ entrance through the mitochondrial Ca2+ uniporter (De Stefani et al. 2011; Liu et al. 2017; Granatiero et al. 2017), can act as local Ca2+ buffers, actively participating in Ca2+ handling (Walsh et al. 2009; Gunter and Sheu 2009; Drago et al. 2012; Morciano et al. 2017). However, mitochondrial Ca2+ overload has been shown to be detrimental in various cell types (Liu et al. 2016; Kostic et al. 2015; Charles et al. 2017), and, specifically, it has been proven to play a mechanistic role in the pathogenesis of HF (Santulli et al. 2015). Therefore, our hypothesis is that whereas transient mitochondrial Ca2+ uptake promotes ATP production, prolonged or excessive Ca2+ uptake can be harmful.
The ability of mitochondria to respond to changes in Ca2+ levels, increasing metabolic outcome, and acting as Ca2+ scavenger, contributes to the adequate contractile response of cardiomyocytes. Indeed, alterations in these processes can lead to increase in cytosolic Ca2+ with potential activation of detrimental pathways that have been associated with HF, including CaMKII (Zhang and Brown 2004). Additionally, alterations in SR-mitochondria contacts have been found in cardiac dysfunction and HF (Lopez-Crisosto et al. 2017).
Whether mitochondrial Ca2+ transients exist during physiological EC coupling, in a beat-to-beat fashion, remains to be determined. Anyway, an important aspect of these phenomena is that Ca2+ proves once again its ability to act as a messenger, communicating to mitochondria the energetic demand of the cell, for adequate contraction.
Acknowledgments
GS is supported by NIH (R00DK107895).
Contributor Information
Jessica Gambardella, Department of Advanced Biomedical Sciences, “Federico II” University, Naples, Italy; Department of Medicine, Surgery and Dentistry, Scuola Medica Salernitana, University of Salerno, Fisciano, Italy.
Bruno Trimarco, Department of Advanced Biomedical Sciences, “Federico II” University, Naples, Italy.
Guido Iaccarino, Department of Medicine, Surgery and Dentistry, Scuola Medica Salernitana, University of Salerno, Fisciano, Italy.
Gaetano Santulli, Department of Advanced Biomedical Sciences, “Federico II” University, Naples, Italy; Department of Medicine, Albert Einstein College of Medicine, 1300 Morris Park Ave, Forch 525, 10461 New York, NY, USA.
References
- Acsai K, Antoons G, Livshitz L, Rudy Y, Sipido KR. Microdomain [ca(2)(+)] near ryanodine receptors as reported by L-type ca(2)(+) and Na+/ca(2)(+) exchange currents. J Physiol. 2011;589(10):2569–2583. doi: 10.1113/jphysiol.2010.202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Nichols CG, Smith GL. The effects of changes in muscle length during diastole on the calcium transient in ferret ventricular muscle. J Physiol. 1988;406:359–370. doi: 10.1113/jphysiol.1988.sp017385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DF, Beattie J, Paul A, Currie S. Interaction of calcium/calmodulin-dependent protein kinase IIdeltaC with sorcin indirectly modulates ryanodine receptor function in cardiac myocytes. J Mol Cell Cardiol. 2007;43(4):492–503. doi: 10.1016/j.yjmcc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Rose J, Aggarwal R, Stuyvers BD, O’Rourke B, Kass DA, et al. Cellular and molecular determinants of altered Ca2+ handling in the failing rabbit heart: primary defects in SR Ca2+ uptake and release mechanisms. Am J Physiol Heart Circ Physiol. 2007;292(3):H1607–H1618. doi: 10.1152/ajpheart.00525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balke CW, Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res. 1998;37(2):290–299. doi: 10.1016/s0008-6363(97)00272-1. [DOI] [PubMed] [Google Scholar]
- Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor) J Biol Chem. 2001;276(23):20144–20153. doi: 10.1074/jbc.M010771200. [DOI] [PubMed] [Google Scholar]
- Benitah JP, Gomez AM, Virsolvy A, Richard S. New perspectives on the key role of calcium in the progression of heart disease. J Muscle Res Cell Motil. 2003;24(4–6):275–283. [PubMed] [Google Scholar]
- Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115(12):3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature. 2017;546(7659):549–553. doi: 10.1038/nature22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunello E, Caremani M, Melli L, Linari M, Fernandez-Martinez M, Narayanan T, et al. The contributions of filaments and cross-bridges to sarcomere compliance in skeletal muscle. J Physiol. 2014;592(17):3881–3899. doi: 10.1113/jphysiol.2014.276196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caremani M, Pinzauti F, Reconditi M, Piazzesi G, Stienen GJ, Lombardi V, et al. Size and speed of the working stroke of cardiac myosin in situ. Proc Natl Acad Sci U S A. 2016;113(13):3675–3680. doi: 10.1073/pnas.1525057113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles E, Hammadi M, Kischel P, Delcroix V, Demaurex N, Castelbou C, et al. The antidepressant fluoxetine induces necrosis by energy depletion and mitochondrial calcium overload. Oncotarget. 2017;8(2):3181–3196. doi: 10.18632/oncotarget.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugun A, Sato O, Takeshima H, Ogawa Y. Mg2+ activates the ryanodine receptor type 2 (RyR2) at intermediate Ca2+ concentrations. Am J Physiol Cell Physiol. 2007;292(1):C535–C544. doi: 10.1152/ajpcell.00275.2006. [DOI] [PubMed] [Google Scholar]
- Clark RB, Tremblay A, Melnyk P, Allen BG, Giles WR, Fiset CT. Tubule localization of the inward-rectifier K(+) channel in mouse ventricular myocytes: a role in K(+) accumulation. J Physiol. 2001;537(3):979–992. doi: 10.1111/j.1469-7793.2001.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RO, Thomas RC. The effect of calcium pump inhibitors on the response of intracellular calcium to caffeine in snail neurones. Cell Calcium. 2001;30(1):41–48. doi: 10.1054/ceca.2001.0209. [DOI] [PubMed] [Google Scholar]
- Colomo F, Poggesi C, Tesi C. Force responses to rapid length changes in single intact cells from frog heart. J Physiol. 1994;475(2):347–350. doi: 10.1113/jphysiol.1994.sp020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocini C, Coppini R, Ferrantini C, Yan P, Loew LM, Poggesi C, et al. T-tubular electrical defects contribute to blunted beta-adrenergic response in heart failure. Int J Mol Sci. 2016;17(9) doi: 10.3390/ijms17091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie S, Smith GL. Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res. 1999;41(1):135–146. doi: 10.1016/s0008-6363(98)00241-7. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A. 2012;109(32):12986–12991. doi: 10.1073/pnas.1210718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebashi S, Ebashi F, Kodama A. Troponin as the Ca++-receptive protein in the contractile system. J Biochem. 1967;62(1):137–138. doi: 10.1093/oxfordjournals.jbchem.a128628. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Phys. 1983;245(1):C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249(3):469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Use of chlorotetracycline fluorescence to demonstrate Ca2+-induced release of Ca2+ from the sarcoplasmic reticulum of skinned cardiac cells. Nature. 1979;281(5727):146–148. doi: 10.1038/281146a0. [DOI] [PubMed] [Google Scholar]
- Fameli N, Ogunbayo OA, van Breemen C, Evans AM. Cytoplasmic nanojunctions between lysosomes and sarcoplasmic reticulum are required for specific calcium signaling. F1000Res. 2014;3:93. doi: 10.12688/f1000research.3720.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y, Komeda M, Matsuoka S. Stoichiometry of Na+ Ca2+ exchange in inside-out patches excised from guinea-pig ventricular myocytes. J Physiol. 2000;523(2):339–351. doi: 10.1111/j.1469-7793.2000.t01-2-00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, et al. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276(24):21425–21433. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- Gambardella J, Sorriento D, Ciccarelli M, Del Giudice C, Fiordelisi A, Napolitano L, et al. Functional role of mitochondria in Arrhythmogenesis. Adv Exp Med Biol. 2017;982:191–202. doi: 10.1007/978-3-319-55330-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Tripathy A, Lu X, Meissner G. Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 1997;412(1):223–226. doi: 10.1016/s0014-5793(97)00781-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez P, Falcon D, Castro MJ, Urena J, Lopez-Barneo J, Castellano A. Hypoxic induction of T-type Ca(2+) channels in rat cardiac myocytes: role of HIF-1alpha and RhoA/ROCK signalling. J Physiol. 2015;593(21):4729–4745. doi: 10.1113/JP271053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granatiero V, De Stefani D, Rizzuto R. Mitochondrial calcium handling in physiology and disease. Adv Exp Med Biol. 2017;982:25–47. doi: 10.1007/978-3-319-55330-6_2. [DOI] [PubMed] [Google Scholar]
- Grossini E, Molinari C, Caimmi PP, Uberti F, Vacca G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: role for mitochondrial K(ATP) channel. Br J Pharmacol. 2009;156(2):250–261. doi: 10.1111/j.1476-5381.2008.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787(11):1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson F, Guarracino F, Schwinger R. The inodilator levosimendan as a treatment for acute heart failure in various settings. Eur Heart J. 2017;19(suppl_C):C2–C7. doi: 10.1093/eurheartj/sux001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachida M, Lu H, Kaneko N, Horikawa Y, Ohkado A, Gu H, et al. Protective effect of JTV519 (K201), a new 1,4-benzothiazepine derivative, on prolonged myocardial preservation. Transplant Proc. 1999a;31(1–2):996–1000. doi: 10.1016/s0041-1345(98)01875-2. [DOI] [PubMed] [Google Scholar]
- Hachida M, Kihara S, Nonoyama M, Koyanagi H. Protective effect of JTV519, a new 1,4-benzothiazepine derivative, on prolonged myocardial preservation. J Card Surg. 1999b;14(3):187–193. doi: 10.1111/j.1540-8191.1999.tb00977.x. [DOI] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270(5):2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Curci S, Machen TE, Schulz IATP. regulates calcium leak from agonist-sensitive internal calcium stores. FASEB J. 1996;10(2):302–308. doi: 10.1096/fasebj.10.2.8641563. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Han S. Gradation of ca(2+)-induced Ca2+ release by voltage-clamp pulse duration in potentiated guinea-pig ventricular myocytes. J Physiol. 1994;480(3):423–438. doi: 10.1113/jphysiol.1994.sp020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TM, Hilgemann DW. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature. 2004;427(6974):544–548. doi: 10.1038/nature02271. [DOI] [PubMed] [Google Scholar]
- Karlstad J, Sun Y, Singh BB. Ca(2+) signaling: an outlook on the characterization of ca(2+) channels and their importance in cellular functions. Adv Exp Med Biol. 2012;740:143–157. doi: 10.1007/978-94-007-2888-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz AM. Regulation of cardiac muscle contractility. J Gen Physiol. 1967;50(6 Suppl):185–196. doi: 10.1085/jgp.50.6.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Kido T, Vogel M, Fink RH, Ishiwata S. Temperature change does not affect force between regulated actin filaments and heavy meromyosin in single-molecule experiments. J Physiol. 2006;574(3):877–887. doi: 10.1113/jphysiol.2006.111708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer J, Levine L. Ryanodine receptor adaptation and Ca2+(−)induced Ca2+ release-dependent Ca2+ oscillations. Biophys J. 1996;71(6):3477–3487. doi: 10.1016/S0006-3495(96)79543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsis RN, Narula J. Introduction-cell death in heart failure. Heart Fail Rev. 2008;13(2):107–109. doi: 10.1007/s10741-008-9080-3. [DOI] [PubMed] [Google Scholar]
- Kohno M, Yano M, Kobayashi S, Doi M, Oda T, Tokuhisa T, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol Heart Circ Physiol. 2003;284(3):H1035–H1042. doi: 10.1152/ajpheart.00722.2002. [DOI] [PubMed] [Google Scholar]
- Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu CT, et al. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 2015;13(2):376–386. doi: 10.1016/j.celrep.2015.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107(22):10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, et al. A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J Biol Chem. 1995;270(44):26511–26522. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- Landoni G, Lomivorotov VV, Alvaro G, Lobreglio R, Pisano A, Guarracino F, et al. Levosimendan for hemodynamic support after cardiac surgery. N Engl J Med. 2017;376(21):2021–2031. doi: 10.1056/NEJMoa1616325. [DOI] [PubMed] [Google Scholar]
- Lehman W, Galinska-Rakoczy A, Hatch V, Tobacman LS, Craig R. Structural basis for the activation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2009;388(4):673–681. doi: 10.1016/j.jmb.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motil. 2013;34(3–4):155–163. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi F, Caniggia A. Nature of myocardial contraction and of action potentials; importance of the cationic gradient. Acta Med Scand. 1953;146(4):300–312. doi: 10.1111/j.0954-6820.1953.tb10244.x. [DOI] [PubMed] [Google Scholar]
- Li H, Lichter JG, Seidel T, Tomaselli GF, Bridge JH, Sachse FB. Cardiac resynchronization therapy reduces subcellular heterogeneity of ryanodine receptors, T-tubules, and Ca2+ Sparks produced by Dyssynchronous heart failure. Circ Heart Fail. 2015;8(6):1105–1114. doi: 10.1161/CIRCHEARTFAILURE.115.002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck B, Schmitz W, Messenger RNA. Expression and immunological quantification of phospholamban and SR-ca(2+)-ATPase in failing and nonfailing human hearts. Cardiovasc Res. 2000;45(1):241–244. doi: 10.1016/s0008-6363(99)00337-5. [DOI] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016;16(6):1561–1573. doi: 10.1016/j.celrep.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Parks RJ, Liu J, Stares J, Rovira II, Murphy E, et al. The in vivo biology of the mitochondrial calcium uniporter. Adv Exp Med Biol. 2017;982:49–63. doi: 10.1007/978-3-319-55330-6_3. [DOI] [PubMed] [Google Scholar]
- Lopez-Crisosto C, Pennanen C, Vasquez-Trincado C, Morales PE, Bravo-Sagua R, Quest AFG, et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol. 2017;14(6):342–360. doi: 10.1038/nrcardio.2017.23. [DOI] [PubMed] [Google Scholar]
- Louch WE, Hake J, Mork HK, Hougen K, Skrbic B, Ursu D, et al. Slow Ca(2)(+) sparks de-synchronize Ca(2)(+) release in failing cardiomyocytes: evidence for altered configuration of Ca(2)(+) release units? J Mol Cell Cardiol. 2013;58:41–52. doi: 10.1016/j.yjmcc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Muriel JM, Bloch RJ. Coupling of excitation to Ca2+ release is modulated by dysferlin. J Physiol. 2017;595:5191. doi: 10.1113/JP274515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Yang D, Lan X, Li K, Li X, Chen J, et al. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium. 2008;43(2):165–174. doi: 10.1016/j.ceca.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545(7652):93–97. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Garcia D, Walklett K. Pharmacogenetics of cardiac inotropy. Pharmacogenomics. 2014;15(14):1807–1821. doi: 10.2217/pgs.14.120. [DOI] [PubMed] [Google Scholar]
- Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106(16):6854–6859. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGowan GA, Kirk JA, Evans C, Shroff SG. Pressure-calcium relationships in perfused mouse hearts. Am J Physiol Heart Circ Physiol. 2006;290(6):H2614–H2624. doi: 10.1152/ajpheart.00979.2005. [DOI] [PubMed] [Google Scholar]
- Maltsev AV, Maltsev VA, Stern MD. Clusters of calcium release channels harness the Ising phase transition to confine their elementary intracellular signals. Proc Natl Acad Sci U S A. 2017;114(29):7525–7530. doi: 10.1073/pnas.1701409114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matecki S, Dridi H, Jung B, Saint N, Reiken SR, Scheuermann V, et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc Natl Acad Sci U S A. 2016;113(32):9069–9074. doi: 10.1073/pnas.1609707113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE randomized trial. JAMA. 2007;297(17):1883–1891. doi: 10.1001/jama.297.17.1883. [DOI] [PubMed] [Google Scholar]
- Mignery GA, Newton CL, Archer BT, 3rd, Sudhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1990;265(21):12679–12685. [PubMed] [Google Scholar]
- Min CK, Yeom DR, Lee KE, Kwon HK, Kang M, Kim YS, et al. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for ca (2)+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem J. 2012;447(3):371–379. doi: 10.1042/BJ20120705. [DOI] [PubMed] [Google Scholar]
- Miragoli M, Cabassi A. Mitochondrial Mechanosensor microdomains in cardiovascular disorders. Adv Exp Med Biol. 2017;982:247–264. doi: 10.1007/978-3-319-55330-6_13. [DOI] [PubMed] [Google Scholar]
- Morciano G, Bonora M, Campo G, Aquila G, Rizzo P, Giorgi C, et al. Mechanistic role of mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 2017;982:169–189. doi: 10.1007/978-3-319-55330-6_9. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ogura T, Protasi F, Franzini-Armstrong C, Allen PD, Beam KG. Functional nonequality of the cardiac and skeletal ryanodine receptors. Proc Natl Acad Sci U S A. 1997;94(3):1019–1022. doi: 10.1073/pnas.94.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanasi PP, Magyar J, Varro A, Ordog B. Beat-to-beat variability of cardiac action potential duration: underlying mechanism and clinical implications. Can J Physiol Pharmacol. 2017 doi: 10.1139/cjpp-2016-0597. [DOI] [PubMed] [Google Scholar]
- Ono K, Yano M, Ohkusa T, Kohno M, Hisaoka T, Tanigawa T, et al. Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal ca(2+) release in heart failure. Cardiovasc Res. 2000;48(2):323–331. doi: 10.1016/s0008-6363(00)00191-7. [DOI] [PubMed] [Google Scholar]
- Oyehaug L, Loose KO, Jolle GF, Roe AT, Sjaastad I, Christensen G, et al. Synchrony of cardiomyocyte Ca(2+) release is controlled by T-tubule organization, SR Ca(2+) content, and ryanodine receptor Ca(2+) sensitivity. Biophys J. 2013;104(8):1685–1697. doi: 10.1016/j.bpj.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page E, McCallister LP, Power B. Sterological measurements of cardiac ultrastructures implicated in excitation-contraction coupling. Proc Natl Acad Sci U S A. 1971;68(7):1465–1466. doi: 10.1073/pnas.68.7.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolini C, Fessenden JD, Pessah IN, Franzini-Armstrong C. Evidence for conformational coupling between two calcium channels. Proc Natl Acad Sci U S A. 2004;101(34):12748–12752. doi: 10.1073/pnas.0404836101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell E, Palmer TM, Yarwood SJ. The future of EPAC-targeted therapies: agonism versus antagonism. Trends Pharmacol Sci. 2015;36(4):203–214. doi: 10.1016/j.tips.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Duke K, Light RB, Jacobs H, Mink SN, Bose D. Impaired sarcoplasmic calcium release inhibits myocardial contraction in experimental sepsis. J Crit Care. 2000;15(2):64–72. doi: 10.1053/jcrc.2000.7902. [DOI] [PubMed] [Google Scholar]
- Piazzesi G, Lombardi V. Simulation of the rapid regeneration of the actin-myosin working stroke with a tight coupling model of muscle contraction. J Muscle Res Cell Motil. 1996;17(1):45–53. doi: 10.1007/BF00140323. [DOI] [PubMed] [Google Scholar]
- Pinali C, Malik N, Davenport JB, Allan LJ, Murfitt L, Iqbal MM, et al. Post-myocardial infarction T-tubules form enlarged branched structures with dysregulation of Junctophilin-2 and bridging integrator 1 (BIN-1) J Am Heart Assoc. 2017;6(5):e004834. doi: 10.1161/JAHA.116.004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer BN, Cutler MJ, Wan X, Laurita KR. Spontaneous calcium oscillations during diastole in the whole heart: the influence of ryanodine reception function and gap junction coupling. Am J Physiol Heart Circ Physiol. 2011;300(5):H1822–H1828. doi: 10.1152/ajpheart.00766.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polzl G, Altenberger J, Baholli L, Beltran P, Borbely A, Comin-Colet J, et al. Repetitive use of levosimendan in advanced heart failure: need for stronger evidence in a field in dire need of a useful therapy. Int J Cardiol. 2017;243:389. doi: 10.1016/j.ijcard.2017.05.081. [DOI] [PubMed] [Google Scholar]
- Pott C, Yip M, Goldhaber JI, Philipson KD. Regulation of cardiac L-type Ca2+ current in Na+−Ca2+ exchanger knockout mice: functional coupling of the Ca2+ channel and the Na+−Ca2+ exchanger. Biophys J. 2007;92(4):1431–1437. doi: 10.1529/biophysj.106.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JN, Madasu Y, Dominguez R. Mechanism of actin filament pointed-end capping by tropomodulin. Science. 2014;345(6195):463–467. doi: 10.1126/science.1256159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy YS, Honig CR. Ca 2+ -binding and Ca 2+-sensitizing functions of cardiac native tropomyosin, troponin, and tropomyosin. Biochim Biophys Acta. 1972;275(3):453–463. doi: 10.1016/0005-2728(72)90226-5. [DOI] [PubMed] [Google Scholar]
- Robertson IM, Baryshnikova OK, Li MX, Sykes BD. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochemistry. 2008;47(28):7485–7495. doi: 10.1021/bi800438k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumberger E, Ahrens U. The effect of ryanodine on the force-frequency-relationship of the heart muscle. Pflugers Arch. 1972;332(Suppl):R36. [PubMed] [Google Scholar]
- Sacherer M, Sedej S, Wakula P, Wallner M, Vos MA, Kockskamper J, et al. JTV519 (K201) reduces sarcoplasmic reticulum ca(2)(+) leak and improves diastolic function in vitro in murine and human non-failing myocardium. Br J Pharmacol. 2012;167(3):493–504. doi: 10.1111/j.1476-5381.2012.01995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Marks AR. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol. 2015;8(2):206–222. doi: 10.2174/1874467208666150507105105. [DOI] [PubMed] [Google Scholar]
- Santulli G, Ciccarelli M, Trimarco B, Iaccarino G. Physical activity ameliorates cardiovascular health in elderly subjects: the functional role of the beta adrenergic system. Front Physiol. 2013;4:209. doi: 10.3389/fphys.2013.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Nakashima R, Yuan Q, Marks AR. Intracellular calcium release channels: an update. J Physiol. 2017a;595(10):3041–3051. doi: 10.1113/JP272781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Lewis DR, Marks AR. Physiology and pathophysiology of excitation-contraction coupling: the functional role of ryanodine receptor. J Muscle Res Cell Motil. 2017b doi: 10.1007/s10974-017-9470-z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobesberger S, Wright P, Tokar S, Bhargava A, Mansfield C, Glukhov AV, et al. T-tubule remodelling disturbs localized beta2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc Res. 2017;113(7):770–782. doi: 10.1093/cvr/cvx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinger RH, Bolck B, Munch G, Brixius K, Muller-Ehmsen J, Erdmann E. cAMP-dependent protein kinase A-stimulated sarcoplasmic reticulum function in heart failure. Ann N Y Acad Sci. 1998;853:240–250. doi: 10.1111/j.1749-6632.1998.tb08272.x. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31(3):479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- Sen L, Cui G, Fonarow GC, Laks H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279(2):H709–H718. doi: 10.1152/ajpheart.2000.279.2.H709. [DOI] [PubMed] [Google Scholar]
- Shacklock PS, Wier WG, Balke CW. Local Ca2+ transients (Ca2+ sparks) originate at transverse tubules in rat heart cells. J Physiol. 1995;487(3):601–608. doi: 10.1113/jphysiol.1995.sp020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Colecraft HM. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res. 2013;98(2):177–186. doi: 10.1093/cvr/cvt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeran FL, Pepe S. Mitochondrial bioenergetics and dysfunction in failing heart. Adv Exp Med Biol. 2017;982:65–80. doi: 10.1007/978-3-319-55330-6_4. [DOI] [PubMed] [Google Scholar]
- Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85(6):3666–3686. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261(28):13333–13341. [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73(1):97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorriento D, Gambardella J, Fiordelisi A, Trimarco B, Ciccarelli M, Iaccarino G, et al. Mechanistic role of kinases in the regulation of mitochondrial fitness. Adv Exp Med Biol. 2017;982:521–528. doi: 10.1007/978-3-319-55330-6_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S, Balakrishnan S, Jyoti T, Mohammed AA, Arasan S, Vijayanand C. Force-frequency relation in frog-ventricle is dependent on the direction of sodium/calcium exchange in diastole. Acta Physiol Scand. 2005;185(3):193–202. doi: 10.1111/j.1365-201X.2005.01487.x. [DOI] [PubMed] [Google Scholar]
- Sutko JL, Airey JA, Welch W, Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol Rev. 1997;49(1):53–98. [PubMed] [Google Scholar]
- Takasago T, Imagawa T, Furukawa K, Ogurusu T, Shigekawa M. Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J Biochem. 1991;109(1):163–170. doi: 10.1093/oxfordjournals.jbchem.a123339. [DOI] [PubMed] [Google Scholar]
- Thevis M, Beuck S, Thomas A, Fussholler G, Sigmund G, Schlorer N, et al. Electron ionization mass spectrometry of the ryanodine receptor-based ca(2+)-channel stabilizer S-107 and its implementation into routine doping control. Rapid Commun Mass Spectrom. 2009;23(15):2363–2370. doi: 10.1002/rcm.4161. [DOI] [PubMed] [Google Scholar]
- Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, et al. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271(34):20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- Torrealba N, Aranguiz P, Alonso C, Rothermel BA, Lavandero S. Mitochondria in structural and functional cardiac remodeling. Adv Exp Med Biol. 2017;982:277–306. doi: 10.1007/978-3-319-55330-6_15. [DOI] [PubMed] [Google Scholar]
- Tunwell RE, Wickenden C, Bertrand BM, Shevchenko VI, Walsh MB, Allen PD, et al. The human cardiac muscle ryanodine receptor-calcium release channel: identification, primary structure and topological analysis. Biochem J. 1996;318(2):477–487. doi: 10.1042/bj3180477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci U S A. 2014;111(42):15250–15255. doi: 10.1073/pnas.1412754111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C, Barrow S, Voronina S, Chvanov M, Petersen OH, Tepikin A. Modulation of calcium signalling by mitochondria. Biochim Biophys Acta. 2009;1787(11):1374–1382. doi: 10.1016/j.bbabio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xu Y, Guth K, Kerrick WG, Troponin C. Regulates the rate constant for the dissociation of force-generating myosin cross-bridges in cardiac muscle. J Muscle Res Cell Motil. 1999;20(7):645–653. doi: 10.1023/a:1005559613516. [DOI] [PubMed] [Google Scholar]
- Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410(6828):592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107(4):520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Santulli G, Guo X, Gao M, Chen BX, Marks AR. Imaging atrial arrhythmic intracellular calcium in intact heart. J Mol Cell Cardiol. 2013;64:120–123. doi: 10.1016/j.yjmcc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, et al. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HB, Rogers K, Qi Y, Kanematsu T, Fleischer S. Three amino acid residues determine selective binding of FK506-binding protein 12.6 to the cardiac ryanodine receptor. J Biol Chem. 1999;274(22):15315–15319. doi: 10.1074/jbc.274.22.15315. [DOI] [PubMed] [Google Scholar]
- Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115(3):556–564. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Chen Z, Santulli G, Gu L, Yang ZG, Yuan ZQ, et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci Rep. 2014;4:7425. doi: 10.1038/srep07425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63(3):476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Zhao ZH, Jin CL, Jang JH, Wu YN, Kim SJ, Jin HH, et al. Assessment of myofilament Ca2+ sensitivity underlying cardiac excitation-contraction coupling. J Vis Exp: JoVE. 2016:114. doi: 10.3791/54057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, January CT. Both T- and L-type Ca2+ channels can contribute to excitation-contraction coupling in cardiac Purkinje cells. Biophys J. 1998;74(4):1830–1839. doi: 10.1016/S0006-3495(98)77893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Hua X, Li D, Zhang J, Xia Q. Rapamycin attenuates mouse liver ischemia and reperfusion injury by inhibiting endoplasmic reticulum stress. Transplant Proc. 2015;47(6):1646–1652. doi: 10.1016/j.transproceed.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Archiv: Eur J Physiol. 2014;466(6):1129–1137. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]