

Abstract
Comparative genomics approaches have identified numerous conserved cis-regulatory sequences near genes in plant genomes. Despite the identification of these conserved noncoding sequences (CNSs), our knowledge of their functional importance and selection remains limited. Here, we used a combination of DNA methylome analysis, microarray expression analyses, and functional annotation to study these sequences in the model tree Populus trichocarpa. Methylation in CG contexts and non-CG contexts was lower in CNSs, particularly CNSs in the 5′-upstream regions of genes, compared with other sites in the genome. We observed that CNSs are enriched in genes with transcription and binding functions, and this also associated with syntenic genes and those from whole-genome duplications, suggesting that cis-regulatory sequences play a key role in genome evolution. We detected a significant positive correlation between CNS number and protein interactions, suggesting that CNSs may have roles in the evolution and maintenance of biological networks. The divergence of CNSs indicates that duplication–degeneration–complementation drives the subfunctionalization of a proportion of duplicated genes from whole-genome duplication. Furthermore, population genomics confirmed that most CNSs are under strong purifying selection and only a small subset of CNSs shows evidence of adaptive evolution. These findings provide a foundation for future studies exploring these key genomic features in the maintenance of biological networks, local adaptation, and transcription.
Introduction
Conserved noncoding sequences (CNSs), DNA sequences conserved across species, are under sequence constraint, probably due to their functional importance. One of the major remaining challenges in functional and evolutionary genomics is to determine the functional importance of many components of the genome, in particular CNSs located near protein-coding genes. Some of these CNSs are thought to be important regulatory sequences that are involved in diverse regulatory functions such as transcription initiation and processing of transcripts. (Haudry et al. 2013; Tompa et al. 2005). About 3% of vertebrate CNSs are under purifying selection, and most of these sequences function as cis-elements that regulate gene transcription in transgenic assays (Cooper et al. 2005; Sanges et al. 2006). During evolution, functional sequences are expected to be under selective constraints and diverge more slowly than non-functional sequences (Haudry et al. 2013). Researchers have proposed that CNSs repress the rearrangement of the surrounding genome, but do not drive retention of duplicate genes (Hufton et al. 2009). Therefore, comparative analyses of CNSs, based on phylogenetic footprinting of conserved DNA sequences, have been used to detect functional elements.
Genome-wide comparisons of ten dicot plants yielded large numbers of CNSs (Velde et al. 2016). These genomic DNA motifs (median size 15 bp) occur in different genic regions (5′-upstream, 5′-UTR, intron, 3′-UTR, 3′-downstream) and have different potential functions. Although the CNSs showed significant patterns of conservation, the evolutionary and functional roles of these CNSs remain unclear. DNA cytosine methylation is an epigenetic modification that has essential roles in cellular processes and development, and methylation levels are often low in DNA–protein interaction sites (Lister et al. 2009). Therefore, investigation of methylation patterns of CNSs should provide information on CNSs with potential protein–DNA interactions.
Most highly conserved CNSs are enriched near genes that regulate transcription and development (Bejerano et al. 2004; Danecek et al. 2011), where the precise and complex regulation caused selection against mutations in cis-regulatory regions. The exceptional constraint on these cis-regulatory sequences may influence the evolution of surrounding genomic regions, raising the possibility that the interdigitation of CNSs may play a key role in maintaining conservation of gene order (Hufton et al. 2009). However, many studies in vertebrates have shown that arrays of extragenic CNSs are linked to their target genes and thus evolutionarily resistant to rearrangement (Hufton et al. 2009; Kikuta et al. 2007; Mongin et al. 2009). The role of intragenic CNSs in genome evolution requires further examination.
Previous studies of CNSs generally concluded that “regulatory genes”, also called “Bigfoot genes”, often including genes with transcription factor activity, are CNS-rich (Freeling and Subramaniam, 2009). Since CNSs are typically annotated as DNA-binding motifs, we expected that genes with more CNSs should be more precisely and strictly regulated and positively correlated with the number of potential DNA–protein interactions. For downstream targets, CNSs were also positively correlated with their interaction partners, for most CNSs are functional sequences involved in gene regulation. However, that does not mean that all CNSs are functional, and artifacts are possible.
Polyploidization, a major evolutionary process that is common in vascular plants, triggers vast genetic and genomic imbalances. Numerous studies in diverse plant taxa have documented that polyploidy may induce rapid changes in gene expression and functional divergence between paralogous gene pairs (Buggs, 2013; Xu et al. 2014). The Populus trichocarpa genome underwent a whole-genome duplication (WGD) that is shared across the Salicaceae, containing approximately 8000 similarly aged paralogous gene pairs (Rodgers-Melnick et al. 2012). The resequencing data of 544 P. trichocarpa individuals revealed a high degree of natural variation and adaptive phenotypic variation in natural populations, suggesting that local adaptation is prevalent (Evans et al. 2014). A major unexplored question is whether the fundamental differences of CNSs between retained duplicate pairs from the Salicoid WGD are connected to patterns of adaptation, phenotypic evolution, and natural selection. Utilizing publicly available CNSs, single-base-resolution methylomes, gene expression, and genome resequencing data, we aimed to accomplish the following: (1) identify signatures of natural selection and adaptive evolution of CNSs, (2) delineate the factors associated with different processes of genome evolution including the paralog retention following a WGD event, synteny and CNSs, and (3) test the hypothesis that genes encoding proteins with a high number of interactions need more complex cis-regulation and thus have more CNSs.
Materials and methods
Identification of syntenic genes and retained paralogs
Retained paralogs of Populus were inferred from a previous study (Rodgers-Melnick et al. 2012), and transferred to the gene symbol of version 3.0. Retained paralogs of three other species (Vitis vinifera, Brassica rapa, and Glycine max) and the synteny blocks between Populus and Arabidopsis were downloaded from Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication) (Lee et al. 2013) and were detected based on the MCscan algorithm (Tang et al. 2008). Then the syntenic genes were excluded from the total gene set, and the remaining genes were defined as non-syntenic genes.
Tissue specificity measurement
The expression profile was examined using a whole-genome microarray constructed by Roche NimbleGen (http://www.nimblegen.com/; Rodgers-Melnick et al. 2012). The array was refined based on Nimblegen’s design guidelines to minimize the noise and increase the positive signals. A total of 14 tissues were obtained for microarray hybridizations and for further co-expression clustering analyses. To measure the tissue specificity, the tissue specificity score (Liao and Zhang, 2006) was computed thus: Let aij be the average expression of gene i in tissue j. Then the tissue specificity of gene i is defined by
where n is the number of tissues. If a gene expressed in only one tissue, the score is one; if the expression of a gene is the same in all tissues, the score is zero.
Correlation of CNS frequency and position with gene pair co-expression
The positions of CNSs in ten dicot plants were downloaded from this investigation (Velde et al. 2016). These CNSs were identified in the sequences 2 kb upstream and 1 kb downstream of the gene. First, the expression profile from 14 whole-genome microarray expression datasets (Rodgers-Melnick et al. 2012) was clustered into 40 co-expression modules by applying K-means clustering with Euclidean distance as the distance metric (Saeed et al. 2003). The K-means algorithm partitions genes into defined set of discrete clusters, attempting to maximize the expression similarity of the genes in each cluster. Thus, this method clustered genes that are possibly co-regulated. Next, we randomly selected ten genes from each module and calculated the Pearson correlation coefficient based on the CNS count (across the full gene or sub-gene region) and their expression value. The mean value of the Pearson correlation coefficient was generated by 10,000 bootstrap replicates, and the significance was determined by randomly selection an identical number of gene vectors from the genome background across 10,000 permutation tests.
Detection of deeply conserved CNSs
Orthologs were determined for each Populus and Arabidopsis gene using the PLAZA Integrative Orthology method (ORTHO; ftp://ftp.psb.ugent.be/pub/plaza/plaza_public_02_5/IntegrativeOrthology/integrative_orthology.ORTHO.csv.gz). Pairwise comparisons of CNSs were generated between all Populus or Arabidopsis CNSs and their orthologous genes for all four sequence types (intronic, UTR, upstream, downstream). CNSs with a maximum length of 21 bp were annotated using the fimo software packaged in the MEME suite (Bailey et al. 2009) with the parameters --verbosity 1 --thresh 1.0E-5, and the motif matrices were derived from the PlantRegMap database. The remaining CNSs were compared with VISTA used the minimum percent identity threshold of 50% and window size of 20 bp. Only CNSs with the same annotation in PlantRegMap or above the threshold of VISTA were considered as deeply conserved sequences.
Identifying transcription factor binding sites and enriched motifs
Transcription factor binding sites were predicted by PlantRegMap (Jin et al. 2016). Motif enrichment analyses were performed using AME integrated in the MEME suite (Bailey et al. 2009). For enrichment analysis, randomly selected genomic regions were selected as control background, and only the motifs with P < 0.05 (Fisher’s exact test) were considered as significantly enriched.
Bisulfite-sequencing for methylomes and transcriptome analyses
Bisulfite-seq data of P. trichocarpa under well-watered (WW) and water-deficit stress (WS) treatments are available under the SRA accessions SRR1171318, SRR11713189, SRR1171316, and SRR1171317 (Liang et al. 2014). Because the data were analyzed based on Populus version 2.0 reference genome, we reanalyzed the bisulfite-seq data using the Populus version 3.0 genome. Quality-trimmed reads were aligned to the reference Populus genome (Phytozome version 10) using bowtie2 (v2.2.9) (Langmead and Salzberg 2012) and analyzed using Bismark (v0.16.3) (Krueger and Andrews 2011) with default parameters. Duplicated reads were removed using the Bismark deduplication tool, and cytosine methylation states were extracted using Bismark methylation extractor. CNS regions were divided in four equally sized bins and the CNS flanking regions were divided in six equally sized bins with 100 bp windows from 5′ to 3′, and the mean mC/C level within each bin for each methylation type was determined. Mean methylation level in each bin was determined based on the number of mC/C sites of a given methylation type (mCpG, mCHG, or mCHH) divided by the total mC/C sites in each bin.
Clean reads were mapped using TopHat (Trapnell et al. 2012) to generate read alignments for each sample. Genomic annotations were obtained from Phytozome (http://www.phytozome.net/). The transcript isoform level and gene level counts were calculated and differential transcript levels were then computed using Cuffdiff (Trapnell et al. 2012). The transcriptome data are available under the SRA accessions SRR1171652 and SRR1030352 (Liang et al. 2014).
Functional annotation
Protein sequences (version 3.0) with the longest transcripts were annotated using (CDD, Coils, CATH-Gene3D, HAMAP, Pfam, PIRSF, PRINTS, ProDom, SMART, SuperFamily, TIGRFAMS, PROSITE, PRINTS) in conjunction with InterProScan (Quevillon et al. 2005). Then the total number of GO terms was mapped to the InterPro annotations using the GO terms database (ftp://ftp.ebi.ac.uk/pub/databases/interpro/interpro2go). As the resulting GO categories were very specific, we used the same method as Rodgers-Melnick et al. (2012) by identifying nodes that were descendants of the following GO categories within the Gene Ontology hierarchy: protein binding (GO:0005515), transcription factor activity (GO:0003700), ion channel activity (GO:0005216), nucleic acid binding (GO:0003676), phosphatase activity (GO:0016791), protein kinase activity (GO:0016301), ligase activity (GO:0016874), protease activity (GO:0008233), receptor activity (GO:0004872), transporter activity (GO:0005215), stress response (GO:0006950), catalytic activity (GO:0003824), and cation binding (GO:0043169).
Estimating protein connectivity
The STRING database (Mering et al. 2003) was used to estimate the number of protein interactions for each gene. The STRING database integrates data from experimentally derived protein–protein interactions through literature curation, and stores computationally predicted interactions; therefore, the data of model organisms are supported by more evidence.
Statistical analysis of gene set associations and trends in synteny and paralog retention
Statistical inference of CNS numbers and protein connectivity was conducted with a permutation test on the mean (permutation test) (Röhmel 1996), and each mean is shown with a bootstrap-based 95% CI (Studentized CI), displaying the extent to which outliers may cause the mean to fluctuate. This method has performed well in skewed populations without assumption of the true distributions (Hufton et al. 2009).
The synteny-CNSs and paralog retention-CNSs trends were tested by permutation. The PPI number on the CNSs was randomly shuffled and the mean value was calculated based on bootstrap-based 95% CI. Pearson’s correlation coefficient and significance were calculated using R package.
Signatures of adaptive evolution and association analysis
We used two methods to examine the signatures of adaptive evolution of CNSs. Differences in allele frequencies among four naturally occurring populations (Columbia, Tahoe, WA/BC, and Willamette) of P. trichocarpa were estimated as Fst throughout the genome. Resequencing data were derived from a previous study (Evans et al. 2014). We compared the single-nucleotide polymorphism (SNP) frequency among different genome elements, including conserved miRNAs, Populus-specific miRNAs, and noncoding regions. MiRNAs were further separated into seed regions, mature regions, hairpin regions (pre-miRNA), and the rest of the miRNA excluding the seed regions (Rest). The conserved miRNAs and Populus-specific miRNA dataset were derived from a previous study (Xie et al. 2017). All of them represent essential genomic noncoding elements. Fst of each SNP was calculated using VCFtools (Danecek et al. 2011). We took signals in the top 5% of Fst values as variations representing high allele frequency differences among populations and thus candidates for experiencing divergent selection. The climate-driven genomic regions across the Populus genome that appear to be affected by divergence/positive selection were previously detected by five metrics (Evans et al. 2014): differentiation (Fst), allele frequency cline steepness across mean annual temperature and precipitation measurements (SPA; Yang et al. 2012), extended haplotype homozygosity around alleles from rapid allele frequency increase (iHS; Voight et al. 2006), and allele frequency clines (bayenv; Günther and Coop 2013) with each of the first two climate PC axes (PC1 and PC2). We mapped the CNSs to the selection genomic regions and examined the overlapping CNSs.
Target prediction of miRNAs
Potential targets of miRNAs were predicted using PSRNATARGET (Dai and Zhao 2011) with the maximum expectation score set to 2.0. The Pearson correlation coefficients between miRNAs and their target genes were calculated using R (v.3.2.3, http://mirror.bjtu.edu.cn/cran/) based on their transcript abundances in six tissues (Xie et al. 2017).
Conservation analyses of CNSs between retained paralogs and ancestral state
To test the conservation of CNSs between retained paralogs and the ancestral state, we defined the orthologs in Arabidopsis as the ancestral state. Orthologs were determined for each species and Arabidopsis gene using the PLAZA Integrative Orthology method (ORTHO; ftp://ftp.psb.ugent.be/pub/plaza/plaza_public_02_5/IntegrativeOrthology/integrative_orthology.ORTHO.csv.gz). If the retained paralog has more than one ortholog in Arabidopsis, the best hit is selected. We first compared the CNS composition between the retained paralogs and the ancestral orthologs. Then, the CNSs of the retained paralogs were compared with the ancestral CNSs using the minimum percent identity threshold of 50% and window size of 20 bp for all four sequence types (intronic, UTR, upstream, downstream). Only CNSs of all four sequence types above the threshold of VISTA were considered as similar to the ancestral state. Retained paralogs were classified into two categories: both display nucleotide differences from the ancestral state and one of the retained paralogs is similar to the ancestral state. Note that genes without CNSs were excluded from this analysis.
Statistical analyses of predictors of different genome evolution categories
A logistic regression and multivariable linear regression model was used to determine significant predictors of the three genome evolution patterns, including genes related to CNS, synteny, and paralog retention.
For genes of synteny and paralog retention, logistic regression was used. Each gene model with evidence of significant expression was given an indicator variable of 1/0 for the presence of genes belonging to the three categories, and the three tests were considered as distinct entities. To measure expression specificity, the tissue specificity score was computed (Xie et al. 2017). Gene length was measured in bases between the start and stop codon, including all intronic sequence. Each gene of GO functional categories was given an indicator variable of 0/1 depending on the presence or absence, respectively. Logistic regression was then carried out using the generalized linear model, binomial family, logit link integrated in R package. The significance of each variable was assessed by F-statistic test.
For genes related to CNS, we used multivariable linear regression model to test the significant predictors, for the number of CNS number was significant related to different functional categories. The significance of each variable was assessed by chisquare test.
All variables were added to the model as main effects, and backward selection was used to choose the best of predictor variables. For each run, a total of 15 variables were given as input, and adjusted R2 was calculated.
Results
Functional inference of CNSs
Populus CNSs were typically short (median length of 15 bp) and had a broader GC content than the genic portion of the genome (Table S1; Figure S1). The 157,567 CNSs were located in upstream regions (49.1%), 5′ UTRs (17.4%), introns (8.8%), 3′ UTRs (12.5%), and downstream regions (12.1%) in the P. trichocarpa genome (PHYTOZOME v.10.0; Figure S2). Examination of their distribution revealed that these sequences had a high frequency of motifs located in the 1-kb promoter regions, with some weak peaks scattered throughout the promoters (Figure S3). After excluding all the CNSs that are known cis-regulatory elements in PlantRegMap database with a maximum length of 21 bp, we identified 31,998 CNSs to be significant binding sites of 356 transcription factors (TFs) (P < = 0.001; Table S2). We observed that upstream regions were more enriched for TF binding motifs than other regions, with 32 motifs specific to upstream regions (Tables S3–S7). For example, the WRKY60-binding motif involved in abscisic acid responses and abiotic stress (Han et al. 2010), and the ABI4 binding motif mediating abscisic acid and temperature responses (Tamminen et al. 2001), were enriched in these regions. These numbers indicate thousands of biological network links where a protein recognizes a conserved binding site and influences expression of its target genes.
Furthermore, we detected 22 CNSs overlapping with miRNA hairpins and 6 CNSs overlapping with miRNA binding sites (Tables S8–S9). Examination of the expression of miRNA/target pairs (genes with CNSs that contain miRNA hairpins or miRNA binding sites only in 5′ and 3′ UTR sequences) revealed significantly negative correlations (Pearson’s correlation <−0.79; P < 0.05; Table S10). These results suggest that CNSs may play essential regulatory roles through interaction with miRNAs.
When focusing on gene expression patterns, we noticed that genes with more CNSs had higher expression than those with fewer CNSs (Figure S4a). We also examined the tissue specificity of genes with different number of CNSs, and found that genes with more CNSs tend to have higher tissue specificity (Figure S4b). All these suggest that CNSs are actually associated with gene regulation, and play essential roles in maintaining the function of genes.
To determine whether there was an association between gene pair co-expression and CNS signatures, we first determined co-expression modules across 14 expression datasets (Rodgers-Melnick et al. 2012). Using the K-means clustering method, 40 co-expression modules were identified (Figure S5). Next, we determined the Spearman’s q rank correlation between CNS frequency and each gene pair normalized expression in each co-expression modules (all rho and P values are listed in Table S11). This analysis revealed significant correlations between gene pair co-expression with sub-gene CNS frequency in all expression datasets and all sub-gene positions examined (Fig. 1). We also found inverse correlations of the same gene sets in different tissues. Significant positive correlations were identified in full gene, 5′ upstream, 5′ UTR and 3′ UTR sub-gene positions, in particular upstream and 5′ UTR CNSs, indicating a broad positive effect of upstream signatures on gene pair co-expression. In contrast, significant negative correlations between co-expression and intron/downstream CNSs, in particular intron CNSs, were examined in most datasets. These data suggest that CNS frequency is correlated with gene pair co-expression, but that the putative underlying expression control mechanisms in sub-gene positions (e.g. 5′ upstream/intron) and tissues may be mixed.
Fig. 1.

Correlation between conserved noncoding sequence (CNS) frequency and gene pair co-expression. CNS counts subdivided into sub-genic regions, including 5′ upstream, 5′ UTR, intron, 3′ UTR, 3′ downstream, or the sum of all five regions (full gene). Each row in one sub-gene position indicates the whole-genome microarray dataset of one tissue (Rodgers-Melnick et al. 2012). Each column indicates one co-expression module
We further examined whether CNSs were enriched for TF binding motifs among different co-expression modules. As a result, we were able to discover many TF binding motifs enriched in CNS sequences, displaying high variety among groups (Table S12). We found that numerous TFs binding the identified motifs are co-expressed with the genes linked to the enriched motifs (Table S13), suggesting that the motifs associated with these TFs are likely to be functional in the CNSs. We also found that the tissue specificity is positively correlated with the diversity of motifs (P < 0.01; Figure S4c), suggesting their regulatory roles in gene expression. DNA methylation can alter the interactions between DNA-binding proteins and their targets, further active/repress expression. To further evaluate this relationship, we compared the average level of methylation of CNSs, throughout various genomic features and in plants under water-deficit stress and in well-watered conditions (Liang et al. 2014). For mCG methylation, we observed lower average methylation levels in 5′ and 3′ UTRs, and the highest methylation in introns (Fig. 2a; Figure S6). The mCHG and mCHH methylation levels also decreased toward the 5′ and 3′ UTRs. Intriguingly, the methylation level of CNSs across five categories were significantly lower than their corresponding genomic contexts (Fig. 2a; permutation test; P < 0.001). For example, the methylation level of 5′ UTR CNSs was lower than that of 5′ UTR regions. Scanning the average methylation level at CNS sites and their flanking regions also revealed a depletion of methylation at CNS sites (Fig. 2b–g; Figure S6). We examined the fold change in methylation level of CNSs surrounding the differentially expressed genes (n = 910; P < 0.05) between the two conditions. After drought treatment, we found that the CG and non-CG methylation levels of CNSs in upstream and downstream regions of the down-regulated genes are increased significantly. A reverse trend was observed within the corresponding regions of the up-regulated genes (Figure S6h). Overall, our results suggest that CNSs were enriched in cis-regulatory elements and the methylation data agree with the hypothesis that CNSs are bound by TFs.
Fig. 2.

The global pattern of Populus DNA methylomes under well-watered conditions. a Distribution of CG, CHG, and CHH methylation levels (mC/total C×100%) in each sequence context of gene-related regions, including upstream, UTR, exon, intron, and downstream. Error bars indicate 95% confidence intervals generated by 1000 bootstrap replicates. b–f The average methylation level at CNS sites and their flanking regions in distinct sequence contexts. g The average methylation level of random selected regions. Average DNA methylation levels in each CNS region were split into four bins and their flanking regions were divided into 12 equally sized bins of 100 bp. Purple, green, and red indicate the distribution of CpG, CHG, and CHH methylation levels, respectively. The methylation data from the previous study (Liang et al. 2014) were reanalyzed for this experiment
Allele frequencies of CNSs reflect local adaptation
Many CNSs are important functional TF binding motifs that determine transcriptional activity, and are under selective pressure comparable to that of protein-coding sequences (Haudry et al. 2013). We traced the evolution of the CNSs and 360 previously identified microRNA (miRNA) sequences by intraspecific sequence comparisons with a collection of 544 P. trichocarpa individuals (Evans et al. 2014). Of the 157,567 CNSs, 19% displayed differences in their sequences, and changes modifying the CNSs occurred less frequently than changes in hairpin sequences of Populus-specific miRNAs, the rest of the miRNA excluding the seed regions, or genomic noncoding sequences, suggesting that CNSs are selectively constrained (Fig. 3). Moreover, most CNSs are present in all accessions, with low levels of minor CNS alleles, indicating a potential fitness benefit of the CNSs (Figure S7).
Fig. 3.

Unique and shared CNSs among five selection scans. a The SNP frequency in distinct sequence contexts. The SNP frequency (no. of SNPs per kb) were compared across different noncoding sequences, including conserved miRNAs (seed regions, mature regions, the rest of the miRNA excluding the seed regions (Rest) and hairpins), Populus-specific miRNAs, and intergenic regions. The miRNA data were derived from a previous study (Xie et al. 2017). b Venn diagram of the number of CNSs through the genome in the top 1% for each selection scan using five different metrics: Fst, SPA, iHS, bayenv C1, and bayenv C2. The detected genomic regions were derived from the previous study (Evans et al. 2014). The CNSs show lower SNP frequency than Populus-specific pre-miR sequences and some of them are located in the genome selection regions, suggesting that a substantial fraction of CNSs experience selection pressure
We first examined variation in the frequencies of CNSs to explore the possibility that differences in allele frequencies between populations of P. trichocarpa (Columbia; Tahoe; WA/BC; Willamette) may reflect local adaptation. We mapped seven million variations onto the CNSs, and identified 30,076 biallelic SNPs. Of the total 157,567 CNSs, 1672 SNPs located in 1535 CNSs were identified as those that surpassed the 5% signal (Fst = 0.1179; Table S14). Next, five metrics were used to detect climate-driven genomic regions across the Populus genome that appear to be affected by divergence/positive selection, and each of these metrics provides a distinct view of selection forces shaping these genomic regions (Evans et al. 2014). These metrics were: Fst, SPA, iHS, bayenv C1, bayenv C2. We then mapped CNSs to the “selection outliers” examined by five distinct measures of natural selection. Of the total 157,567 CNSs, we found 6020 CNSs in the top 1% from all measures and 332 detected by at least two of the selection metrics (Table S15). The targets of the 332 CNSs have a variety of annotations, including MYB106, RGA, terpene synthase, and disease resistance genes (Table S16). Most of the selection outlier regions were uniquely detected among selection scan metrics, suggesting that each metric provides a distinct view of selection forces or local adaptation. Other lines of evidence for local adaptation include the examination of significant SNPs for phenotypic associations as determined by the emmax mixed model analysis. We mapped 2987 genome-wide significant trait-associated SNPs to the CNS sequences and detected 16 significant associations (Table S17). These analyses collectively suggest that natural selection drives the rapid evolution of small fraction of CNSs.
The divergence of CNSs supports the duplication–degeneration–complementation model
The duplication–degeneration–complementation (DDC) model predicts that degenerative mutations in regulatory elements may knock out independent subfunctions of duplicated genes, and thus could increase rather than decrease the probability of duplicate gene preservation (Force et al. 1999). Consistent with this, a recent study showed that nearly half of retained paralogs have diverged in expression (Pophaly and Tellier 2015). Under the DDC model, mutations could act upon both retained paralogs, thereby increasing long-term opportunities for the evolution of new gene functions. More than 87% of the retained paralog pairs in four species displayed CNS composition or nucleotide differences from the ancestral state following a pattern consistent with a DDC process, wherein regulatory sequences randomly degenerate, eventually leading to complete subfunctionalization (Table 1). The remaining retained pairs appeared to support the classical model, with one member usually degenerating within a few million years by accumulating deleterious mutations, while the other duplicate retains the original function (Force et al. 1999). Overall, the CNS divergence pattern suggested that DDC may be a major mechanism in paralog retention.
Table 1.
Comparison of the associated genes of CNSs in five species
| Species | WGD paralogsa | Singletonsb | CNS-associated paralogs displaying differencesc | CNS-associated singletons displaying differencesd | P-valuee |
|---|---|---|---|---|---|
| Populus trichocarpa | 15,822 | 25,513 | 11,831 | 12,122 | 1.59E-168 |
| Arabidopsis thaliana | 8502 | 18,914 | 6157 | 8902 | 1.74E-92 |
| Glycine max | 37,048 | 17,133 | 20,930 | 8566 | 4.91E-15 |
| Medicago truncatula | 7338 | 40,561 | 4136 | 16,224 | 4.59E-56 |
| Vitis vinifera | 3684 | 22,662 | 949 | 4532 | 2.42E-10 |
aNumber of WGD (whole-genome duplication) paralogs in the gene set
bNumber of singletons in the gene set
cNumber of CNS-associated WGD paralogs displaying differences in their sequences
dNumber of CNS-associated singletons displaying differences in their sequences
eP-value was derived from Fisher’s exact test
Deeply CNSs are strongly associated with synteny and paralog retention. The selection constraint on CNSs may influence the evolution of their surrounding genomic regions, raising the possibility that the interdigitating of CNSs may play a key role in conservation of gene order. To systematically study the association of CNSs with genomic synteny and paralog retention, we used deeply CNSs, those conserved in at least six dicot plant species, for downstream analysis (Table S18). We observed that gene families that are syntenic and show paralog retention have more CNSs than the genome average and randomly selected genes (Fig. 4a). In contrast, non-syntenic genes have fewer CNSs than the genome average. Notably, genes with paralog retention and synteny show significantly more associated CNSs than can be explained by gene duplication or synteny alone (permutation test; P < 0.01). A simple explanation for this observation is that it is the presence of duplicates increases the association between synteny and CNSs, suggesting that conserved cis-regulatory sequences could preserve gene order and paralog retention. Supporting this, we observed that CNSs are strongly associated with synteny and paralog retention (Fig. 4b, c; P < 0.05; Pearson’s correlation test). Conversely, CNSs are significantly negatively associated with conservation of non-syntenic genes (Fig. 4d; P < 0.05; Pearson’s correlation test). Additional CNSs increase the probability of paralog retention and synteny, such that when genes have 19 CNSs, more than 78% of Populus and 80% of Arabidopsis genes have evidence of synteny. Similarly, when genes have 19 CNSs, more than 58% of Populus and 45% of Arabidopsis paralogs are retained.
Fig. 4.

Retained paralogs and syntenic genes have more CNSs. a The mean number of CNSs linked to different syntenic, non-syntenic, and whole-genome duplication gene sets. In Populus: all (n = 41,335), synteny (n = 11,867), non-syntenic (n = 29,468), paralog retention (n = 18,164), paralog retention and synteny (n = 7804); and in Arabidopsis: all (n = 27,206), synteny (n = 9694), non-syntenic (n = 17,512), paralog retention (n = 8504), paralog retention and synteny (n = 4067) samples were used for the experiment. b The association between CNSs and synteny in Populus and Arabidopsis for CNSs. Each data point is calculated from at least 50 genes. c The association between CNSs and paralog retention in Populus and Arabidopsis for CNSs. d The association between CNSs and non-syntenic genes in Populus and Arabidopsis for CNSs. Each data point is calculated from at least 50 genes. Error bars, bootstrap-based 95% confidence intervals on the mean estimates. Both of two trends b, c represent a significant positive correlation between CNSs number and synteny/paralog retention, and d represent a significant negative correlation between CNSs number and non-syntenic genes conservation. where R is the Pearson correlation coefficient, and P is the upper tail probability that the true correlation is greater than zero
Our results indicate that genes with synteny and paralog retention are positively associated with increased frequency of CNSs, while non-syntenic genes are negatively associated with increased frequency of CNSs. However, the functions of genes associated with these genomic processes often overlap (Hufton et al. 2009). Thus, the association between intragenic CNSs and genome evolution processes needs rigorous examination. We used a multivariable logistic regression model to identify significant predictors of the three genome evolution processes (see Materials and methods). Candidate variables included 13 gene ontology (GO) functional categories, expression specificity, and the genomic length of the gene. The nine variables we tested were significant predictors of paralog retention (P ⩽ 0.05), seven were significant predictors of CNS-linked genes (P ⩽ 0.03), and ten were significant predictors of syntenic genes (Table 2). Notably, 11 common predictors were positively associated with the odds of the three evolutionary patterns and two categories showed contrasting effects in either of three processes (Fig. 5). As expected, nine significant predictors were negatively associated with the odds of non-syntenic genes, such as transcription factor and protein binding functions, suggesting that non-syntenic genes are less likely to be associated with these gene functions. All these results suggest that CNSs may play roles in genome evolution, possibly by influencing the integrity of genes with transcription and binding functions.
Table 2.
Significant predictors for retention of WGD, syntenic, non-syntenic, and CNS-associated genes
| WGD | CNS | Synteny | Non-syntenic | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimatea | SEb | Pr− > |z|c | Estimate | SE | Pr− > |z| | Estimate | SE | Pr− > |z| | Estimate | SE | Pr− > |z| | |
| -Intercept | −0.75 | 0.04 | 0 | 0.63 | 0.05 | 0.63 | −0.2 | 0.04 | 0 | −1.46 | 0.07 | 0 |
| ExpSpecific | 1.3 | 0.06 | 0 | 1.82 | 0.08 | 0 | 1.47 | 0.07 | 0 | −0.2 | 0.01 | 0 |
| Length | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | −1.86 | 0.72 | 0 |
| Phosphatase activity | 0.72 | 0.31 | 0.02 | N/A | N/A | N/A | 1.83 | 0.73 | 0.01 | 0 | N/A | N/A |
| Transporter activity | N/A | N/A | N/A | 0.48 | 0.18 | 0.01 | −0.49 | 0.14 | 0 | 0 | N/A | N/A |
| Ion channel activity | N/A | N/A | N/A | N/A | N/A | N/A | −1.07 | 0.37 | 0 | 0 | N/A | N/A |
| Protein kinase activity | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 0 | N/A | N/A |
| Catalytic activity | N/A | N/A | N/A | N/A | N/A | N/A | −0.29 | 0.08 | 0 | 0.24 | 0.25 | 0.01 |
| Stress response | 0.46 | 0.24 | 0.05 | 0.68 | 0.32 | 0.03 | N/A | N/A | N/A | 0 | N/A | N/A |
| Receptor activity | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | −0.55 | 0.11 | 0 |
| Nucleic acid binding | 0.17 | 0.08 | 0.03 | N/A | N/A | N/A | 0.53 | 0.11 | 0 | 0 | N/A | N/A |
| Protease activity | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | −10.69 | 74.51 | 0.01 |
| Ligase activity | 1.01 | 0.57 | 0.08 | N/A | N/A | N/A | N/A | N/A | N/A | −1.53 | 0.6 | 0.01 |
| Cation binding | 0.65 | 0.29 | 0.02 | 0.94 | 0.38 | 0.01 | 1.8 | 0.6 | 0 | −1.7 | 0.18 | 0 |
| Transcription factor | 1.12 | 0.09 | 0 | 0.49 | 0.13 | 0 | 1.69 | 0.18 | 0 | −0.11 | 0.05 | 0.02 |
| Protein binding | 0.17 | 0.03 | 0 | 0.17 | 0.05 | 0 | 0.09 | 0.05 | 0.04 | −1.46 | 0.07 | 0 |
Logistic regression was used to identify significant predictors of occurrence of different gene categories, including genes related to CNS, synteny, and paralog retention. Candidate variables included gene ontology (GO) functional categories, gene length, and the tissue specificity score (ExpSpecific). Gene length was measured in bases between the start and stop codon, including all intron sequence
aEstimate indicates the contribution of individual predictors, representing the change in the logit for each unit change in the predictor
bSE indicates the standard error of regression coefficients estimation
cThe significance of each variable
Fig. 5.
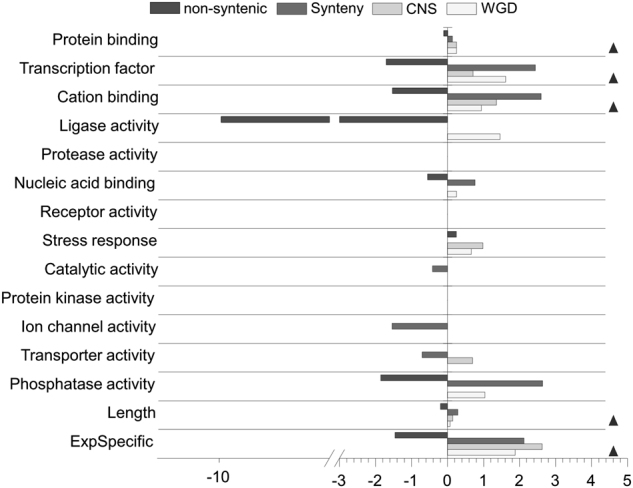
The exponentiated logistic regression coefficients for categories significant for syntenic, non-syntenic, CNS-associated genes, and WGD. Black triangles indicate categories that were associated with similar trends of the three genome evolution categories, including WGD, CNS, and Synteny. ExpSpecific indicate the tissue specificity
Protein interaction was highly associated with numbers of CNSs
In this study, we proposed that genes encoding proteins with a high number of interactions may need more complex cis-regulation and, therefore, more CNSs, compared with genes encoding proteins with fewer interactions. To test this, we first estimated the number of protein–protein interactions (PPI) for all the genes and compared the number of interactions for genes in different evolutionary categories (Fig. 6). Retained paralogs had significantly more PPI than the genome mean and randomly selected genes. Indeed, genes whose proteins have many potential interaction partners, such as transcriptional regulators and signaling complexes, are more likely to be retained after a WGD event (Rodgers-Melnick et al. 2012). Genes with synteny also showed protein connectivity values similar to the genomic mean, and syntenic genes with CNS had more connections than the genomic mean (Fig. 6). Genes with CNSs were significantly over-connected in both species. The PPI data of Arabidopsis may better represent the true associations due to the fact that Arabidopsis genome has a more thorough functional annotation and more evidence for interactions than Populus, with 30% more interactions attributed to genes. The high connections of paralog retention-CNS and synteny-CNS gene sets are due to the fact that these gene sets have overlap in the gene functions of transcription and protein binding (Table 2). Consistent with this, genes encoding proteins with specific functions as transcription factors, and in macromolecule complexes, which are known to have more connections than genes with other functions, are highly overrepresented in the synteny-CNS genes (Table S19). Next, we examined the correlation of CNS number and PPI. We observed a significant positive correlation (P < 0.05; Fig. 6) between the connections and CNS numbers in both species. Overall, this analysis revealed that genes that have more protein interactions need precise and complex regulation, and thus associate with more CNSs.
Fig. 6.

Genes of different genome evolution patterns have different PPIs. a The mean number of CNSs linked to different synteny and duplication related gene sets. The ‘*’ above each column indicates P < 0.001. b The association between CNSs and protein–protein interactions in Populus and Arabidopsis for CNSs. The trend represents a significant positive correlation between CNS number and interactions, where R is the Pearson correlation coefficient, and P is the upper tail probability that the true correlation is greater than zero. Error bars, bootstrap-based 95% confidence intervals on the mean estimates. Statistical inference is conducted with a permutation test on the mean (perm.test in the R package exact RankTests)
Discussion
Examination of plant CNSs suggests their regulatory function predominantly acts at the DNA level
Alignment-based approaches discovered thousands of CNSs that are conserved in multiple plants and animals (Haudry et al. 2013; Kikuta et al. 2007; Van et al. 2014). One important aspect is to associate CNSs with function. A specific category of CNSs represents sequences that function by binding or blocking some molecule (Subramaniam and Freeling, 2012). Based on their proximity to protein-coding genes, mammalian lineage-specific CNSs were suggested to be important for the regulation of their surrounding genes (Babarinde and Saitou, 2013; Takahashi and Saitou, 2012). Computational and experimental analyses have shown the involvement of certain CNSs in the regulation of the surrounding genes (Bhatia et al. 2014; Sumiyama et al. 2002). To date, numerous studies have concluded that CNSs are regulatory sequences. As expected, after excluding all the CNSs that are known cis-regulatory elements in PlantRegMap with a maximum length of 21 bp, we identified 31,998 CNSs to be significant binding sites of 356 TFs. Several motifs with unknown or known function were strongly enriched in upstream regions, but not elsewhere, hinting at a role in transcription initiation. We further focused on the potential CNS control of gene expression profile with a brief examination of 14 expression datasets based on permutation tests. Despite the relatively large amount of “noise” conferred by the transcriptomes in our examination, we detected significant positive/negative correlations between CNS frequency and gene expression in forty modules. These results suggest a high association between CNSs and the expression of genes. Moreover, all the CNS sites at different genomic features displayed a lower methylation level in CG and non-CG contexts. The depletion of DNA methylation at CNS sites in each treatment or cell type indicates the maintenance of these CNSs in a lower methylated level or unmethylated state, potentially preventing interference in the process of protein–DNA interaction at these sites. Our analysis reveal CNS regulatory complexity, in that CNSs may be involved in multiple mechanisms of transcription control based on their sub-gene position, methylated state, as well as tissue-specific control, possibly mixed.
We also detected 22 CNSs overlapping with miRNA hairpins and 6 CNSs overlapping with miRNA binding sites. Negative correlations have been detected between miRNA and genes with CNS that contain miRNA hairpins or miRNA binding sites. All these results suggest that a considerable amount of CNS function as binding sites acts at the DNA level, through which TFs control the spatio-temporal expression of target genes. Supporting this, many detected CNSs function as regulatory elements and drive expression, often in tissue-specific patterns (Hufton et al. 2009). Also, significant correlations between CNS counts and α-pair co-expression were detected in Arabidopsis thaliana (Spangler et al. 2011), suggesting that CNSs may participate in the maintenance of gene regulatory patterns.
Local adaptation and adaptive trait associations of CNSs
In multicellular organisms, in addition to the integrity of protein-coding sequences, phenotypic diversity and evolution are accompanied and perhaps facilitated, by dramatic increases in or maintenance of the complexity of gene regulatory elements, which lie at considerable distance from the protein-coding sequences. Allelic variants interrupt the function of these CNSs and have functionally significant consequences for phenotypic diversity, physiology, and evolutionary divergence within and between species. Though some fraction of of CNSs were shown to be under adaptive selection in previous studies (Baxter et al. 2012; Velde et al. 2016), they have not established a clear functional effect of alleles predicted to be under selection. We provide comprehensive analyses of the adaptive evolution and trait-associated sites of CNSs. Our analysis only detected 19% of CNSs as displaying differences in their sequences, suggesting that these sites may often experience strong selective constraints, for genetic variations at these sites can disrupt their function. In agreement, previous studies provided evidence for strong purifying selection acting on CNSs (Drake et al. 2005; Haudry et al. 2013). Because mutation rate differences do not affect frequency and deleterious mutations rarely rise to detectable frequency (Fay et al. 2001), frequency spectra may better reflect whether CNSs are functionally relevant. In agreement, our results show that evolutionary pressure has suppressed CNS minor allele frequencies.
Although the markedly lower mutation rate, reflected in SNP density, proves that selection forces shaping CNSs acted on the Populus genome, it does not demonstrate that CNSs are mutation cold spots. Conservation could be explained if new mutations arising in CNSs are often deleterious, while some are more likely to become fixed in the population, with the notion that new, advantageous mutations spread rapidly to fixation. Supporting this, detection of Fst outliers revealed that many allelic variations present in CNSs showed strong population differentiation that may interrupt the predicted regulatory functions, which is suggestive of directional selection. We then found strong signals of climate-driven, divergent/positive selection on genome regions among populations of P. trichocarpa detected by Evans et al. (2014). We found 332 CNSs that were detected by at least two of the selection scan metrics. Such CNSs provide excellent noncoding candidates for functional analyses aimed at elucidating the drivers of local adaptation in Populus, with the consideration that mutations of CNSs appear to have plastic effects on gene function (Mayo et al. 2006). Regulatory mutations must be one of the primary factors that contribute to phenotypic differences, compared with the divergence in gene sequence, as a perfectly good gene product can be useless if synthesized under the wrong conditions (Wray, 2007). Supporting this, association analysis identified 16 SNPs in CNSs that have produced adaptive climate-driven modifications of traits (bud set, height, bud flush). Also, experimental evidence showed that point mutations in cis-regulatory regions make qualitative contributions to interesting and ecologically significant differences in morphology, physiology, and behavior (Wray, 2007). These results indicate that naturally occurring mutations could disrupt or enhance protein–DNA interactions, and thus have the potential to alter the regulatory function of CNSs with consequent modulation of TF–target interactions.
CNSs conserve biological networks
Genetic regulatory networks consist of a set of complex cis-regulatory-binding sites interacting with a finite number of TFs, and give a global overview of how transcriptional control is steered in the cell through TF activity. They are typically organized in nested, multi-layered, interacting hierarchical regulatory modules and function-specific building blocks (Longabaugh et al. 2005). Some biological functions require a large number of coordinated genes that act alone or that have multiple sets of interaction neighbors, appearing as orphan co-expression modules. However, TFs do not have a consistent group of interacting partners, which largely depend on biological conditions or cell context (Stuart et al. 2003). Therefore, the expression of TFs or targets with more partners will be more rigorously regulated compared with factors with fewer interacting partners, for complex regulation is achieved through cooperative binding of different TFs, adding an extra combinatorial level of regulation. Supporting this, we observed a significant positive correlation between CNSs and PPI, suggesting genes encoding proteins with many interactions have more complex cis-regulation and more CNSs, allowing them to be regulated and interact with their neighbors precisely.
Though enrichment analyses have been done of CNSs-associated genes in previous studies (Baxter et al. 2012; Spangler et al. 2011), there are no uniform comparisons among gene sets of distinct genome evolution categories, such as WGD genes and non-syntenic genes. In our study, five significant functional categories were identified to be the common predictors of synteny, CNS targets, and paralog retention. Observation of common factors of paralog retention and CNS targets is not surprising considering that genes with many interaction partners experience paralog retention. In fact, empirical data in yeast and Arabidopsis demonstrate that genes involved in signal transduction and transcription, which are very often “bigfoot” genes, are more likely to undergo paralog retention (Maere et al. 2005; Rodgers-Melnick et al. 2012). For syntenic genes, “transcription factors” and “protein binding” categories are highly enriched, leading to a significant increase of PPI of the synteny-CNS overlapping gene set. Our results indicated strong associations between noncoding sequences and synteny and paralog retention, possibly by influencing the integrity of genes with transcription and binding functions.
Because of the large ratio of such hub genes in genomes, the biological pathways could self-organize to construct biological networks, in which nodes represents TFs or “protein binding” genes. The participation of these hub genes becomes one of the driving forces for the emergence of the network scale-free topology; thus CNSs near such genes contribute to the stability of the biological networks, enable the network to adapt quickly to environmental changes, and mitigate gene expression fluctuations. In a network, the precise expression of hub genes is important because their transcript abundances are highly coupled with the expression of their target genes, implying that CNSs experience strong selection forces. This study provides new insight for understanding the underlying evolution and function of plant species that have natural populations spanning large geographical regions, such as Populus. Conserved noncoding elements are also essential targets of natural selection and genome-wide association studies will provide further information about such genetic markers that affect import ecological traits for local adaptation.
Electronic supplementary material
Acknowledgements
We thank Prof. Scott Allen Jackson from University of Georgia, USA for specific suggestions and detailed comments to improve the manuscript. This work was supported by the Project of the State “13.5” Key Research Program of China (No. 2016YFD0600102) and the Project of the National Natural Science Foundation of China (No. 31600537).
Authors contributions
JX collected and analyzed the data, and wrote the manuscript; DZ designed the experiment; JS provided valuable suggestions to the manuscript; KQ, XL, DC, and DZ revised the manuscript; and DZ obtained funding and is responsible for this article. All authors read and approved the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0055-4) contains supplementary material, which is available to authorized users.
References
- Babarinde IA, Saitou N. Heterogeneous tempo and mode of conserved noncoding sequence evolution among four mammalian orders. Genome Biol Evol. 2013;5:2330–2343. doi: 10.1093/gbe/evt177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter L, Jironkin A, Hickman R, Moore J, Barrington C, Krusche P, et al. Conserved noncoding sequences highlight shared components of regulatory networks in dicotyledonous plants. Plant Cell. 2012;24:3949–3965. doi: 10.1105/tpc.112.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Bhatia S, Monahan J, Ravi V, Gautier P, Murdoch E, Brenner S, et al. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev Biol. 2014;387:214. doi: 10.1016/j.ydbio.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell. 2004;16:1679. doi: 10.1105/tpc.021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs RJ. Unravelling gene expression of complex crop genomes. Heredity. 2013;110:97–98. doi: 10.1038/hdy.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero-Paulet L, Fares MA. Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol Biol Evol. 2012;29:3541–3551. doi: 10.1093/molbev/mss162. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, Depristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JA, Bird C, Nemesh J, Thomas DJ, Newtoncheh C, Reymond A, et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2005;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- Evans LM, Slavov GT, Rodgers-Melnick E, Martin J, Ranjan P, Muchero W, et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat Genet. 2014;46:1089–1096. doi: 10.1038/ng.3075. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu C. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Subramaniam S. Conserved noncoding sequences (CNSs) in higher plants. Curr Opin Plant Biol. 2009;12:126–132. doi: 10.1016/j.pbi.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Günther T, Coop G. Robust identification of local adaptation from allele frequencies. Genetics. 2013;195:205–220. doi: 10.1534/genetics.113.152462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics. 2004;20:3643–3646. doi: 10.1093/bioinformatics/bth397. [DOI] [PubMed] [Google Scholar]
- Han C, Lai Z, Shi J, Yong X, Chen Z, Xu X. Roles of arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010;10:281. doi: 10.1186/1471-2229-10-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudry A, Platts AE, Vello E, Hoen DR, Leclercq M, Williamson RJ, et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet. 2013;45:891–898. doi: 10.1038/ng.2684. [DOI] [PubMed] [Google Scholar]
- Hufton AL, Mathia S, Braun H, Georgi U, Lehrach H, Vingron M, et al. Deeply conserved chordate noncoding sequences preserve genome synteny but do not drive gene duplicate retention. Genome Res. 2009;19:2036–2051. doi: 10.1101/gr.093237.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, He K, Tang X, Li Z, Lv L, Zhao Y, et al. An Arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol Biol Evol. 2015;32:1767–1773. doi: 10.1093/molbev/msv058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016;45:D1040–D11045. doi: 10.1093/nar/gkw982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engström PG, Fredman D, et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007;17:545–555. doi: 10.1101/gr.6086307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Tang H, Wang X, Paterson AH. PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 2013;41:1152–1158. doi: 10.1093/nar/gks1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Zhang Z, Wu H, Huang C, Shuai P, Ye CY, et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014;15:1–11. doi: 10.1186/1471-2156-15-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Zhang J. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol Biol Evol. 2006;23:530–540. doi: 10.1093/molbev/msj054. [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longabaugh WJR, Davidson EH, Bolouri H. Computational representation of developmental genetic regulatory networks. Dev Biol. 2005;283:1–16. doi: 10.1016/j.ydbio.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Maere S, Bodt SD, Raes J, Casneuf T, Montagu MV, Kuiper M, et al. Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA. 2005;102:5454–5459. doi: 10.1073/pnas.0501102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo AE, Setty Y, Shavit S, Zaslaver A, Alon U. Plasticity of the cis-regulatory input function of a gene. PloS Biol. 2006;4:e45. doi: 10.1371/journal.pbio.0040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mering CV, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin E, Dewar K, Blanchette M. Long-range regulation is a major driving force in maintaining genome integrity. BMC Evol Biol. 2009;9:1–16. doi: 10.1186/1471-2148-9-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pophaly SD, Tellier A. Population level purifying selection and gene expression shape subgenome evolution in maize. Mol Biol Evol. 2015;32:3226–3235. doi: 10.1093/molbev/msv191. [DOI] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:116–120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhmel J. Precision intervals for estimates of the difference in success rates for binary random variables based on the permutation principle. Biom J. 1996;38:977–993. doi: 10.1002/bimj.4710380810. [DOI] [Google Scholar]
- Rodgers-Melnick E, Mane SP, Dharmawardhana P, Slavov GT, Crasta OR, Strauss SH, et al. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 2012;22:95–105. doi: 10.1101/gr.125146.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Sanges R, Kalmar E, Claudiani P, D’Amato M, Muller F, Stupka E. Shuffling of cis-regulatory elements is a pervasive feature of the vertebrate lineage. Genome Biol. 2006;7:1–19. doi: 10.1186/gb-2006-7-7-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serluca FC, Sidow A, Mably JD, Fishman MC. Partitioning of tissue expression accompanies multiple duplications of the Na+/K+ATPase alpha subunit gene. Genome Res. 2001;11:1625. doi: 10.1101/gr.192001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler JB, Subramaniam S, Freeling M, Feltus FA. Evidence of function for conserved noncoding sequences in Arabidopsis thaliana. New Phytol. 2011;193:241–252. doi: 10.1111/j.1469-8137.2011.03916.x. [DOI] [PubMed] [Google Scholar]
- Stadler PF, Hofacker IL, Lorenz R, Bernhart SH, Tafer H, Siederdissen CHNZ, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Freeling M (2012) Conserved noncoding sequences in plant genomes (Springer Vienna) pp 113–122
- Sumiyama K, Irvine SQ, Stock DW, Weiss KM, Kawasaki K, Shimizu N, et al. Genomic structure and functional control of the Dlx3-7 Bigene cluster. Proc Natl Acad Sci USA. 2002;99:780. doi: 10.1073/pnas.012584999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Saitou N. Identification and characterization of lineage-specific highly conserved noncoding sequences in mammalian genomes. Genome Biol Evol. 2012;4:641. doi: 10.1093/gbe/evs035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminen I, Mäkelä P, Heino P, Palva ET. Ectopic expression of ABI3 gene enhances freezing tolerance in response to abscisic acid and low temperature in Arabidopsis thaliana. Plant J. 2001;25:1–8. doi: 10.1046/j.1365-313x.2001.00927.x. [DOI] [PubMed] [Google Scholar]
- Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18:1944. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M, Nan L, Bailey TL, Church GM, Moor BD, Eskin E, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van dVJ, Heyndrickx KS, Vandepoele K, Vandepoele K. Inference of transcriptional networks in Arabidopsis through conserved noncoding sequence analysis. Plant Cell. 2014;26:2729–2745. doi: 10.1105/tpc.114.127001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velde JVD, Bel MV, Eechoutte DV, Vandepoele K. A collection of conserved non-coding sequences to study gene regulation in flowering plants. Plant Physiol. 2016;171:2586–2598. doi: 10.1104/pp.16.00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PloS Biol. 2006;4:e147. doi: 10.1371/journal.pbio.0040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Xie J, Yang X, Song Y, Du Q, Li Y, Chen J, et al. Adaptive evolution and functional innovation of Populus‐specific recently evolved microRNAs. N Phytol. 2017;213:206–219. doi: 10.1111/nph.14046. [DOI] [PubMed] [Google Scholar]
- Xu C, Bai Y, Lin X, Zhao N, Hu L, Gong Z, et al. Genome-wide disruption of gene expression in allopolyploids but not hybrids of rice subspecies. Mol Biol Evol. 2014;31:1066–1076. doi: 10.1093/molbev/msu085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WY, Novembre J, Eskin E, Halperin E. A model-based approach for analysis of spatial structure in genetic data. Nat Genet. 2012;44:725–731. doi: 10.1038/ng.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.