

OVERVIEW
An understanding of the genetic causes and molecular pathways of hereditary cancer syndromes has historically informed our knowledge and treatment of all types of cancers. For this review, we focus on three rare syndromes and their associated genetic mutations including BAP1, TP53, and SDHx (SDHA, SDHB, SDHC, SDHD, SDHAF2). BAP1 encodes an enzyme that catalyzes the removal of ubiquitin from protein substrates, and germline mutations of BAP1 cause a novel cancer syndrome characterized by high incidence of benign atypical melanocytic tumors, uveal melanomas, cutaneous melanomas, malignant mesotheliomas, and potentially other cancers. TP53 mutations cause Li-Fraumeni syndrome (LFS), a highly penetrant cancer syndrome associated with multiple tumors including but not limited to sarcomas, breast cancers, brain tumors, and adrenocortical carcinomas. Genomic modifiers for tumor risk and genotype-phenotype correlations in LFS are beginning to be identified. SDH is a mitochondrial enzyme complex involved in the tricarboxylic acid (TCA) cycle, and germline SDHx mutations lead to increased succinate with subsequent paragangliomas, pheochromocytomas, renal cell carcinomas (RCCs), gastrointestinal stromal tumors (GISTs), and other rarer cancers. In all of these syndromes, the molecular pathways have informed our understanding of tumor risk and successful early tumor surveillance and screening programs.
Hereditary cancer syndromes may account for up to 5% to 10% of new-onset adult cancers.1 In children, the hereditary cancer syndromes may explain at least 29% of cancer diagnoses.2–3 Historically, these syndromes have informed our knowledge and treatment of patients with both familial cancer risk and spontaneous, or de novo, cancer. Herein we describe the molecular pathways and recent advances in three rare hereditary cancer syndromes affecting both children and adults. The clinical translation of this information to effective screening and prevention programs is also discussed.
BAP1 CANCER SYNDROME: PREDISPOSITION TO MESOTHELIOMA AND MELANOCYTIC TUMORS
Recently, there has been an upsurge of interest in BAP1 (BRCA1-associated protein-1), an ubiquitin carboxyterminal hydrolase (UCH) first described by Rauscher and colleagues.4 The BAP1 gene was found to be homozygously deleted in two lung carcinoma cell lines; this and other biochemical studies provided the first evidence implicating BAP1 as a tumor suppressor gene.4 Nearly 15 years later, Harbor and colleagues reported a high frequency of BAP1 mutations in metastasizing uveal melanoma (UM), including one that was germline in origin.5 Subsequently, two groups simultaneously reported germline mutations in four different high-risk cancer families, suggesting a BAP1-related cancer susceptibility syndrome.6–7 In one report, germline inactivating mutations of BAP1 were discovered in two families with high incidence of malignant mesothelioma (MM), UM, and other cancers. In the second report, two families were described in which germline mutations in BAP1 predisposed to multiple melanocytic tumors ranging from epithelioid nevi to atypical melanocytic tumors, with some mutation carriers developing UM or cutaneous melanoma (CM).7 Below, we focus on the involvement of BAP1 mutations in a new cancer predisposition disorder, now known as the BAP1 cancer syndrome. Notably, however, somatic BAP1 mutations have also been reported in various sporadic tumors including MM,6,8 RCC,9 and a rare, distinct subset of melanocytic tumors known as atypical Spitz tumors (ASTs).10
Mesotheliomas are aggressive tumors causally linked to exposure to asbestos or related carcinogenic fibers such as erionite. Although only a small percentage of asbestos-exposed individuals develop MM, clustering of MM occurs in some families,6 consistent with the idea that genetic factors play a role in its development. Historically, the understanding of MM pathogenesis was understood in the context of somatic genetic losses within chromosome arms 3p, 9p, and 22q, the latter two specifically affecting CDKN2A and NF2, suggesting a multistep cascade involving the inactivation of multiple tumor suppressors. The field was revolutionized recently by the discovery of the elusive 3p MM gene and the finding of multiple MMs in the context of a hereditary tumor syndrome. Bott and colleagues reported inactivating mutations in BAP1, a tumor suppressor gene located at 3p21.1, in 22% of sporadic MMs.8 Independently, Testa and colleagues similarly discovered somatic mutations of BAP1 in 22% of sporadic MMs.6 Moreover, heterozygous germline mutations of BAP1 were identified in two high-risk cancer families in which the predominant malignancy was MM; notably, one family also had two cases of UM,6 a tumor type previously shown to be associated with somatic BAP1 mutations.5 In some cases, tumor tissue was also available, which showed biallelic inactivation of BAP1. From the perspective of environmental exposure, it is noteworthy that members of neither of these MM families had been exposed occupationally to asbestos. Moreover, only traces of asbestos were found in their homes, raising the question of whether a genetic factor alone is sufficient for MM development in these families. In one family, a germline splice acceptor site mutation in BAP1 was identified in all six family members who developed MM as well as in several others who developed variable carcinomas, including one RCC. The mutation resulted in an aberrant splice product and a frameshift predicted to lead to a premature stop codon. In the second family, the germline BAP1 mutation (a C/G to T/A transition in exon 16 creating a stop codon and premature truncation of the BAP1 protein) was associated with development of various cancers, including seven MMs and two UMs (one occurring in an individual who later developed MM). Intriguingly, germline BAP1 mutations were also found in 2 of 26 sporadic MMs tested, and both patients with mutant BAP1 were previously diagnosed with UM.
Concurrent work by Wiesner and colleagues revealed inactivating germline BAP1 mutations in two families with multiple benign melanocytic tumors7; some affected individuals developed UM or CM,7 and one family member was subsequently diagnosed with MM.10 The existence of a BAP1-related melanocytic disorder was confirmed by a report of germline BAP1 inactivation in families with metastatic UM or with both UM and CM; and some carriers also had atypical melanocytic tumors.11 Another group reported a germline BAP1 mutation in a family with a wide variety of cancers, including three MMs, three UMs, and three CMs.12 More recently, a germline BAP1 mutation was identified in a family in which MM was found in four members, none with a history of asbestos exposure; one member also had multiple melanocytic tumors.10 Another recent study uncovered three cases of UM in a family with no aggregation of other cancer diagnoses.13 The full tumor spectrum associated with germline BAP1 mutations has yet to be established, as suggested by recent report of a germline BAP1 splice mutation and truncating frameshift in a family with UM, CM, suspected MM, as well as paraganglioma.14 Somatic loss of the wild-type BAP1 allele was documented in the paraganglioma, potentially extending the cancer predisposition spectrum.
Ocular tumors known as UMs arise from melanocytes residing within the uvea. Familial UM is rare, comprising fewer than 1% of all UM patients.15 Unlike MM, the question of whether a specific environmental carcinogen plays an etiologic role is unresolved. In contrast to CM, epidemiologic evidence regarding a potential association between sunlight/UV exposure and UM is considered weak and contradictory.15 UM is characterized by a strong proclivity for lethal metastasis to the liver.5 UMs are separated into class 1 tumors, which have low metastatic potential, and class 2 tumors, which exhibit high metastatic risk. Harbour and colleagues used exome capture coupled with next-generation sequencing to search for metastasis-related mutations in highly metastatic UMs.5 Inactivating somatic mutations of BAP1 were identified in 26/31 (84%) metastasizing tumors, including one that was germline in origin.
Collectively, these findings suggest a BAP1 cancer syndrome in which affected families are predisposed to MM, UM, atypical melanocytic tumors, CM, and possibly other cancers. The germline mutations observed in these families are spread throughout the BAP1 coding region, spanning all of the known functional and protein-protein interaction domains (Fig. 1). All mutations result in predicted loss of the nuclear localization signal, thereby abolishing BAP1 function in the nucleus. Biallelic inactivation of BAP1 has been documented in multiple tumors from these high-risk families, strongly suggesting that BAP1 acts as a classical tumor suppressor gene.
FIG 1. Distribution of BAP1 germline mutations in families with BAP1 cancer syndrome.5–7,10–14 Figure depicts BAP1 protein and various domains, with symbols indicating amino acids corresponding to frameshift, nonsense, and splice site mutation sites.
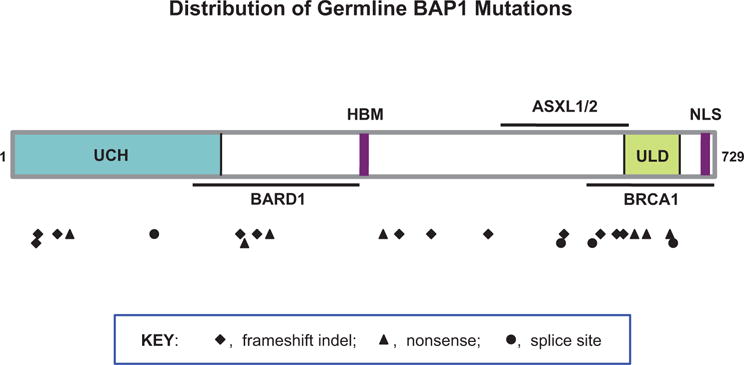
Abbreviations: ASXL1/2, additional sex combs-like proteins 1 and 2 interaction domain: BARD1, BRCA1-associated RING finger domain protein 1 interaction motif; BRCA1, BRCA1 interaction domain; HBM, HCF1-binding motif; NLS, nuclear localization signal: UCH, ubiquitin C-terminal hydrolase: ULD, UCH37-like domain.
BAP1 Function
BAP1 is one of four UCH members involved in the rescue of poly-ubiquitinated substrates from proteosomal degradation. BAP1 is the sole nuclear-localized member, suggesting a role in gene regulation.4 When BAP1 was first discovered as a BRCA1-interacting protein and later shown to bind to BARD1, the focus was on DNA repair and genomic instability. Although there is some evidence that BAP1 may play a role in BRCA1-mediated DNA repair pathways, that connection remains unresolved. The more established function for BAP1 is its role as a core catalytic component of human polycomb-like multiprotein complexes that regulate gene expression. This complex contains various proteins, including polycomb proteins ASXL1/2, YY1, and OGT. The core BAP1-ASXL1/2 complex interacts with host cell factor-1 (HCF-1) and histone-modifying complexes during cell division.16 Studies in which BAP1 cellular levels or enzymatic activity were altered experimentally have revealed defects in cell cycle progression at G1/S, and BAP1 is thought to influence cell proliferation by coregulating transcription from HCF-1/E2F-governed promoters.17 BAP1 has also been shown to interact with ASXL1/2 to form the polycomb group repressive deubiquitinase complex (PR-DUB), which is involved in stem cell pluripotency and development.18 Mutations of BAP1 and potentially genes encoding other PR-DUB components may alter the function of the holo-complex leading to tumorigenesis. Germane to this, somatic ASXL1/2 alterations have been detected in human myelodysplastic disorders and solid tumors, and a conditional, systemic knockout model in which Bap1 was homozygously deleted in adult mice recapitulated features of human myelodysplastic syndrome.19
Unresolved Questions and Conclusions
While the first two reports of germline inactivating BAP1 mutations focused on different disease entities—that is, one on families with a high incidence of MM,6 and the other on families with multiple melanocytic tumors7—both studies found recurrent UMs as well. As proposed by Goldstein,20 current evidence supports the notion that these initial reports of germline BAP1 mutations were describing a single syndrome consisting of a range of tumors with varying penetrance.
Although there is mounting evidence for the existence of a novel BAP1 cancer syndrome, many questions remain. For example, what is the gamut of tumor types connected with the disorder? It is clear that MM, UM, CM, and benign melanocytic skin lesions are part of the spectrum of tumors associated with the BAP1 syndrome; however, genetic epidemiologic studies will be required to determine if there is significant susceptibility to other tumor types (paraganglioma, RCC, lung, and breast carcinomas), each of which has been reported in some BAP1 carriers. Furthermore, why are two distinct tumor types—MM, derived from mesothelial lining, and atypical melanocytic tumors/UM/CM, derived from melanocytes—associated with this syndrome? Is tumor type specificity influenced by the subunit compositions of PR-DUB complexes in each precursor tissue? Similarly, the tumor spectrum connected with Li-Fraumeni syndrome extends from epithelial cancers to sarcomas to brain tumors. Moreover, why do BAP1 mutations predispose to cancer when present in the germ line, yet act as a late, somatic event in connection with UM metastasis? It is also unclear if variations in the number of melanocytic tumors and incidence of MM among BAP1 mutation carriers reflect differences in genetic background of affected individuals or differences in exposure to carcinogens.
Collectively, these new findings will help to identify individuals at high risk of MM, CM, and potentially other cancers who could be targeted for early intervention. BAP1 mutation carriers should be regularly monitored for evidence of early malignancy, and preventive measures such as avoidance of exposure to asbestos and sun should be implemented.
LI-FRAUMENI SYNDROME: GENETIC BASIS FOR CLINICAL SURVEILLANCE
In 1969, Li and Fraumeni described four families in whom soft tissue sarcoma was associated with early-onset breast cancer in close female relatives.21 Subsequent analysis of LFS families has demonstrated breast cancer (most often invasive ductal carcinoma) to be the most common tumor (24% to 31%) followed by soft tissue sarcomas (~11.5% to 18%), brain tumors (3.5% to 14%), osteosarcomas (12.5%-13.5%); and adrenocortical carcinomas (ACCs) (6.5% to 10%).22 In 1990, germline TP53 mutations were discovered to be the underlying cause of the majority of LFS cases.23 p53 is a potent transcription factor that mediates cellular stress responses and initiates DNA repair, cell-cycle arrest, senescence, and apoptosis. Approximately 1,800 different somatic and germline TP53 mutations have been reported, with most localized to the DNA binding domain (DBD) (http://p53.iarc.fr). Mutant p53 loss-of-function, dominant-negative and gain-of-function properties are all important for tumorigenesis in humans, with gain-of-function activities being particularly relevant.24 Currently, 80% of LFS families that fulfill the classical clinical criteria (Table 1) harbor TP53 germline mutations25—most of which reside in the DBD. Causal mutations in genes other than TP53 have not been reproducibly observed, although it is anticipated the new platforms for whole genome and whole exome sequencing may uncover candidate genes.
TABLE 1.
Clinical Criteria for Classic LFS, LFS-like Criteria, and Chompret Criteria
| Classic LFS Criteria:21 |
|---|
| • Proband diagnosed with a sarcoma before age 45 |
| • A first-degreea relative with cancer diagnosed before age 45 |
| • A first- or second-degreeb relative on the same side of the family with cancer diagnosed before age 45 or a sarcoma at any age |
| LFS-like Criteria Birch:84 |
| • Proband with any childhood cancer or sarcoma, brain tumor, or adrenocortical carcinoma diagnosed before age 45 |
| • First- or second-degree relative with a typical LFS cancer (sarcoma, breast cancer, brain tumor, leukemia, or adrenocortical carcinoma) diagnosed at any age and |
| • A first- or second-degree relative on the same side of the family with any cancer diagnosed under age 60 |
| Eeles definition of LFL:84 |
| • Two first- or second-degree relatives with LFS related malignancies at any age |
| Chompret criteria for LFS:29 |
| • Proband diagnosed with a narrow-spectrum cancer (sarcoma, brain tumor, breast cancer, or adrenocortical carcinoma) before age 36 and at least one first- or second-degree |
| • A proband with multiple primary tumors, two of which belong to the narrow spectrum and the first of which occurred before age 36, regardless of family history |
| • A proband with adrenocortical carcinoma, regardless of age at diagnosis or family history |
Abbreviations: LFS, Li-Fraumeni syndrome: LFL, Li-Fraumeni-like.
First-degree relative is defined as a parent, sibling, or child.
Second-degree relative is defined as a grandparent, aunt, uncle, niece, nephew, or grandchild.
LFS accounts for 17% of all genetically defined familial cancer cases. Over 500 families have been reported worldwide with complete or partial (LFS-like) phenotypes, and many more families have been identified and not reported. The TP53 carrier rate is about 1 in 5,000 births.26 LFS is a highly penetrant disorder with the lifetime risk of developing cancer being 93% in men and 68% in women.26 The gender difference cannot be explained by breast or sex-related cancers, since the difference remained after exclusion of breast, ovary and prostate cancer. Females also exhibit an earlier average age of onset (29 years vs. 40 years in men).27 The age distribution at cancer diagnosis is strikingly young, with up to 56% of diagnoses under 30 years of age and 78% under 50 years (http://p53.iarc.fr).
Specific TP53 mutant genotype may influence age of onset and tumor spectrum. Birch and colleagues28 reported a significantly higher incidence and earlier age at cancer diagnosis for breast (p = 0.006) and brain cancers (p = 0.05) in families who carry missense mutations in the DBD. Conversely, whereas families in whom ACCs occur together with a wider spectrum of cancers harbor the usual spectrum of germline mutations, those with isolated ACC or apparently de novo mutations are commonly found to occur outside the DNA-binding loops. Furthermore, nonsense, frameshift, and splice mutations are associated with particularly early tumor onset.22 However, these genotype:phenotype correlations are not consistent, as numerous families carrying the same mutation express widely divergent clinical manifestations of age of onset and cancer type.
The difficulty in clarifying an absolute clinical definition for LFS has likely led to under-reporting of cases. Various clinical definitions of LFS as well as incomplete phenotypes—LF-like (LFL) and incomplete LFS (LFSI)—have been suggested in order to more accurately represent the TP53 genotype:phenotype correlation.29–31 Table 1 highlights the comparative phenotypic characteristics of the classical and related definitions of LFS. The sensitivity and specificity of the Chompret criteria are 82% and 58%, respectively, making it perhaps the most rigorous and relevant definition to justify TP53 mutation analysis. ACC represents 14% of all cancers in LFS and occurs primarily in children. Bone sarcomas occur mainly in young adolescents. Brain tumors and soft tissue sarcomas exhibit a biphasic age distribution, with a first peak in very early childhood (< 5 years) and a second peak between 20 and 40 years. Approximately 10% of patients with LFS develop gliomas, typically before the age of 45 years. Up to 5% of patients develop supratentorial primary neuroectodermal tumors, choroid plexus carcinoma (CPCs), and medulloblastomas. Almost 50% of children with CPC harbor germline TP53 mutations even in the absence of a typical LFS family history. Therefore, children with CPC or ACC should be considered for TP53 mutation analysis.
Approximately 20% to 30% of tumors in TP53 mutation carriers do not belong to the classical LFS tumor spectrum32: Wilms tumor and phyllodes tumors of the breast are strongly associated, pancreatic cancer moderately associated, and neuroblastoma weakly associated with TP53 mutation carrier status. Carcinomas of the lung and gastrointestinal tract, lymphomas, and other neoplasms have been shown to occur in TP53 mutation carriers or first-degree relatives of carriers at much earlier ages than seen in the general population.
Approximately 1% of heritable breast cancers arise due to a germline TP53 mutation. However, 7% of women who develop breast cancer under 30 years of age and have no first- or second-degree relatives with cancer carry a TP53 mutation; presence of first- or second-degree relatives with cancer raises this likelihood to well over 75%.33
Occurrence of multiple metachronous or synchronous primary cancers is one of the remarkable phenotypes observed in LFS.34 Age and tumor diagnosis influence the risk of second malignant neoplasms (SMN), with the highest risk being in those who developed their first tumor in the first two decades of life or those diagnosed with rhabdomyosarcoma (RMS).34 In TP53 mutation carriers, the cumulative probability of a second cancer is 57% (±10%) at 30 years.34 The relative risk in individuals whose first cancer was diagnosed before age 20 is 83, (95% CI 37-187) decreasing to 9.7 (95% CI 4.9-20) for ages 20 to 44, and to 1.5 (95% CI 0.5-4.2) for age 45 and older at first cancer diagnosis.34
A unique R337H mutation exclusively found in LFS families from southeastern Brazil is a risk allele for pediatric ACC and, until recently, was thought to represent a unique example of a tissue-specific predisposing mutation. The carrier frequency in southeastern Brazil is 0.3%.35 Nonbiased ascertainment of families with the R337H mutation shows that the cancer risk encompasses the full spectrum of tumors associated with LFS, in particular early-onset breast cancer (mean age at diagnosis below 40 years)36 and CPC.37 Additionally, current data on overall cancer penetrance suggest that the age-related cancer risk is somewhat lower than in LFS associated with other TP53 mutations, with tumors detected in approximately 25% of carriers at the age of 30 and a lifetime risk of approximately 80%.38
Some TP53 polymorphisms do not appear to be phenotypically silent. The PIN3 (16 bp duplication in intron 3) polymorphism has been associated with an increase in age of onset of tumors in TP53 mutation carriers. A single nucleotide polymorphism (SNP) at codon 72 in exon 4 involves the substitution of an arginine for a proline base. The current consensus from a large number of studies is that R72 is more effective in inducing apoptosis than P72.39 The effect of this polymorphism in determining clinical phenotype or outcome is not known. On the other hand, the SNP309, rs2279744 T/G polymorphic substitution in the MDM2 gene appears to confer an earlier age of onset in LFS patients.40
In LFS, decrease in age of cancer onset and increase in cancer incidence in successive generations have both been observed.41 Interestingly, telomeres in peripheral blood leukocytes of TP53 mutation carriers are shorter than in normal individuals of corresponding age. This difference was more pronounced in children (34% decrease) than in adults (19% decrease).42 The accelerated telomere attrition in TP53 mutation carriers is postulated to lead to greater genomic instability and earlier age of cancer onset in successive generations.43 Global copy number variation frequency and total structural variation are significantly increased in individuals with germline TP53 mutations (p = 0.01). In addition, among families with a history of cancer, offspring were significantly more likely to have an increase in CNVs when compared with their mutation carrier parent.44 These findings, together with the accelerated telomere attrition data, support the notion that TP53 mutation carriers have inherently unstable genomes and harbor other genetic and genomic alterations that can directly modify the age phenotype. Recent evidence suggests that the “early” germline presence of TP53 mutations in a cell may induce early critical telomere length shortening, which in turn may be involved in chromothripsis—an event of catastrophic chromosome rearrangement that is frequently seen in LFS-associated tumors.45 What the other genetic events are that modify the “driver” genotype conferred by the germline TP53 mutation are being actively explored and may ultimately lead to the development of more precise predictive algorithms of cancer phenotype and disease risk.
Screening and Surveillance in LFS
For both at-risk children and adults, lifestyle counseling to avoid ionizing radiation and other DNA-damaging agents is particularly relevant. For female TP53 mutation carriers, the role of prophylactic mastectomy has not been carefully evaluated. Prenatal or preimplantation genetic testing can be offered to fertile couples affected by LFS. Ultimately, determining the exact nature of the TP53 mutation may be of prognostic significance and therefore important for the clinical management of these patients.25 Most importantly, a high index of suspicion should be maintained for known LFS carriers who complain of persisting but unexplained symptoms.
The diversity of tumors occurring in LFS families has prompted investigators to evaluate the use of biochemical and imaging modalities to identify tumors. Although FDG-PET/CT can identify new primary cancers in TP53 mutation carriers,46 repeated radiation exposure may accelerate the risk of secondary malignancies. With this in mind, a recent study by Villani and colleagues47 suggests that an aggressive multimodality approach to clinical surveillance using a combination of biochemical markers of disease, abdominal-pelvic ultrasound, rapid-sequence whole-body MRI, and dedicated brain MRI in both children and adults does identify presymptomatic cancers. Colonoscopy and breast MRI are also included in the adult surveillance protocol. Many of the tumors detected are low-grade or low stage, and their early detection appears to be correlated with improved outcome when compared against TP53 mutation carriers who did not undergo surveillance.47
Studies to improve the predictive value of genetic and genomic modifier effects on the mutant TP53-associated phenotype will inform development of more refined tumor screening protocols and lead to improved understanding of the biologic mechanisms of tumor formation in these patients.
SDHx GENE MUTATIONS: A NEW CLINICAL SYNDROME
Paragangliomas are benign tumors that often occur in the head and neck region, along the parasympathetic chain. Also called glomus tumors, paragangliomas can develop along the glomus jugulare, tympanicum, vagale, or caroticum. These paragangliomas that develop along the parasympathetic chain usually do not secrete catecholamines. Similar tumors also can develop along the sympathetic chain, often referred to as pheochromocytomas when they are located within the adrenal gland. These sympathetic tumors also can be extra-adrenal, occurring alongside the aortapulmonary vasculature, the organ of Zuckercandl, or even the bladder and vas deferens. Sympathetic paragangliomas and pheochromocytomas often secrete catecholamines, which can be useful for early tumor screening/detection. Paragangliomas and pheochromocytomas are quite rare in the general population, occurring at an estimated incidence of 1 in 30,000 to 1 in 1 million.48,49 However, in the presence of an underlying germline SDHx mutation, the tumor rate may be extraordinarily high with a penetrance approaching 80%.50,51
Succinate dehydrogenase (SDH) is part of respiratory complex II in the mitochondria, and this enzyme complex is responsible for converting succinate to fumarate as part of the TCA, or Krebs, cycle. SDH is composed of four distinct SDH proteins called SDHA, SDHB, SDHC, and SDHD corresponding to the genes that encode them.52 A fifth gene, SDHAF2, encodes SDH Assembly Factor 2, which is responsible for assembling the other four SDH proteins into a fully functioning enzyme complex.53 Over the past several years, we have learned that germline mutations in each of these SDHx genes may lead to development of paragangliomas or pheochromocytomas.54 Lack of a functioning SDH complex leads to increased succinate, with subsequent increases in HIF signaling and possible histone deregulation. Tumorigenesis caused by increased succinate may be partially explained by the Warburg effect, the observation of a tumor’s unexpectedly high rate of glycolysis with a concurrent defect in mitochondrial respiration.55 Germline mutations in other genes such as NF1, VHL, RET, TMEM127, and MAX also have been associated with the development of paragangliomas and pheochromocytomas. Based on gene expression and pathway analysis, these tumors can be divided into two clusters corresponding to their underlying gene mutations: Cluster 1 (Cluster 1A: SDHx, Cluster 1B: VHL), associated with pseudohypoxia and aberrant VEGF signaling, and Cluster 2 (RET/NF1/TMEM127/MAX), associated with aberrant kinase signaling pathways.56–59
In the absence of SDHx mutations, paragangliomas (and sometimes pheochromocytomas) are benign and very slow growing tumors. Many head and neck surgeons choose simply to observe these tumors and remove them only if they start to grow and physically impinge on vital structures. However, when such tumors occur because of germline mutations in SDHx genes, paragangliomas and pheochromocytomas can transform to become highly aggressive and metastatic. Once metastatic, these tumors become very difficult to cure. A recent study from the National Institutes of Health demonstrated that nearly 30% of nonmetastatic paragangliomas and pheochromocytomas are due to germline SDHx mutations, and that 44% of adults and 81% of children with metastatic disease are due to germline SDHx mutations.60 In fact, this study did not test for all of the known SDHx genes, so the actual prevalence of germline SDHx mutations in this disease may be higher.61 Patients with metastatic paraganglioma or pheochromocytoma should be considered for genetic referral to test for underlying germline mutations, including the known SDHx genes.
Each SDHx gene mutation leads to a slightly different disease phenotype and clinical presentation (see Table 2).52 Interestingly, PGL-1 disease related to germline SDHD mutations is inherited in a maternally imprinted fashion with only the children of fathers— but not mothers— developing disease. Nevertheless, mothers can still pass SDHD mutations to their children. This is also true for germline SDHAF2 mutations (PGL-2), which are also inherited as a maternally imprinted disease. Disease caused by germline SDHB mutations (PGL-4) seems to be the most common and malignant of the clinical phenotypes in “Familial Paraganglioma and Pheochromocytoma Syndrome.”50–51,60–62 Every germline SDHx mutation (except SDHAF2) has been associated with GISTs.63,64 The association of paragangliomas with GISTs in the same patient has been called Carney-Stratakis syndrome, and germline SDHx genes have been found to be mutated in these patients.50,51 Previously, clinical testing for the specific inherited SDHx mutations was based on disease presentation. For instance, a patient with metastatic paraganglioma that secretes catecholamine would first be tested for SDHB mutations, whereas a young patient with just familial head and neck paragangliomas might first be screened for SDHD or SDHC mutations. However, as technology moves toward hereditary gene panel testing, it is likely that all of these genes will be tested simultaneously in the future.
TABLE 2.
Germline SDHx Mutations and Their Clinical Associations
| SDHA | SDHB | SDHC | SDHD | SDHAF2 | |
|---|---|---|---|---|---|
| Presentation Type | PGL-5 | PGL-4 | PGL-3 | PGL-1 | PGL-2 |
| Chromosome Location | 5p15 | 1p35-36.1 | 1q21 | 11q23 | 11q11.3 |
| Inheritance Pattern | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal dominant, maternal imprinting | Autosomal dominant, maternal imprinting |
| Catecholamine Secreting | Yes | Frequent | Occasionally | Occasionally | Unknown |
| Malignant/Metastatic | Yes | Frequent | Unknown | Occasionally | Unknown |
| Head and Neck PGLs | Yes | Yes | Frequent | Frequent | Frequent |
| PCC (Any Abdominal) | Frequent | Frequent | Occasionally | Occasionally | Unknown |
| Associated with GIST | Frequent | Yes | Yes | Yes | Unknown |
| Associated with Thyroid Cancer | Unknown | Yes | Unknown | Yes | Unknown |
| Associated with Renal Tumors (RCC and Oncocytoma) | Unknown | Yes | Yes | Unknown | Unknown |
| Associated with NBL | Unknown | Yes | Unknown | Unknown | Unknown |
Abbreviations: PGL, paraganglioma: PCC, pheochromocytoma: GIST, gastrointestinal stromal tumor; RCC, renal cell carcinoma; NBL, neuroblastoma.
SDHB Immunohistochemistry
In order to identify patients at risk for underlying germline SDHx mutations, it was observed several years ago that tumor tissues could be stained by immunohistochemistry (IHC) for the SDHB protein.65,66 Lack of SDHB staining indicates that the SDH protein complex is not intact, which could be due to a defect in any one of the SDHx genes. In one prospective study, SDHB IHC for identification of SDH-related tumors had sensitivity of 100% (95% CI 87-100), and identification of non-SDH-related tumors had specificity of 84% (95% CI 60-97).65 It has been suggested that IHC for SDHB should be used on any paraganglioma or pheochromocytoma not meeting the clinical criteria for MEN2, NF1, or VHL disease. If SDHB staining is absent, then SDHx genetic testing can be considered. Clinical laboratories are now beginning to offer SDHB IHC testing, but its ultimate sensitivity and specificity still needs to be determined in larger prospective trials. Currently, SDHB IHC seems to be most useful in the research setting.
Surveillance
Like LFS, we have learned that scheduled surveillance can detect early tumors in patients with underlying germline SDHx mutations. This is important so that smaller, asymptomatic SDH-deficient tumors can be removed before they transform to malignant and metastatic disease. Although no formalized screening guidelines exist, many clinicians perform annual physical examinations, blood pressure checks (for hypertension due to increased catecholamines), and blood work for serum metabolites. Previously, urine catecholamines were examined from 24-hour urine specimens, but many have eliminated urine screening in favor of serum testing. This blood work often includes fractionated catecholamines (epinephrine, norepinephrine, dopamine), fractionated free metanephrines (metanephrines, normetanephrine), and chromogranin A, all of which can be secreted by SDH-related tumors, given their sympathetic and parasympathetic origins.67 Fractionated plasma metanephrines have been reported to be the most sensitive and specific serum test for detecting secreting paragangliomas and pheochromocytomas.70 Fractionated metanephrines are nearly always abnormal in individuals with a hereditary syndrome characterized by secreting tumors (elevated metanephrines for RET and NF1 mutations and elevated normetanephrines for SDHx and VHL mutations). Increased methoxytyramine, a metabolite of dopamine, may be helpful for predicting the likelihood of metastatic disease and for distinguishing SDH-related tumors from VHL-related tumors.68,69 However, testing of methoxytyramine remains difficult to obtain clinically.
Regular imaging has been demonstrated to be very effective for identifying SDH-related tumors, especially in the setting of negative biochemical results.70–73 Screening approaches have included CT scans, MRI scans, [18F]fluorodeoxyglucose PET scans, and [123I]-metaiodobenzylguanidine scintigraphy. At the University of Utah, a recent prospective observational study of whole body, rapid sequence MRI in SDHx mutation carriers demonstrated MRI sensitivity of 88% and specificity of 95% to detect new tumors compared to biochemical testing sensitivity of 38% and specificity of 95% (K. Jasperson, personal communication, submitted). The University of Utah now recommends whole body MRI (5-mm slices from skull base to pelvis) at least every two years for patients with underlying SDHx mutations, followed by PET scans for patients with abnormal MRI results.
GISTs and Other SDH-Deficient Tumors
As testing for SDHx mutations has become more widespread, we have learned more about the spectrum of other SDH-related tumors, including GISTs, renal tumors (RCC, oncocytoma), papillary thyroid cancer, pituitary tumors, and even neuroblastoma.51,74,75 In fact, wild-type GISTs lacking somatic KIT or PDFRA mutations have been shown to be 100% SDH-deficient as measured by SDHB IHC.77 Although rare, NIH has established a Pediatric and Wildtype GIST Clinic and through this effort has helped to better clinically define these SDH-deficient GISTs, which are often multifocal at diagnosis, 75% female, and more indolent. These SDH-deficient GISTs are often epithelioid or have mixed histology, grow in the muscularis propria in a microplexiform pattern, and commonly show lymph node metastases.77–78 Many recent reports have demonstrated germline SDHA mutations associated with SDH-deficient (wild-type KIT/PDFRA) GISTs,79,80 which can be detected by SDHA IHC.81,83 Germline SDHx mutations still account for less than half of SDH-deficient GISTs, and the search for underlying SDH-related genes continues for the majority of patients with SDH-deficient GISTs. It is now recommended that patients with SDH-deficient GISTs be referred for genetic evaluation for underlying SDHx mutations. If germline SDHx mutations are identified in patients with GISTs, they should be considered for biochemical and imaging surveillance due to risk for other tumors.
The Future of SDH Tumor Syndrome
As familial tumors due to inherited SDHx mutations become better recognized, we will begin to learn more about the genetic, epigenetic, and metabolic alterations related to cancer risk. Modifying genes will be identified, along with potential targets for novel prevention and therapeutic strategies. With a large enough cohort of patients, early tumor screening approaches can be assessed for effectiveness and ability to improve outcome. As with other rare cancer syndromes, collaboration and consortiums will be key to advancing our understanding of and treatment for SDH-deficient tumors.
CONCLUSION
Although rare, hereditary cancer syndromes provide an unparalleled opportunity to study the origin of a variety of cancers. As described above, this knowledge can be used to better understand tumorigenesis and develop novel approaches to the treatment of patients with cancer. The recognition of hereditary cancer syndromes in families may lead to enrollment in early prevention and cancer screening trials. When dealing with individuals with such high risk for tumor development, the most effective interventions have been the early identification and removal of tumors. As we learn more about the specific molecular pathways altered in these hereditary cancer syndromes, even more targeted approaches can be developed to benefit patients either with or without familial cancers.
KEY POINTS.
BAP1 encodes a catalyzing enzyme that removes ubiquitin from protein substrates, and germline BAP1 mutations cause a novel cancer syndrome characterized by high incidence of benign atypical melanocytic tumors, uveal melanoma, cutaneous melanoma, malignant mesothelioma, and potentially other cancers.
BAP1 mutation carriers should have regular medical examinations in order to diagnose associated malignancies at an early, more treatable stage.
TP53 mutations cause Li-Fraumeni syndrome with extremely high risk for sarcomas, breast cancer, brain tumors, adrenocortical carcinomas, and other tumors. Genomic modifiers for disease and genotype-phenotype correlations are beginning to be identified.
Biochemical screening and regular imaging have contributed to improved outcomes for patients with Li-Fraumeni syndrome, and larger surveillance studies are now being done.
SDH is a mitochondrial enzyme complex involved in the tricarboxylic acid cycle, and SDHx mutations (SDHA, SDHB, SDHC, SDHD, and SDHAF2) lead to increased succinate and high risk for paragangliomas, pheochromocytomas, renal cell carcinoma (RCC), gastrointestinal stromal tumors (GISTs), and other tumors.
Regular biochemical screening and imaging in patients with germline SDHx mutations can detect early cancer to improve patient outcome.
Acknowledgments
Grant Support: J.R.T. supported by NCI grants CA148805 and CA-06927, a grant from the Mesothelioma Applied Research Foundation, an appropriation from the Commonwealth of Pennsylvania, and a gift from the Local #14 Mesothelioma Fund of the International Association of Heat and Frost Insulators & Allied Workers in memory of Hank Vaughan and Alice Haas. D.M. is funded in part through grants from the Canadian Cancer Society (#701377) and the Canadian Institutes for Health Research. J.D.S. is a Damon Runyon Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI#56-11), Alex’s Lemonade Stand Foundation, CureSearch for Children’s Cancer, and by NCI grant CA161780-01A1.
Footnotes
Employment or Leadership Position: None.
Consultant or Advisory Role: None.
Stock Ownership: None.
Honoraria: Joshua D. Schiffman, Affymetrix.
Research Funding: None.
Expert Testimony: None.
Other Remuneration: None.
Disclosures of Potential Conflicts of Interest
Relationships are considered self-held and compensated unless otherwise noted. Relationships marked “L” indicate leadership positions. Relationships marked “I” are those held by an immediate family member; those marked “B” are held by the author and an immediate family member. Relationships marked “U” are uncompensated.
References
- 1.Garber JE, Offıt K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23:276–292. doi: 10.1200/JCO.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 2.Knapke S, Nagarajan R, Correll J, et al. Hereditary cancer risk assessment in a pediatric oncology follow-up clinic. Pediatr Blood Cancer. 2012;58:85–89. doi: 10.1002/pbc.23283. [DOI] [PubMed] [Google Scholar]
- 3.Schiffman JD. Hereditary cancer syndromes: if you look, you will find them. Pediatr Blood Cancer. 2012;58:5–6. doi: 10.1002/pbc.23336. [DOI] [PubMed] [Google Scholar]
- 4.Jensen DE, Proctor M, Marquis ST, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- 5.Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiesner T, Fried I, Ulz P, et al. Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J Clin Oncol. 2012;30:e337–e340. doi: 10.1200/JCO.2011.41.2965. [DOI] [PubMed] [Google Scholar]
- 11.Njauw CN, Kim I, Piris A, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneousocular melanoma families. PLoS One. 2012;7:e35295. doi: 10.1371/journal.pone.0035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Höiom V, Edsgärd D, Helgadottir H, et al. Hereditary uveal melanoma: a report of a germline mutation in BAP1. Genes Chromosomes Cancer. doi: 10.1002/gcc.22035. Epub 2013 Jan 23. [DOI] [PubMed] [Google Scholar]
- 14.Wadt K, Choi J, Chung JY, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012;25:815–818. doi: 10.1111/pcmr.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin N Am. 2005;18:75–84. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Misaghi S, Ottosen S, Izrael-Tomasevic A, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol. 2009;29:2181–2192. doi: 10.1128/MCB.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eletr ZM, Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys. 2011;60:3–11. doi: 10.1007/s12013-011-9184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheuermann JC, de Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dey A, Seshasayee D, Noubade R, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337:1541–1546. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein AM. Germline BAP1 mutations and tumor susceptibility. Nat Genet. 2011;43:925–926. doi: 10.1038/ng.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 22.Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 23.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, Sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 24.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 25.Royds JA, Iacopetta B. p53 and disease: when the guardian angel fails. Cell Death Differentiation. 2006;13:1017–1026. doi: 10.1038/sj.cdd.4401913. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez KD, Nolter KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 27.Hwang SJLG, Amos CI, Strong LC. Germline p53 mutations: the Li Fraumeni syndrome. Biochime 75- 2003;82:2002. [Google Scholar]
- 28.Birch JM, Blair V, Kelsey AM, et al. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene. 1998;17:1061–1068. doi: 10.1038/sj.onc.1202033. [DOI] [PubMed] [Google Scholar]
- 29.Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li-Fraumeni syndrome. J Clin Oncol. 2009;27:e108–e109. doi: 10.1200/JCO.2009.22.7967. [DOI] [PubMed] [Google Scholar]
- 30.Eeles R. Germline mutations in the TP53 gene. Cancer Surv. 1995;25:101–124. [PubMed] [Google Scholar]
- 31.Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 32.Ruijs MW, Verhoef S, Rookus MA, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421–428. doi: 10.1136/jmg.2009.073429. [DOI] [PubMed] [Google Scholar]
- 33.McCuaig JM, Armel SR, Novokmet A, et al. Routine TP53 testing for breast cancer under age 30: ready for prime time? Fam Cancer. 2012;11:607–613. doi: 10.1007/s10689-012-9557-z. [DOI] [PubMed] [Google Scholar]
- 34.Hisada M, Garber JE, Fung CY, et al. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 35.Palmero EL, Schüler-Faccini L, Caleffı M, et al. Detection of R337H, a germline TP53 mutation predisposing to multiple cancers, in asymptomatic women participating in a breast cancer screening program in Southern Brazil. Cancer Lett. 2008;261:21–25. doi: 10.1016/j.canlet.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 36.Achatz MI, Olivier M, Le Calvez F, et al. The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett. 2007;245:96–102. doi: 10.1016/j.canlet.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 37.Custodio G, Taques GR, Figueiredo BC, et al. Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLoS One. 2011;6:e18015. doi: 10.1371/journal.pone.0018015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Achatz MI, Hainaut P, Ashton-Prolla P. Highly prevalent TP53 mutation predisposing to many cancers in the Brazilian population: a case for newborn screening? Lancet Oncol. 2009;10:920–925. doi: 10.1016/S1470-2045(09)70089-0. [DOI] [PubMed] [Google Scholar]
- 39.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bougeard G, Baert-Desurmont S, Tournier I, et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet. 2006;43:531–533. doi: 10.1136/jmg.2005.037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trkova M, Hladikova M, Kasal P, et al. Is there anticipation in the age at onset of cancer in families with Li-Fraumeni syndrome? J Hum Genet. 2002;47:381–386. doi: 10.1007/s100380200055. [DOI] [PubMed] [Google Scholar]
- 42.Trkova M, Prochazkova K, Krutilkova V, et al. Telomere length in peripheral blood cells of germline TP53 mutation carriers is shorter than that of normal individuals of corresponding age. Cancer. 2007;110:694–702. doi: 10.1002/cncr.22834. [DOI] [PubMed] [Google Scholar]
- 43.Tabori U, Nanda S, Druker H, et al. Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome. Cancer Res. 2007;67:1415–1418. doi: 10.1158/0008-5472.CAN-06-3682. [DOI] [PubMed] [Google Scholar]
- 44.Shlien A, Tabouri U, Marshall CR, et al. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc Natl Acad of USA. 2008;105:11264–11269. doi: 10.1073/pnas.0802970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masciari S, Van den Abbeele AD, Diller LR, et al. F18-fluorodeoxyglucose-positron emission tomography/computed tomography screening in Li-Fraumeni syndrome. JAMA. 2008;299:1315–1319. doi: 10.1001/jama.299.11.1315. [DOI] [PubMed] [Google Scholar]
- 47.Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12:559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 48.Petropoulos AE, Luetje CM, Camarata PJ, et al. Genetic analysis in the diagnosis of familial paragangliomas. Laryngoscope. 2000;110:1225–1229. doi: 10.1097/00005537-200007000-00030. [DOI] [PubMed] [Google Scholar]
- 49.Strosberg JR. Update on the management of unusual neuroendocrine tumors: pheochromocytoma and paraganglioma, medullary thyroid cancer and adrenocortical carcinoma. Semin Oncol. 2013;40:120–33. doi: 10.1053/j.seminoncol.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med. 2009;266:19–42. doi: 10.1111/j.1365-2796.2009.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 52.Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion. 2010;10:393–401. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gimenez-Roqueplo AP, Tischler AS. Pheochromocytoma and Paraganglioma: progress on all fronts. Endocr Pathol. 2012;23:1–3. doi: 10.1007/s12022-011-9190-7. [DOI] [PubMed] [Google Scholar]
- 55.Bayley JP, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev. 2010;20:324–329. doi: 10.1016/j.gde.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–2391. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cascon A, Tennant DA. From transcriptional profiling to tumor biology in pheochromocytoma and paraganglioma. Endocr Pathol. 2012;23:15–20. doi: 10.1007/s12022-012-9195-x. [DOI] [PubMed] [Google Scholar]
- 58.Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol. 2012;23:21–33. doi: 10.1007/s12022-012-9199-6. [DOI] [PubMed] [Google Scholar]
- 59.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- 60.King KS, Prodanov T, Kantorovich V, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29:4137–4142. doi: 10.1200/JCO.2011.34.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schiffman JD. No child left behind in SDHB testing for paragangliomas and pheochromocytomas. J Clin Oncol. 2011;29:4070–4072. doi: 10.1200/JCO.2011.37.8695. [DOI] [PubMed] [Google Scholar]
- 62.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 63.Hoekstra AS, Bayley JP. The role of complex II in disease. Biochim Biochim Biophys Acta. 2012;S0005-2728:1080–1088. doi: 10.1016/j.bbabio.2012.11.005. Epub 2012 Nov 20. [DOI] [PubMed] [Google Scholar]
- 64.Stratakis CA, Carney JA. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med. 2009;266:43–52. doi: 10.1111/j.1365-2796.2009.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisenhofer G, Tischler AS, de Krijger RR. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratification. Endocr Pathol. 2012;23:4–14. doi: 10.1007/s12022-011-9188-1. [DOI] [PubMed] [Google Scholar]
- 67.Eisenhofer G, Pacak K, Huynh TT, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer. 2011;18:97–111. doi: 10.1677/ERC-10-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–20. doi: 10.1373/clinchem.2010.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eisenhofer G, Lenders JW, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48:1739–1749. doi: 10.1016/j.ejca.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Timmers HJ, Chen CC, Carrasquillo JA, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst. 2012;104:700–708. doi: 10.1093/jnci/djs188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PG-L.EVA Investigators. J Clin Endocrinol Metab. 2013;98:E162–73. doi: 10.1210/jc.2012-2975. [DOI] [PubMed] [Google Scholar]
- 72.King KS, Chen CC, Alexopoulos DK, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab. 2011;96:2779–2785. doi: 10.1210/jc.2011-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miederer M, Fottner C, Rossmann H, et al. High incidence of extraadrenal paraganglioma in families with SDHx syndromes detected by functional imaging with [(18)F]fluorodihydroxyphenylalanine PET. Eur J Nucl Med Mol Imaging. doi: 10.1007/s00259-013-2346-6. Epub 2013 Feb 2. [DOI] [PubMed] [Google Scholar]
- 74.Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer. 2012;19:C33–40. doi: 10.1530/ERC-12-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am J Med Genet A. 2010;152A:1531–1535. doi: 10.1002/ajmg.a.33384. [DOI] [PubMed] [Google Scholar]
- 76.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–1721. doi: 10.1097/PAS.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janeway KA, Weldon CB. Pediatric gastrointestinal stromal tumor. Semin Pediatr Surg. 2012;21:31–43. doi: 10.1053/j.sempedsurg.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 79.Belinsky MG, Rink L, Flieder DB, et al. Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2013;52:214–224. doi: 10.1002/gcc.22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oudijk L, Gaal J, Korpershoek E, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–463. doi: 10.1038/modpathol.2012.186. [DOI] [PubMed] [Google Scholar]
- 81.Dwight T, Benn DE, Clarkson A, et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol. 2013;37:226–233. doi: 10.1097/PAS.0b013e3182671155. [DOI] [PubMed] [Google Scholar]
- 82.Miettinen M, Killian JK, Wang ZF, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol. 2013;37:234–240. doi: 10.1097/PAS.0b013e3182671178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Birch JM. Li-Fraumeni syndrome. Eur J Cancer. 1994;30A:1935–1941. doi: 10.1016/0959-8049(94)00383-g. [DOI] [PubMed] [Google Scholar]
- 84.Eeles RA. Germline mutations in the TP53 gene. Cancer Surv. 1995;25:101–124. [PubMed] [Google Scholar]