

Abstract
Background
The introduction of northern snakehead (Channa argus; Anabantiformes: Channidae) and their subsequent expansion is one of many problematic biological invasions in the United States. This harmful aquatic invasive species has become established in various parts of the eastern United States, including the Potomac River basin, and has recently become established in the Mississippi River basin in Arkansas. Effective management of C. argus and prevention of its further spread depends upon knowledge of current population structure in the United States.
Methods
Novel methods for invasive species using whole genomic scans provide unprecedented levels of data, which are able to investigate fine scale differences between and within populations of organisms. In this study, we utilize 2b-RAD genomic sequencing to recover 1,007 single-nucleotide polymorphism (SNP) loci from genomic DNA extracted from 165 C. argus individuals: 147 individuals sampled along the East Coast of the United States and 18 individuals sampled throughout Arkansas.
Results
Analysis of those SNP loci help to resolve existing population structure and recover five genetically distinct populations of C. argus in the United States. Additionally, information from the SNP loci enable us to begin to calculate the long-term effective population size ranges of this harmful aquatic invasive species. We estimate long-term Ne to be 1,840,000–18,400,000 for the Upper Hudson River basin, 4,537,500–45,375,000 for the Lower Hudson River basin, 3,422,500–34,225,000 for the Potomac River basin, 2,715,000–7,150,000 for Philadelphia, and 2,580,000–25,800,000 for Arkansas populations.
Discussion and Conclusions
This work provides evidence for the presence of more genetic populations than previously estimated and estimates population size, showing the invasive potential of C. argus in the United States. The valuable information gained from this study will allow effective management of the existing populations to avoid expansion and possibly enable future eradication efforts.
Keywords: 2b-RAD sequencing, Aquatic invasive species, Population structure, Effective population size, Genomic analyses, Single-nucleotide polymorphism
Introduction
Channa argus (Anabantiformes: Channidae; Northern snakehead, Cantor 1842) is one of numerous fish species that have been introduced to the United States and is threatening native populations of organisms. This large, freshwater, piscivorous fish is native to China, Manchuria, southern Siberia, and Korea (Courtenay & Williams, 2004). They were imported to the US for the live food fish market and their introduction is mainly due to unauthorized intentional release of live individuals (Courtenay & Williams, 2004). Northern Snakehead were first reported in California in 1997, and the first established, reproducing population was documented in 2002 in a Maryland pond close to a Chesapeake bay tributary (Fuller & Benson, 2017). They spread rapidly due to their aggressive predation, rapid maturation rate, and ability to tolerate a wide range of environmental conditions (Orrell & Weigt, 2005; Wegleitner et al., 2016; Fuller & Benson, 2017). Since first detection approximately 20 years ago, C. argus populations have become established throughout the Potomac River, Chesapeake Bay, Hudson River and Delaware River basins (Fuller & Benson, 2017). Additionally, C. argus was discovered in Arkansas in April 2008 and persists following a failed eradication effort using rotenone (Fuller & Benson, 2017).
Biological invasions, such as the introduction and spread of northern snakehead in the United States, are occurring at an alarming rate and are a major contributor to loss of biodiversity, environmental change, and are economically expensive (Leung et al., 2002; Lodge et al., 2006). As the abundance of a non-indigenous species increases over time, eradication becomes increasingly more difficult and management becomes exceedingly more expensive. Successful removal of an invasive species is difficult, if not impossible, and expensive, especially after it becomes established (Lodge et al., 2006). Therefore, early detection and understanding of the population dynamics of the invasive species is important when only small, localized populations exist in a region (Lodge et al., 2006).
Genomic tools have become very important for monitoring and even early detection of rare species (Darling & Mahon, 2011). The data obtained through genomic methods can provide valuable information about the population structure of a species that is rare in a system and is especially advantageous because small sample sizes have been shown to be capable of determining fine scale population structure that accurately represent populations (Pujolar et al., 2013; Jeffries et al., 2016). Advances in high-throughput sequencing technology have decreased sequencing costs, but sequencing whole genomes can still be very expensive (Wang et al., 2012; Gautier et al., 2013; Andrews et al., 2016). Microsatellites have been a common tool for population studies because they are highly polymorphic, occur throughout the genome, and have a high mutation rate (Schlötterer, 2004; Wegleitner et al., 2016). However, microsatellite markers’ mutation pattern is complex, they can have high genotyping error rates, and while they occur throughout the genome, their density is low (DeFaveri et al., 2013). As a result, novel tools in the form of reduced representation genomic techniques have been developed that sample randomly, from throughout the genome (Davey et al., 2011). An example of this type of method involves restriction site associated DNA sequencing (RADseq). RADseq utilizes restriction enzyme anchored positions in the genome, allowing for the identification of thousands of single nucleotide polymorphism (SNP) loci (Davey & Blaxter, 2011). A specific type of RADseq, called 2b-RAD, developed by Wang et al. (2012), has made the process even more efficient by using type IIb restriction enzymes, such as BsaXI and AlfI, which allows for custom reduction schemes to be tailored to the study in question. Thousands of SNP loci that are evenly distributed across the genome at high densities are recovered from 2b-RAD sequencing and provide robust results even when sample sizes are small (Pujolar et al., 2013; Nazareno et al., 2017). Jeffries et al. (2016) found that when compared to a microsatellite dataset, the RADseq approach produced more robust results and recovered finer population structure when they used a small sample size that included a large number of SNP loci. Additionally, Nazareno et al. (2017) were able to recover sufficient within-population genomic diversity using only 6–8 individuals, and only needed two individuals per population to be able to estimate population genetic structure, because they used a large number of polymorphic SNP loci. These and other studies (e.g., Willing, Dreyer & Van Oosterhout, 2012 and Senn et al., 2013) have shown that any limitations that may occur from small sample sizes can be overcome with an appropriate number of SNP loci. Restriction-site associated DNA digestion sequencing methods, such as 2b-RAD are inexpensive, rapid methods of analysis that generate a tremendous amount of data (e.g., hundreds to thousands of SNP loci from throughout the organism’s entire genome) that can be utilized to investigate population structure, estimate effective population size, estimate number of breeding individuals in a population, and to answer phylogeographic questions about populations (Pujolar et al., 2013; Pecoraro et al., 2016; Galaska et al., 2017a; Galaska et al., 2017b).
For C. argus, effective management and prevention of its further spread in North American waters depends upon knowledge of current population structure in the United States. Previous studies using microsatellite markers for this species have shown that a minimum of two genetically distinct populations are present in the eastern United States (Wegleitner et al., 2016). Using their microsatellite data, fish sampled from the Chesapeake Bay and Potomac River system comprise one population, whereas the fish present in the Hudson River basin comprise a genetically distinct population (Orrell & Weigt, 2005; Wegleitner et al., 2016). No Arkansas fish were included in previous population level studies. Therefore, the goal of this study was to determine the relationship of the Arkansas C. argus population to the eastern United States populations. Additionally, we chose to use a 2b-RAD sequencing method for genome scanning, to examine populations within the United States and these data will allow us to estimate the effective population size of established populations of C. argus’ sampled ranges. We hypothesize that fish in the established Arkansas region were introduced from existing populations in the eastern United States. Additionally, we anticipate the Arkansas population size of C. argus to be smaller than the population sizes in the eastern United States, due to an eradication attempt that occurred soon after their discovery. Resulting data will inform management agencies and aid in decisions regarding control of the populations, in order to avoid expansion and new introductions.
Methods
Sample collection and preparation
Muscle tissue and fin clips (fresh, frozen, or preserved in 95% non-denatured ethanol) were collected from 165 C. argus from established populations in the United States by various individuals and management agencies (Fig. 1). This included collections from seven regions: Catlin Creek, part of the Upper Hudson River basin, Meadow and Willow Lakes in Queens, NY, part of the Lower Hudson River basin, fish from a market in Chinatown, in NYC, FDR park in Philadelphia, eight locations in the Potomac River, three rivers that are part of the Chesapeake Bay basin, and five locations in Arkansas (Table 1; see Table S1 for additional collection information). Genomic DNA was extracted from the muscle tissue and fin clips using the Qiagen DNeasy® Blood and Tissue kit (Qiagen, Valencia, CA, USA), following manufacturer’s protocols.
Figure 1. Collection locations for Channa argus individuals in the eastern United States and Arkansas.
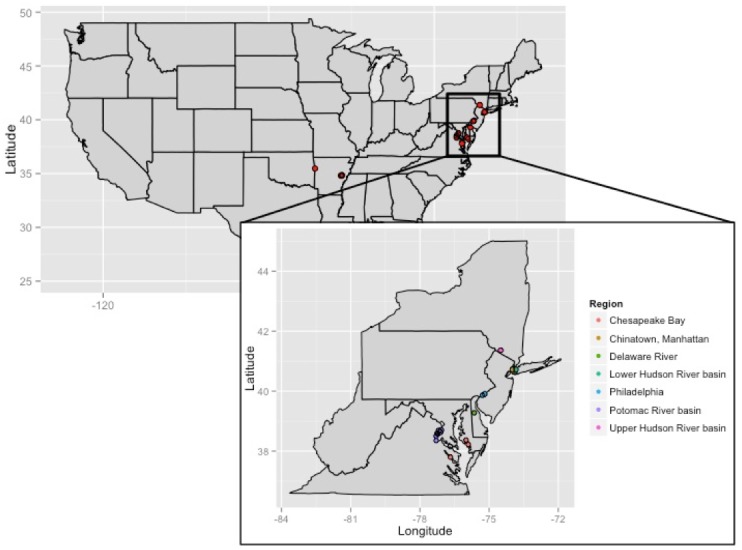
Table 1. Sample collection regions for Channa argus.
| Location sampled | Number of individuals |
|---|---|
| Upper Hudson River basin | 59 |
| Lower Hudson River basin | 10 |
| Chinatown, Manhattan | 3 |
| Philadelphia | 21 |
| Potomac river | 50 |
| Chesapeake bay | 4 |
| Arkansas | 18 |
Genomic data collection
Genomic DNA preparation followed the 2b-RAD protocol from Wang et al. (2012). Digestion of genomic DNA utilized the restriction enzyme AlfI. Two site-specific ligation adaptors (NC/NN) were used to employ a reduction scheme. The adaptors bind to the two base-pair sticky end products of the AlfI restriction. The reduction scheme was chosen based on an approximate genome size of C. argus of 616–861 Mb to target approximately 2,500 SNP loci (A Libertini & F Krapp, 2017, unpublished data. Accessed from http://genomesize.com). Samples were dual barcoded with unique combinations before being sent for sequencing at HudsonAlpha Institute for Biotechnology Genome Services Laboratory (Huntsville, AL, USA) on an Illumina Hi-Seq 2500 using v4 chemistry, generating 50 bp single-end reads.
Data analyses
Raw Illumina reads were demultiplexed by sample, quality filtered and aligned against a custom de novo reference sequence as outlined in Wang et al. (2012) using scripts from Dr. Eli Meyer (Oregon State University) (https://github.com/Eli-Meyer) and the software package Stacks v.1.35 (Catchen et al., 2011). The 2b-RAD data were filtered by loci to include samples with a minimum coverage of 20X. Homozygotic SNP loci were defined to have a maximum variance of 1% and heterozygotic loci had a minimum of 25% variance. Loci had to occur in 75% or more individuals within a sampling locality and be present in at least two localities to be processed. Loci not meeting these requirements were discarded prior to analyses.
Discriminant Analysis of Principal Components (DAPC) in the Adegenet v2.0.1 package (Jombart, 2008; Jombart, Ahmed & Bateman, 2011) in the R v3.4.1 statistical program (R Core Team, 2015) was used to analyze the SNP data to determine population structure of the putative C. argus populations. Adegenet conducts a series of Principal Component Analyses (PCA) on SNP data that is then retained to perform a Discriminant Analysis on all PCAs. Optimal clusters (K), likely representing populations, were identified through Bayesian information criterion (BIC) likelihood values from retained principal components. Visualization of the DAPC analyses was performed within the Adegenet package.
Population structure and admixture were assessed using the Landscape and Ecological Associations (LEA) v1.8.1 package in R (Frichot & François, 2015). K was estimated with the cross-entropy criterion and least squares estimates were used to calculate ancestry proportions (Frichot & François, 2015). Admixture was visualized by individual as bar charts and by locality as pie charts.
Further analyses involved generating summary statistics and analyzing genetic differentiation using the R package HIERFSTAT (ver. 0.04-22; Goudet, 2005), as well as calculating the effective population size (Ne) of each of the C. argus populations. Initially, SNP data were analyzed assuming two populations (East Coast and Arkansas), based on the geographical distance between the eastern United States and Arkansas. Discrete populations were identified using both DAPC and LEA analyses. These analyses showed that the original seven sampling regions were not distinct genetic populations, but instead Chesapeake Bay and Potomac River were one genetic population (Potomac River basin population), and the Lower Hudson River basin and Chinatown fish were also one genetic population (Lower Hudson River basin population). Additional summary statistics were performed on each of the five putative populations and pairwise Fst differences were calculated to assess genetic differentiation between the newly identified populations. The effective population size of the East Coast and Arkansas populations were calculated using the following equation: π = 4∗Ne∗μ, where π represents nucleotide diversity, Ne represents the effective population size, and μ represents the mutation rate of the SNPs (Tajima, 1983; Pujolar et al., 2013). Lastly, the effective number of breeders (Nb) was calculated, using the following equation: Nb = Ne(0.485 + 0.758∗log (adult lifespan∕age of first reproduction)) (Ruzzante et al., 2016).
Results
A total of 23,695 single nucleotide polymorphism loci were recovered from 165 C. argus; quality filtering and SNP calling resulted in 1,007 independent SNP loci being retained in the final dataset.
Results of the DAPC analyses support five geographically and genomically distinct clusters or populations of C. argus in the United States (Fig. 2, Fig. S1). Cluster 1 contains 100% of the fish from Arkansas. Cluster 2 contains 100% of the fish from the Lower Hudson River basin and the Chinatown, Manhattan fish market. Cluster 3 contains 52 individuals, 96% of the fish collected from the Potomac River basin, and 1.7% of the fish collected from the Upper Hudson River basin. Cluster 4 contains 59 individuals, 96.6% of the fish from the Upper Hudson River basin, and 3.8% of the fish from the Potomac River basin. Cluster 5 contains 100% of the fish from Philadelphia, the only fish collected in the Delaware River, part of the Potomac River basin, and 1.7% of the fish from the Upper Hudson River basin.
Figure 2. Discriminant Analysis of Principal Components for Channa argus single-nucleotide polymorphism data.

Cluster 1 contains all 18 individuals from Arkansas. Cluster 2 contains 13 individuals: all 10 from Lower Hudson River basin and all three from the Chinatown, Manhattan fish market. Cluster 3 contains 52 individuals: 51 from Potomac River basin and one from Upper Hudson River basin. Cluster 4 contains 59 individuals: 57 from Upper Hudson River basin and two from Potomac River basin. Cluster 5 contains 23 individuals: one from Potomac River basin (Delaware River), one from Upper Hudson River basin, and all 21 individuals from Philadelphia.
Additionally, the results of the LEA analyses also support the existence of five populations (K = 5, Fig. S2). Each of the populations is partially admixed, although the amount of gene flow varies among the populations (Figs. 3–4). The individuals from the fish market in Chinatown are statistically from the same population as most of the individuals in the Lower Hudson River basin, indicating they came from the same source (Fig. 3). One of the Upper Hudson River basin individuals and one of the Potomac River basin individuals share genotypes, determined statistically and shown in Fig. 3, with the Philadelphia fish. In contrast, the rest of the fish in the Potomac River basin are comprised primarily of a different ancestral genotype. The main ancestral genotype for all of the Arkansas fish is rare in the individuals from all of the East Coast populations.
Figure 3. Admixture for C. argus populations (K = 5) in the United States.
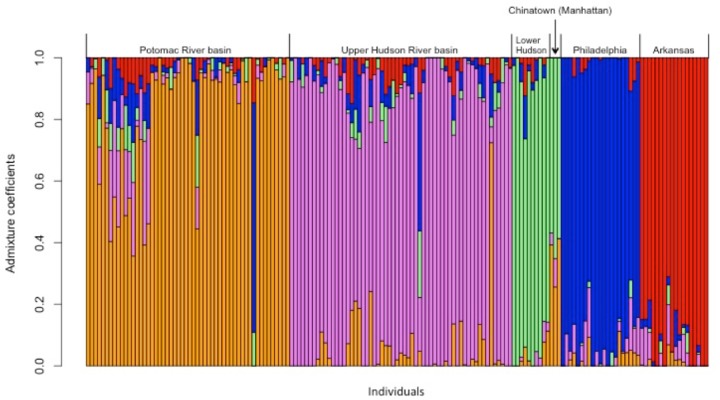
Each bar represents an individual (X axis) from each of the collection locations (top of figure). Individual admixture coefficients are represented in each column (Y axis).
Figure 4. Admixture results by collection location.
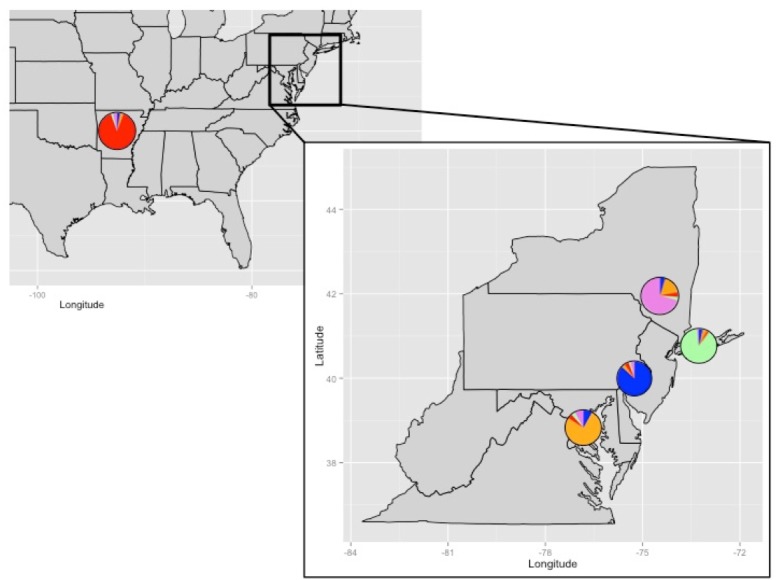
Pie charts represent the average admixture from each geographically distinct putative population (Arkansas, Potomac River Basin, Upper Hudson River Basin, Lower Hudson River basin, Philadelphia) as noted in the admixture plot of Fig. 3.
Summary statistics of genetic diversity and genetic distances between the putative C. argus populations are given in Tables 2 and 3. The smallest genetic distance (0.08369683) is between the Upper Hudson River basin and the Philadelphia fish, whereas the largest genetic distance (0.18527692) is between the Potomac River and Arkansas populations. Heterozygosity is low for all putative populations. For the Potomac River basin population, the observed heterozygosity is lower than expected, whereas observed heterozygosity is the same as expected for the Upper Hudson River basin, Philadelphia, and Arkansas putative populations. Observed heterozygosity is higher than expected for only the Lower Hudson River basin putative population.
Table 2. Summary statistics for single nucleotide polymorphism loci.
Individuals from Chinatown fish market excluded.
| Putative population | Ho | Var | StdErr | He | Var | StdErr | Fis | Var | StdErr |
|---|---|---|---|---|---|---|---|---|---|
| Potomac River basin | 0.1167 | 0.0296 | 0.0078 | 0.1313 | 0.0326 | 0.0082 | 0.1429 | 21.3512 | 0.1226 |
| Upper Hudson River basin | 0.0662 | 0.0197 | 0.0069 | 0.0715 | 0.0187 | 0.0067 | 0.1185 | 25.9216 | 0.1698 |
| Lower Hudson River basin | 0.1643 | 0.0324 | 0.0059 | 0.1634 | 0.0252 | 0.0052 | 0.0266 | 15.0182 | 0.0329 |
| Philadelphia | 0.0862 | 0.0223 | 0.0049 | 0.0884 | 0.0223 | 0.0049 | 0.0094 | 32.6641 | 0.0524 |
| Arkansas | 0.0889 | 0.0301 | 0.0064 | 0.0893 | 0.0268 | 0.0060 | 0.0099 | 35.4852 | 0.0538 |
Table 3. Pairwise genetic distances between putative populations of Channa argus.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| 2 | 0.17150707 | |||
| 3 | 0.09671192 | 0.09378427 | ||
| 4 | 0.16979238 | 0.08369683 | 0.09108509 | |
| 5 | 0.18527692 | 0.10232123 | 0.10871429 | 0.09207021 |
Notes.
- 1
- Potomac River/Chesapeake Bay
- 2
- Upper Hudson River basin
- 3
- Lower Hudson River basin/Chinatown
- 4
- Philadelphia
- 5
- Arkansas
The long-term effective population sizes (Ne) for each of the C. argus populations are shown in Table 4. The Chinatown fish market individuals are of questionable origin (i.e., not from a field site but from a captive market), thus they were excluded from this calculation. The Upper Hudson River basin Ne is the smallest, with an estimated range of 1,840,000–18,400,000 individuals (π = 0.0722 and the SNP mutation rate (μ) is 1 ×10−8–1 × 10−9 mutations per generation). The number of breeding individuals (Nb) is estimated to be 2,287,120–22,871,200 individuals based on an adult lifespan of 10 years and age of first reproduction of one year (Odenkirk & Owens, 2007; Odenkirk et al., 2013). In contrast, the Lower Hudson River basin putative population has the largest Ne, with an estimated range of 4,537,500–45,375,000 individuals (π = 0.1705), and an estimated range of 5,640,113–56,401,125 breeding individuals (Nb). The Ne of Arkansas population is estimated to be 2,580,000–25,800,000 individuals (π = 0.0921) and Nb estimated range of 3,206,940–32,069,400 individuals.
Table 4. Nucleotide diversity and long term Ne and Nb values.
Nucleotide diversity (π), long-term Ne and Nb estimates for each of the populations identified by DAPC. Chinatown samples have been excluded because they came from a fish market.
| Population | π | Ne | Nb |
|---|---|---|---|
| Upper Hudson River basin | 0.0736 | 1,840,000–18,400,000 | 2,287,120–22,871,200 |
| Lower Hudson River basin | 0.1815 | 4,537,500–45,375,000 | 5,640,113–56,401,125 |
| Potomac River | 0.1369 | 3,422,500–34,225,000 | 4,254,168–42,541,675 |
| Philadelphia | 0.1086 | 2,715,000–27,150,000 | 3,374,745–33,747,450 |
| Arkansas | 0.1032 | 2,580,000–25,800,000 | 3,206,940–32,069,400 |
Discussion
The SNP data support the presence of five genetically distinct putative populations of C. argus: four in the eastern United States, and a distinct group in Arkansas. However, the Upper Hudson River basin population is not an active population. It was eradicated in 2008–2009 and considered successfully removed, based on subsequent monitoring by the New York State Department of Environmental Conservation. The tissues were collected as part of that effort and have been used here to attempt to determine if the Arkansas population originated from the Upper Hudson River basin. When compared to the East Coast snakehead populations that were sampled, the Arkansas population is distinct and mainly comprised of a unique ancestral genotype. Additionally, heterozygosity and the inbreeding coefficient is low for the Arkansas population, so there is little genetic variability, and inbreeding is occurring, showing that the population is geographically and genetically isolated from the east coast populations. This supports the possibility that the Arkansas population originated from a different source population than the source(s) of the eastern United States introductions. Thorough geographic sampling provides a clear distinction between the East Coast populations and the Arkansas population. The Arkansas population is the one that is the most genetically differentiated from the fish in the Potomac River and Lower Hudson River basins. It cannot be completely ruled out that the Arkansas population originated from an unidentified East Coast population not sampled in our study. Adding samples from C. argus native ranges could help to determine the source of the North American introductions, however, those samples were not available for the current study.
One of the principal goals of this study was to use a higher resolution method to answer the questions that remained unresolved from a previous study using microsatellites that sought to determine the number of genetic populations of C. argus in the eastern United States, as well as determine the source of the C. argus population in the Upper Hudson River basin. Wegleitner et al. (2016) suggested that two genetic populations exist in the eastern United States, and this was likely as a result of two separate introductions of C. argus. The results of this genomic scan study show a more complex story, providing evidence for the presence of five distinct populations of C. argus. Based on the shared ancestry and lack of genetic differentiation in the Potomac River basin population and fish from the Chinatown fish market, the fish in both geographic regions likely came from the same source. One individual sampled from the Potomac River basin shows strong genomic similarity to the C. argus in Philadelphia, as well as one individual from the Hudson River basin, suggesting introduction from one source, and subsequent population expansion. This is not surprising because of the close proximity of the Potomac River basin to Philadelphia (approx. 306 kilometers). However, because of the admixture that has occurred in each of the populations, it is not possible with the current dataset to determine how many introductions occurred.
Conclusions
An accurate assessment of current population size is crucial for conservation efforts and management, along with making accurate models of population growth. This is the first estimate of long-term effective population size for the Arkansas and East Coast populations using SNP data and these estimates are based off the estimation method used by Pujolar et al. (2013) that calculates Ne based on nucleotide diversity in a population. Since they were first reported in Arkansas in 2008, it’s possible that individuals from the original source population could skew the Ne and Nb estimates if any of the fish used in this study are from the original introduction event. Channa argus has a relatively long lifespan and high fecundity in both native and introduced ranges. Previous studies have used C. argus individuals from the Potomac River basin that were estimated to be as old as 10.1 years (Odenkirk & Owens, 2007) and have been documented to have a maximum lifespan of 15 years in their native environment (Jiao et al., 2009). Adult female fecundity estimates are 22,000–51,000 in their native range (Amur River basin; Nikol’skiy, 1956) to 28,600–115,000 in an introduced population (Syr Dar’ya basin, Turkmenistan/Uzbekistan; Dukravets & Machulin, 1978; Fuller & Benson, 2017). Furthermore, they may spawn up to five times a year in their native range, while in an introduced population in Kazakhstan, they spawned 1–3 times per year (Courtenay & Williams, 2004). In addition to producing moderately sized egg clutches, C. argus guard their nests and young for up to nine weeks (Ling, 1977; Landis, Lapointe & Angermeier, 2011). Landis & Lapointe (2010) observed both male and female C. argus guarding eggs and fry for at least four weeks in a Potomac River tributary, including protecting them when they left the nest. Based on their SNP data, Pujolar et al. (2013) also estimated much higher values for long-term effective population size of the European eel than had previously been reported using microsatellite data. Common programs that provide Ne estimates using microsatellite data make assumptions that oversimplify microsatellite mutation patterns, which could result in low Ne values. Unfortunately, there are no other Ne estimates for C. argus populations in North America using microsatellites, nor does census information exist. Therefore, it is difficult to assess the accuracy of our Ne estimates. However, given the properties of SNP markers, the time since established in the United States (at least 15 years), the high genetic diversity in these populations, high reproductive potential, and extensive protection of offspring, it is not surprising that the long term effective population size estimates are so high.
According to the US Army Corps of Engineers (Grippo et al., 2017), C. argus has a low overall risk rating for the probability of establishment in the Great Lakes basin because they are currently located in Arkansas in low numbers and known to only occur in one river system. These results show that there are likely more fish in Arkansas than previously known, they may occur in more than one river system, and likely have become established. Despite a decrease in population size following an eradication attempt by the Arkansas Fish and Game Commission in 2009, the Arkansas C. argus effective population size is not the smallest of the five populations. Channa argus is an extremely environmentally tolerant species, and therefore was able to rebound from the eradication attempt, and currently has an effective population size within the range of the other populations, demonstrating its potential to quickly increase in abundance. Furthermore, Kramer et al. (2017) used global species distribution models and information about local habitats to assess the suitability of the Great Lakes for C. argus establishment and determined that the climate conditions throughout most of the Great Lakes would be suitable for their establishment. These results show that C. argus has established large effective populations along the East Coast of the United States and in Arkansas. Other aquatic invasive species with similar life history traits and habitat preferences, such as grass carp, quickly expanded their ranges after being introduced in Arkansas, spreading into the Mississippi River basin, and the Great Lakes basin within 50 years of their introduction. These results illustrate the potential for C. argus to quickly expand its range, like the grass carp and its relatives. Expansion could have major economic and environmental impacts on commercial and recreational fisheries, and native fish populations (Courtenay & Williams, 2004; Grippo et al., 2017).
The ability to develop effective management strategies to avoid further C. argus expansion and subsequent harm to the Mississippi River and Great Lakes basin ecosystems is enhanced by the results of this study. Source population information would aid in the development of a complete picture and the best resulting management strategies. That information in currently not available, but important conclusions and management recommendations can still be made based on our current dataset. These data provide evidence of gene flow between the Potomac River basin and Philadelphia populations. Previous studies have shown that this will make eradication efforts difficult due to the increased likelihood of recolonization (Abdelkrim, Pascal & Smadi, 2017; Farrington et al., 2017). However, the C. argus population in Arkansas is isolated from the East Coast populations, indicating recent establishment and little to no gene flow between Arkansas and the East Coast. Additionally, the main ancestral genotype present in the Arkansas population is rare in the East Coast populations, providing evidence for a different source population. If, as our data suggest, the Arkansas fish originated from a different source than the East Coast populations, then it is important to monitor and prevent possible means of introduction into Arkansas. Therefore, management efforts should focus on the entire population in Arkansas and should be handled differently than the East Coast populations. The isolation of the Arkansas population increases the likelihood of successful eradication, which is crucial to avoid expansion and subsequent harm to the Great Lakes basin.
Supplemental Information
Acknowledgments
We thank Michael Flaherty, Josh Newhard, Joe Love, Melissa Cohen, Allen Temple, Paul Overbeck, Gabriela Hogue, John Odenkirk, and Justin Homan for contributing C. argus tissue samples used in this study.
Funding Statement
This work was supported by a grant to Andrew R. Mahon from the US Fish and Wildlife Service Southeast Region (Grant Agreement F12AP01114). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Carlee A. Resh conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Matthew P. Galaska analyzed the data, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
Andrew R. Mahon conceived and designed the experiments, analyzed the data, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
Animal Ethics
The following information was supplied relating to ethical approvals (i.e., approving body and any reference numbers):
No IACUC approval was required for this project as we utilized museum collected tissue samples. No live animals were utilized.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
SRA sequence data are available in Genbank at the following accession numbers: SAMN08331753–SAMN08331787.
References
- Abdelkrim, Pascal & Smadi (2017).Abdelkrim J, Pascal M, Smadi S. Establishing causes of eradication failure based on genetics: case study of ship rat eradication in Ste. Anne archipelago. Conservation Biology. 2017;21:719–730. doi: 10.1111/j.1523-1739.2007.00696.x. [DOI] [PubMed] [Google Scholar]
- Andrews et al. (2016).Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics. 2016;17(2):81–92. doi: 10.1038/nrg.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen et al. (2011).Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. Genes Genomes Genetics. 2011;1(3):171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtenay & Williams (2004).Courtenay WR, Williams JD. Snakeheads (Pisces, Channidae): a biological synopsis and risk assessment. US Geological Survey, Restonhttps://pubs.usgs.gov/circ/2004/1251/report.pdf Circular. 1251. 2004
- Darling & Mahon (2011).Darling JA, Mahon AR. From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environmental Research. 2011;111(7):978–988. doi: 10.1016/j.envres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Davey & Blaxter (2011).Davey JW, Blaxter ML. RADseq: next-generation population genetics. Briefings in Functional Genomics. 2011;9(5–6):416–423. doi: 10.1093/bfgp/elq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey et al. (2011).Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- DeFaveri et al. (2013).DeFaveri J, Viitaniemi H, Leder E, Merilä J. Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Molecular Ecology Resources. 2013;13(3):377–392. doi: 10.1111/1755-0998.12071. [DOI] [PubMed] [Google Scholar]
- Dukravets & Machulin (1978).Dukravets GM, Machulin AI. The morphology and ecology of the Amur snakehead, Ophiocephalus argus warpachowskii, acclimatized in the Syr Dar’ya Basin. Journal of Ichthyology. 1978;16:203–208. [Google Scholar]
- Farrington et al. (2017).Farrington HL, Edwards CE, Bartron M, Lance RF. Phylogeography and population genetics of introduced Silver Carp (Hypophthalmichthys molitrix) and Bighead Carp (H. nobilis) in North America. Biological Invasions. 2017;19:2789–2811. doi: 10.1007/s10530-017-1484-3. [DOI] [Google Scholar]
- Frichot & François (2015).Frichot E, François O. LEA: an R package for landscape and ecological association studies. Methods in Ecology and Evolution. 2015;6(8):925–929. doi: 10.1111/2041-210X.12382. [DOI] [Google Scholar]
- Fuller & Benson (2017).Fuller PF, Benson AJ. Channa argus. Gainesville, FL: USGS Nonindigenous Aquatic Species Database [online] 2017. https://nas.er.usgs.gov/queries/FactSheet.aspx?speciesID=2265. [6 December 2017]. https://nas.er.usgs.gov/queries/FactSheet.aspx?speciesID=2265
- Galaska et al. (2017a).Galaska MP, Sands CJ, Santos SR, Mahon AR, Halanych KM. Geographic structure in the Southern Ocean circumpolar brittle star Ophionotus victoriae (Ophiuridae) revealed from mtDNA and single-nucleotide polymorphism data. Ecology and Evolution. 2017a;7(2):475–485. doi: 10.1002/ece3.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaska et al. (2017b).Galaska MP, Sands CJ, Santos SR, Mahon AR, Halanych KM. Crossing the divide: admixture across the Antarctic polar front revealed by the brittle star Astrotoma agassizii. Biological Bulletin. 2017b;232(3):198–211. doi: 10.1086/693460. [DOI] [PubMed] [Google Scholar]
- Gautier et al. (2013).Gautier M, Foucaud J, Gharbi K, Cézard T, Galan M, Loiseau A, Thomson M, Pudlo P, Kerdelhué C, Estoup A. Estimation of population allele frequencies from next-generation sequencing data: pool-versus individual-based genotyping. Molecular Ecology. 2013;22(14):3766–3779. doi: 10.1111/mec.12360. [DOI] [PubMed] [Google Scholar]
- Goudet (2005).Goudet J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Molecular Ecology Notes. 2005;5(1):184–186. doi: 10.1111/j.1471-8286.2004.00828.x. [DOI] [Google Scholar]
- Grippo et al. (2017).Grippo MA, Hlohowskyj I, Fox L, Herman B, Pothoff J, Yoe C, Hayse J. Aquatic nuisance species in the great lakes and Mississippi river basin—a risk assessment in support of GLMRIS. Environmental Management. 2017;59(1):154–173. doi: 10.1007/s00267-016-0770-7. [DOI] [PubMed] [Google Scholar]
- Jeffries et al. (2016).Jeffries DL, Copp GH, Lawson HL, Olsén KH, Sayer CD, Hänfling B. Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Molecular Ecology. 2016;25(13):2997–3018. doi: 10.1111/mec.13613. [DOI] [PubMed] [Google Scholar]
- Jiao et al. (2009).Jiao Y, Lapointe NWR, Angermeier PL, Murphy BR. Hierarchical demographic approaches for assessing invasion dynamics of non-indigenous species: an example using northern snakehead (Channa argus) Ecological Modelling. 2009;220(13):1681–1689. doi: 10.1016/j.ecolmodel.2009.04.008. [DOI] [Google Scholar]
- Jombart (2008).Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jombart, Ahmed & Bateman (2011).Jombart T, Ahmed I, Bateman A. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27(21):3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer et al. (2017).Kramer AM, Annis G, Wittmann ME, Chadderton WL, Rutherford ES, Lodge DM, Mason L, Beletsky D, Riseng C, Drake JM. Suitability of Laurentian Great Lakes for invasive species based on global species distribution models and local habitat. Ecosphere. 2017;8(7):e01883. doi: 10.1002/ecs2.1883. [DOI] [Google Scholar]
- Landis & Lapointe (2010).Landis AMG, Lapointe NWR. First record of a northern snakehead (Channa argus Cantor) nest in North America. Northeastern Naturalist. 2010;17(2):325–332. doi: 10.1656/045.017.0214. [DOI] [Google Scholar]
- Landis, Lapointe & Angermeier (2011).Landis AMG, Lapointe NW, Angermeier PL. Individual growth and reproductive behavior in a newly established population of northern snakehead (Channa argus), Potomac River, USA. Hydrobiologia. 2011;661(1):123–131. doi: 10.1007/s10750-010-0509-z. [DOI] [Google Scholar]
- Leung et al. (2002).Leung B, Lodge DM, Finnoff D, Shogren JF, Lewis MA, Lamberti G. An ounce of prevention or a pound of cure: bioeconomic risk analysis of invasive species. Proceedings of the Royal Society B: Biological Sciences. 2002;269(1508):2407–2413. doi: 10.1098/rspb.2002.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling (1977).Ling SW. Aquaculture in Southeast Asia: a historical overview. University of Washington Press; Seattle: 1977. [Google Scholar]
- Lodge et al. (2006).Lodge DM, Williams S, MacIsaac HJ, Hayes KR, Leung B, Reichard S, Mack RN, Moyle PB, Smith M, Andow DA, Carlton JT, McMichael A. Biological invasions: recommendations for US policy and management. Ecological Applications. 2006;16(6):2035–2054. doi: 10.1890/1051-0761(2006)016[2035:BIRFUP]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Nazareno et al. (2017).Nazareno AG, Bemmels JB, Dick CW, Lohmann LG. Minimum sample sizes for population genomics: an empirical study from an Amazonian plant species. Molecular Ecology Resources. 2017;17:1136–1147. doi: 10.1111/1755-0998.12654. [DOI] [PubMed] [Google Scholar]
- Nikol’skiy (1956).Nikol’skiy GV. Fish of the Amur basin (Ryby basseyna Amura) Izd-vo AN USSR; Moscow: 1956. [Google Scholar]
- Odenkirk et al. (2013).Odenkirk J, Lim C, Owens S, Isel M. Insight into age and growth of Northern Snakehead in the Potomac river. North American Journal of Fisheries Management. 2013;33(4):773–776. doi: 10.1080/02755947.2013.806382. [DOI] [Google Scholar]
- Odenkirk & Owens (2007).Odenkirk J, Owens S. Expansion of a Northern Snakehead population in the Potomac river system. Transactions of the American Fisheries Society. 2007;136(6):1633–1639. doi: 10.1577/T07-025.1. [DOI] [Google Scholar]
- Orrell & Weigt (2005).Orrell TM, Weigt L. The Northern Snakehead Channa argus (Anabantomorpha: Channidae), a non-indigenous fish species in the Potomac river, U.S.A. Proceedings of the Biological Society of Washington. 2005;118(2):407–415. doi: 10.2988/0006-324X(2005)118[407:TNSCAA]2.0.CO;2. [DOI] [Google Scholar]
- Pecoraro et al. (2016).Pecoraro C, Babbucci M, Villamor A, Franch R, Papetti C, Leroy B, Ortega-Garcia S, Muir J, Rooker J, Arocha F, Murua H, Zudaire I, Chassot E, Bodin N, Tinti F, Bargelloni L, Cariani A. Methodological assessment of 2b-RAD genotyping technique for population structure inferences in yellowfin tuna (Thunnus albacares) Marine Genomics. 2016;25:43–48. doi: 10.1016/j.margen.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Pujolar et al. (2013).Pujolar JM, Jacobsen MW, Frydenberg J, Als TD, Larsen PF, Maes GE, Zane L, Jian JB, Cheng L, Hansen MM. A resource of genome-wide single-nucleotide polymorphisms generated by RAD tag sequencing in the critically endangered European eel. Molecular Ecology Resources. 2013;13(4):706–714. doi: 10.1111/1755-0998.12117. [DOI] [PubMed] [Google Scholar]
- R Core Team (2015).R Core Team . Vienna: R Project for Statistical Computing; 2015. [Google Scholar]
- Ruzzante et al. (2016).Ruzzante DE, McCracken GR, Parmelee S, Hill K, Corrigan A, MacMillan J, Walde SJ. Effective number of breeders, effective population size and their relationship with census size in an iteroparous species, Salvelinus fontinalis. Proceedings of the Royal Society B: Biological Sciences. 2016;283(1823):20152601. doi: 10.1098/rspb.2015.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer (2004).Schlötterer C. The evolution of molecular markers—just a matter of fashion? Nature Reviews Genetics. 2004;5(1):63–69. doi: 10.1038/nrg1249. [DOI] [PubMed] [Google Scholar]
- Senn et al. (2013).Senn H, Ogden R, Cezard T, Gharbi K, Iqbal Z, Johnson E, Kamps-Hughes N, Rosell F, McEwing R. Reference-free SNP discovery for the Eurasian beaver from restriction site-associated DNA paired-end data. Molecular Ecology. 2013;22(11):3141–3150. doi: 10.1111/mec.12242. [DOI] [PubMed] [Google Scholar]
- Tajima (1983).Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al. (2012).Wang S, Meyer E, McKay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nature Methods. 2012;9(8):808–810. doi: 10.1038/nmeth.2023. [DOI] [PubMed] [Google Scholar]
- Wegleitner et al. (2016).Wegleitner BJ, Tucker A, Chadderton WL, Mahon AR. Identifying the genetic structure of introduced populations of northern snakehead (Channa argus) in Eastern USA. Aquatic Invasions. 2016;11(2):199–208. doi: 10.3391/ai.2016.11.2.09. [DOI] [Google Scholar]
- Willing, Dreyer & Van Oosterhout (2012).Willing EM, Dreyer C, Van Oosterhout C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLOS ONE. 2012;7(8):e42649. doi: 10.1371/journal.pone.0042649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.