

Abstract
Restimulation-induced cell death (RICD) is an apoptotic program that regulates effector T cell expansion, triggered by repeated stimulation through the T cell receptor (TCR) in the presence of interleukin-2 (IL-2). Although CD4+ regulatory T cells (Tregs) consume IL-2 and experience frequent TCR stimulation, they are highly resistant to RICD. Resistance in Tregs is dependent on the forkhead box P3 (FOXP3) transcription factor, although the mechanism remains unclear. T cells from patients with X-linked lymphoproliferative disease (XLP-1), that lack the adaptor molecule SLAM-associated protein (SAP), are also resistant to RICD. Here we demonstrate that normal Tregs express very low levels of SAP compared to conventional T cells. FOXP3 reduces SAP expression by directly binding to and repressing the SH2D1A (SAP) promoter. Indeed, ectopic SAP expression restores RICD sensitivity in human FOXP3+ Tregs. Our findings illuminate the mechanism behind FOXP3-mediated RICD resistance in Tregs, providing new insight into their long-term persistence.
Keywords: Restimulation-induced cell death, FOXP3, apoptosis, regulatory T cells, SAP
1. Introduction
An efficient adaptive immune response relies on the rapid clonal expansion of effector T cells, followed by the timely disposal of these cells to avoid nonspecific damage to host tissues. The adaptive immune system utilizes specific apoptosis programs to regulate T cell expansion and contraction and maintain homeostasis [1]. One of these programs is restimulation-induced cell death (RICD). RICD helps to keep effector T cell expansion in check, triggered by T cell receptor (TCR) restimulation in the presence of interleukin-2 (IL-2) [2-5]. Recent work from our lab has demonstrated that RICD sensitivity can be tuned by alterations in cell metabolism or diacylglycerol kinase (DGK) activity [6, 7]. A better understanding of the underlying mechanisms that influence RICD can therefore lead to novel strategies for controlling the magnitude and potency of a T cell response. However, our knowledge of the molecular signals that govern RICD sensitivity remains inadequate, especially with regard to different T cell populations with variable longevity.
The physiological relevance of RICD is best illustrated in patients with X-linked lymphoproliferative disease (XLP-1), who experience life-threatening, uncontrolled accumulation of CD8+ T cells in response to Epstein-Barr virus (EBV) infection [8]. A marked defect in RICD contributes to unconstrained CD8+ T cell expansion in this setting, resulting in severe immunopathology [9]. XLP-1 patients harbor null mutations in the signaling adaptor SLAM-associated protein (SAP), which is required for RICD [9, 10]. Indeed, work from our lab and others has shown that SAP associates with the signaling lymphocyte activation molecule (SLAM) family receptor NTB-A to potentiate TCR signal strength by displacing SHP-1 and recruiting LCK instead of FYN, which facilitates downstream signaling for apoptosis after TCR re-engagement [11-13]. SAP also represses DGKα to maintain a sufficient pool of diacylglycerol for robust TCR signaling [7, 14], further highlighting SAP as a central player in promoting RICD of effector T cells.
CD4+ regulatory T cells (Tregs) are a specialized subset of T cells that modulate the immune response and suppress the proliferation of lymphocyte effectors. Given their essential role in maintaining immune homeostasis and preventing autoimmunity [15], Tregs are of special interest regarding their therapeutic potential for tolerance induction in a variety of clinical settings [16, 17]. However, the processes that ultimately maintain Treg cell survival remain unclear [18]. For example, Tregs are extremely resistant to RICD despite frequent stimulation (often via self-antigen), susceptibility to FAS-induced death, and strict dependency on IL-2 derived from effector T cells [19, 20].
The gene encoding SAP, SH2D1A, was identified as a FOXP3 target in a genome-wide chromatin immunoprecipitation (chIP) study of human Tregs [21]. We therefore hypothesized that in human Tregs, FOXP3 restricts RICD by directly suppressing SAP expression. Using primary, activated human Tregs and conventional CD4+ T cells (Tcons) derived from healthy human donors, we confirmed that RICD resistance in Tregs is FOXP3 dependent [19]. We now demonstrate that SAP expression is markedly reduced in activated Tregs, and that FOXP3 confers resistance to RICD through repression of the SH2D1A promoter. These findings further elucidate the mechanism of RICD resistance in Tregs, providing new insights into Treg homeostasis.
2. Materials and Methods
2.1 Cell isolation and culture conditions
Peripheral blood mononuclear cells (PBMC) were obtained from buffy coats donated by healthy human donors at the National Institutes of Health (NIH) Blood Bank. Access to Blood Bank donors was kindly provided by Dr. Michael Lenardo. CD4+ T cells were purified from PBMC by immunomagnetic negative selection using the EasySep Human CD4+ T cell enrichment kit (Stem Cell Technologies). Cells were then stained on ice for 30 minutes with the following Abs: anti CD4-FITC (clone RPA-T4), anti-CD25-PE-Cy7 (clone BC96), and anti-CD127-PE (clone A019D5) (Tonbo Biosciences). Tregs and Tcons were sorted on a BD FACSAria cell sorter. The gating strategy is shown in Figure 1, where Tregs were defined as CD4+ CD25hi CD127lo and Tcons were defined as CD4+ CD25lo CD127hi [22].
Figure 1. Gating strategy to sort human Tregs and Tcons.
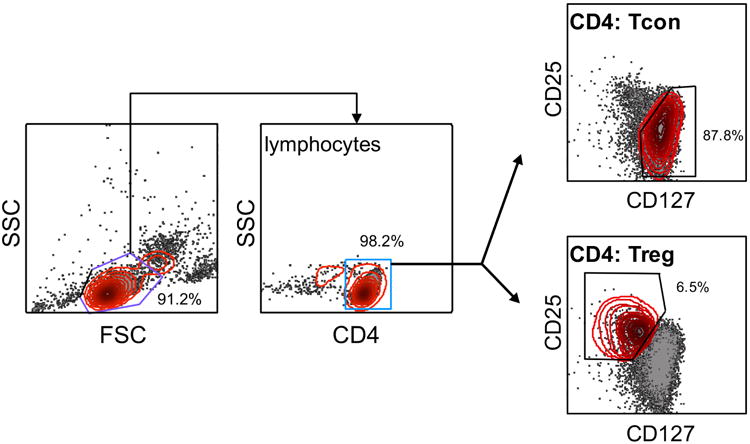
CD4+ T cells were isolated from healthy human blood donors by negative selection and stained with CD4, CD25, and CD127 antibodies before sorting. Lymphocytes were delineated by forward/side scatter gating, and CD4+ cells were further separated as CD25hi CD127lo Tregs or CD25lo CD127hi Tcons. A representative sort is shown; % of CD4+ Tcons vs. Tregs are labeled for each gate.
Sorted cells were activated with anti-CD2/CD3/CD28 antibody-bound biotin beads (Human T cell Activation/Expansion Kit, Miltenyi) in complete RPMI (RPMI 1640 (Life Technologies) + 10% fetal calf serum (FCS) (HyClone) + 1% penicillin/streptomycin (Lonza) + 2 μM ODN [23] for 3 days. Activated T cells were then washed in PBS and subsequently cultured in media as described above with 200 U/mL rIL-2 (PeproTech) and 2 μM ODN at 1×106 cells/mL, replacing the media every 3 days. Jurkat T cells were obtained from the American Type Culture Collection (clone E6.1) and cultured in complete RPMI at 37°C and 5% CO2.
2.2 Flow cytometry and apoptosis assays
RICD assays were performed as described previously [24]. Briefly, 1×105 effector T cells were restimulated with 1 μg/ml anti-CD3 mAb (clone OKT3) plus protein A (2 μg/ml) in triplicate wells for 24 hours. Cells were stained with 50nM TO-PRO-3 (Thermo Fisher) to distinguish live and dead cells, and analyzed on a BD Accuri C6 flow cytometer. Death was quantified as percent cell loss, based on quantification of viable cells collected under constant time, where % cell loss = (1 – [number of viable cells (treated) / number of viable cells (untreated)]) × 100.
For surface receptor staining, cells were washed in PBS + 1% FBS + 0.01% sodium azide and incubated with antibodies against CD3, CD25, NTB-A, CD95 (FAS) and CD69 (BD Biosciences) on ice for 30 minutes. Intracellular staining was performed using the FOXP3 intracellular staining kit with anti-FOXP3-APC Ab (eBioscience). All flow cytometry analysis was performed with FlowJo version 10.
2.3 Western blotting
Cells were lysed in 1% Nonidet P-40 (NP-40) lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 1 mM Na3VO4, 1 mM NaF) + complete protease inhibitors (Roche) for 30 minutes on ice. Lysates were cleared by centrifugation and boiled in 2× sample buffer (Laemmli buffer + 50 μM 2-βME) and separated on SDS-PAGE gels (Bio-Rad). Using the Trans-Blot Turbo system (Bio-Rad), proteins were transferred to nitrocellulose membranes and subsequently blocked with 2% Tropix I-Block (Applied Biosystems). Blots were probed with the following antibodies: anti-FOXP3 (Novus Biologicals NB600-245), anti-SAP, anti-LCK (Cell Signaling Technology), anti-β-actin (Sigma-Aldrich). After washing in TBS/0.1% Tween20, blots were incubated with horseradish peroxidase-conjugated secondary Abs (Southern Biotech), washed again, and developed using enhanced chemiluminescence (SuperSignal, ThermoFisher).
2.4 Quantitative RT-PCR
Total RNA was isolated from T cells using QIAshredder and RNeasy Mini Plus columns with DNase digestion (Qiagen). cDNA was prepared using the i Script cDNA kit for RT-qPCR (Bio-Rad), and qPCR was performed with Maxima SYBR Green/ROX qPCR Master Mix (ThermoFisher) using a two-step cycling protocol: 95°C for 1 minute, 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds, followed by a final elongation step at 60°C. The primers used were: SAP forward 5′-tctgtatgaaccctgtgttgg-3′, SAP reverse 5′-acaggatgttgtctacttgcc-3′, FOXP3 forward 5′-gatggtacagtctctggagc-3′, FOXP3 reverse 5′-gggaatgtgctgtttccatgg-3′, RPL30 forward 5′-gaatggcatggtcttgaagcc-3′, and RPL30 reverse 5′-ggccaccttcttgtgaatgc-3′.
2.4 SAP promoter cloning and luciferase assays
The pGL3 Firefly luciferase basic vector (Promega) was digested with KpnI and XhoI in order to insert SH2D1A promoter inserts of 1000 bp (maximum activity) versus 100bp (minimal activity) as previously published [25]. Segments of the SH2D1A promoter were amplified using these primers: SAPprom-F1: 5′ atttgcattaatacagtttagcctcaatcgaag-3, SAPprom-F2: 5′ -atttgcggtaccgttgttggggtgcttctctc-3′, SAPprom-R: 5′-ttactcaccggtggcctggtggactcttgg-3′.
Expression cassettes encoding WT FOXP3 or GFP were PCR amplified and cloned into the pmax vector (Lonza) after cutting with KpnI and XhoI. PCR primers used were: pmaxFOXP3-F 5′-atttgcggtaccgccgccaccatgcccaaccccaggcctg-3′, pmaxFOXP3-R 5′-ttactcctcgagtcaggggccaggtgtaggg-3′, The loss of function mutation in the DNA binding domain of FOXP3 (R397W) was introduced by site-directed mutagenesis using DpnI digestion and the following primers for amplification: R397W-F 5′-tgctttgtgTgggtggagagcgagaagg-3′, R397W-R 5′-gctctccacccAcacaaagcacttgtgcagac-3′. For luciferase assays, Jurkat T cells were triple transfected with 5 μg pmax-GFP or FOXP3 expression vector, 5 μg pSAP-GL3 firefly luciferase vector, 0.5 μg pRL-TK renilla luciferase vector, and the pmax vector and assayed after 24 hours. Luciferase activity was quantified using a Dual Luciferase Assay Kit (Promega) and a Synergy H1 microplate reader (Bio-Tek).
2.5 Chromatin immunoprecipitation
To perform chromatic immunoprecipitation (chIP) of Jurkat T cells and/or primary T cells, we used the Simple ChIP Plus enzymatic chromatin IP kit (Cell Signaling Technology) with the addition of anti-FOXP3 antibody Clone 206D (Biolegend) or the Mouse IgG1 isotype κ (BD) control antibody. DNA eluted from IPs was amplified by PCR (Apex 2.0 × Taq RED mastermix) with SAP promoter primers SAP-F 5′-aagttattcctggtggcctc-3′, and SAP-R 5′-ttggttcgatcgagcttgcc-3′. Cycling conditions were 95°C for 180 seconds, 32 cycles of 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, followed by 72°C for 5 minutes. PCR products were then electrophoresed on a 1% agarose gel (1× TAE) with ethidium bromide for visualization.
2.5 Lentiviral infections and siRNA transfections
pCLS-YFP-2A-FOXP3 and GFP lentiviral vectors were kindly provided by Dr. James Riley [26]. YFP-2A-FOXP3 and GFP expression cassettes were subcloned into the pWPT lentiviral transfer vector using BamHI and SalI. Lentiviruses were produced by co-transfecting the lentiviral plasmid with dR8.2 and VSV-G packaging vectors into 293T cells using calcium-phosphate. Two viral collections were made 48 to 72 h post-transfection, and viral supernatants were pooled and filtered.
For lentiviral transduction of primary T cells, 24 well plates were coated with retronectin overnight at 4°C at 20 μg/ml (Clontech Laboratories). Wells were blocked with 2% BSA in PBS for 30 minutes, and then viral supernatants (or media alone) were added to the plate and centrifuged for 2 hours at 2000 × g at 32°C. Unbound virus was washed from the wells with PBS, and then cells in complete RPMI + 200 U/mL rIL-2 were transferred to the plate and centrifuged at 2000 × g for 15 minutes at 32°C. GFP/YFP expression was measured 24 hours post-infection by flow cytometry to measure transduction efficiency. RICD assays were analyzed on cells counted from gated GFP/YFP+ populations.
T cells were electroporated with siRNAs against FOXP3 (FOXP3HSS121456) and SAP (SH2D1AHSS106218) (ThermoFisher) using the Amaxa Nucleofection 4D system and the P3 Primary Cell kit (Lonza). Stealth RNAi negative control medium GC duplex (ThermoFisher) siRNA was used as a non-specific (NS) control, and all assays were conducted 4 days post electroporation for peak knockdown efficiency.
2.6 Statistics
To assess significance in RICD assays, Student t-tests were used in assays comparing two groups, and two-way ANOVA tests were performed for multiple comparisons (Tukey's multiple comparisons test). For all figures, p-values <0.05 are indicated by *, and values <0.01 by **. Prism 7 software (GraphPad) was used for all statistical analyses.
3. Results
3.1 Activated human Tregs display minimal SAP expression and are resistant to RICD
Previous work has shown that human Tregs are resistant to RICD, which is dependent on FOXP3 expression [20]. However, moderate FOXP3-mediated suppression of FASL alone does not completely account for the profound RICD insensitivity in Tregs [19]. To explore this further, we first verified that primary CD25hiCD127lo Tregs sorted from healthy human donors (Figure 1) were resistant to RICD compared to Tcons. As expected, activated Tcons were significantly more sensitive to RICD than Tregs in every donor tested (Figure 2A). However, Tcons and Tregs were equally sensitive to death by direct ligation of the FAS death receptor (Figure 2B). Importantly, apoptosis sensitivity in Tregs could not be explained by alterations in the expression of the T cell receptor (CD3) or FAS, because expression levels were similar in Tcons and Tregs (Figure 2C). The SLAM family receptor NTB-A, which is required for optimal RICD sensitivity [9, 11], also exhibited similar expression in Tregs and Tcons (Figure 2C). As expected, Tregs maintained elevated expression of the high affinity IL-2 receptor CD25 compared to Tcons (Figure 2C).
Figure 2. Human Tregs are resistant to RICD, but not FAS-induced death.
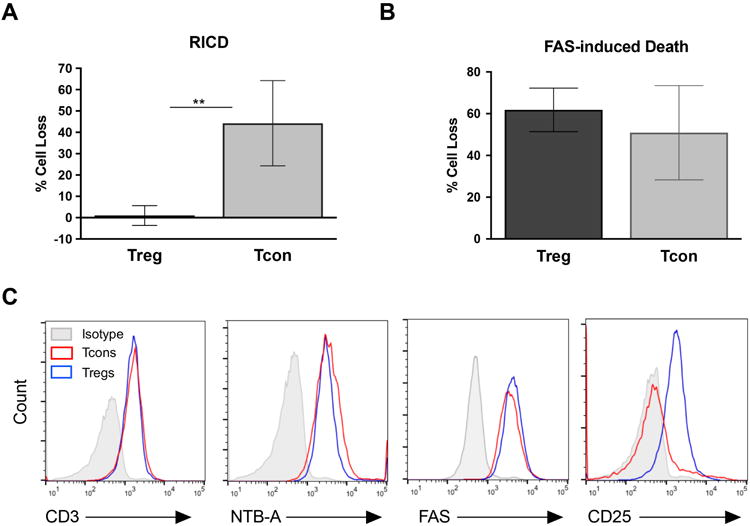
(A ) Sorted Treg and Tcons were activated with anti-CD2/3/28 beads and expanded in culture with IL-2. Cells were then restimulated with OKT3 antibody (1 μg/ml) plus Protein A (2 μg/ml) for 24 hours in triplicate. Cell death was measured by flow cytometry using TO-PRO-3 live/dead staining. Data are average ± SD of 4 independent experiments using separate donors; significance was assessed by an unpaired t-test. (B) Cells cultured as in (A) were treated with 200 ng/ml APO1.3 antibody plus Protein A for 24 hours. Cell death was measured as above; results data are average ± SD of 3 independent experiments. (C) Cells were stained with antibodies against CD3, NTB-A, FAS, and CD25, or an isotype control antibody, and analyzed by flow cytometry. Results are representative of 3 independent experiments.
T cells deficient in SAP expression, such as in XLP-1 patients, are highly resistant to RICD [11]. We therefore asked whether human Tregs express less SAP compared to Tcons. Indeed, genome-wide ChIP analysis of human Tregs uncovered SH2D1A as a FOXP3 target gene, although this particular finding was not subjected to further validation [21]. We therefore hypothesized that FOXP3 confers RICD resistance in activated Tregs by repressing the SH2D1A promoter and reducing SAP expression. We observed that SAP mRNA levels were markedly reduced in activated Tregs compared to Tcons, exhibiting an inverse relationship with FOXP3 expression (Figure 3A). As expected, this correlated with decreased SAP protein in Treg cell lysates (Figure 3B). Overall, our survey of cells from multiple human donors indicated that activated Tregs contain very little SAP expression compared to Tcons, and are extremely resistant to RICD, similar to XLP-1 patient-derived SAP-deficient T cells.
Figure 3. Inverse correlation between FOXP3 and SAP expression in human Tregs.
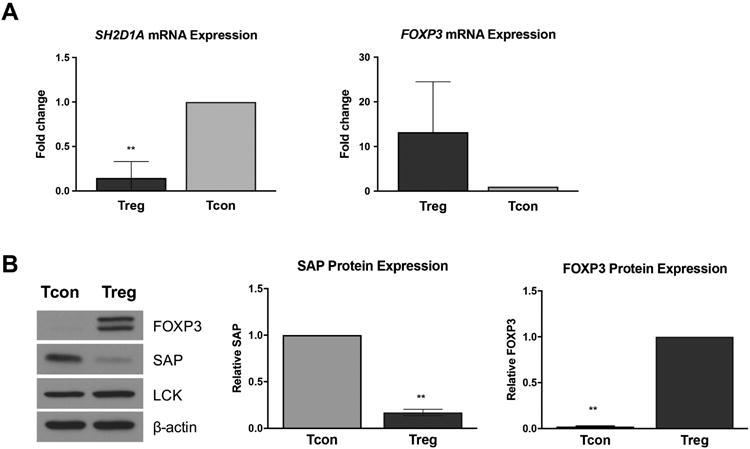
(A) RNA was extracted from activated cells, and qRT-PCR was performed with primers specific for FOXP3 and SAP mRNA. RPL30 was used as a housekeeping gene for normalization. Data are average ± SD of 3 independent experiments; significance was assessed by unpaired t tests. (B) Protein lysates were subjected to SDS-PAGE and Western blot analysis for FOXP3, SAP, and LCK. β-actin was used as a loading control. One representative blot is shown (left). Results from 3 independent experiments were quantified by spot densitometry (right) and assessed for significance by two-tailed t tests.
3.2 Expression of FOXP3 suppresses SH2D1A promoter activity
We next tested whether ectopic FOXP3 expression can repress SH2D1A gene promoter activity. In silico analysis of a ∼1000 bp sequence of the proximal SH2D1A promoter revealed at least three putative FOXP3 binding sites [21, 25] (Figure 4A). In order to measure SH2D1A promoter activity +/- FOXP3 expression, we employed a luciferase reporter system in Jurkat T cells. Both wild-type (WT) and a loss-of-function mutant of FOXP3 (R397W, defective in DNA binding [27]) were cloned into the pmax expression vector; GFP served as a negative control. The ∼1000 bp fragment of the proximal SH2D1A promoter was cloned into the pGL3 firefly luciferase vector to create a reporter plasmid of SH2D1A promoter activity (∼1000bp), with a smaller ∼100 bp promoter fragment with minimal activity serving as a negative control. As predicted, the ∼1000 bp promoter sequence displayed ∼3-fold higher activity than the minimal ∼100 bp sequence (Figure 4B). With concomitant transfection of GFP or FOXP3 expression plasmids, we found that robust transient expression of WT FOXP3 substantially reduced luciferase expression driven by the 1000bp SH2D1A promoter (Figure 4B). In contrast, FOXP3 R397W failed to repress SAP promoter activity despite comparable overexpression (Figure 4B).
Figure 4. FOXP3 represses SH2D1A promoter activity.
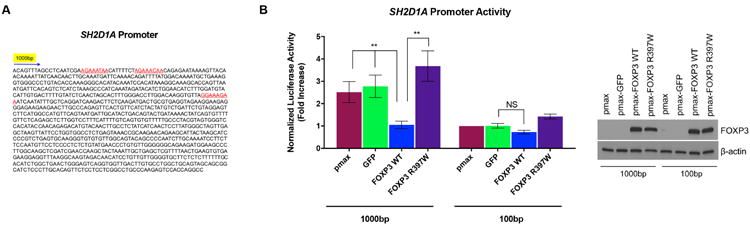
(A) Sequence of the proximal SH2D1A promoter (∼1000 bp) cloned into the pGL3 luciferase vector. Putative FOXP3 binding sites are highlighted in red. (B) Jurkat T cells were triple transfected with pmax-GFP or FOXP3 expression vectors, firefly luciferase SH2D1A promoter constructs, (1000bp vs. 100bp), and a renilla luciferase control plasmid. Promoter activity was measured using a dual luciferase assay at 24 hours post-transfection. Luciferase activity (firefly/renilla ratio) was normalized to pmax empty vector control cells (left). Expression of FOXP3 was confirmed by Western blot analysis (right); β-actin served as a loading control. Data are average ± SD of 3 independent experiments. Significance was determined by ordinary one-way ANOVA with Tukey's multiple comparisons test.
3.3 FOXP3 directly binds to the SH2D1A promoter
The aforementioned results strongly suggested that FOXP3 can directly suppress SH2D1A promoter activity through its DNA binding function. To assay for direct FOXP3 binding to the SH2D1A gene, we first derived Jurkat T cells stably expressing FOXP3, as opposed to the transient overexpression system described above. Lentiviruses encoding YFP-2A-FOXP3 (allowing for bicistronic expression) or GFP [26] were used to transduce Jurkat T cells. Robust expression of GFP/YFP was maintained over several weeks in culture, indicating stable integration (Figure 5A). FOXP3 protein expression was also readily detected by Western blot (Figure 5B), although at reduced levels compared to transient Jurkat transfections (Figure 4C). As expected, endogenous SAP expression was reduced in Jurkat T cells that stably expressed FOXP3 compared to the GFP control (Figure 5B).
Figure 5. FOXP3 binds directly to the SH2D1A promoter.
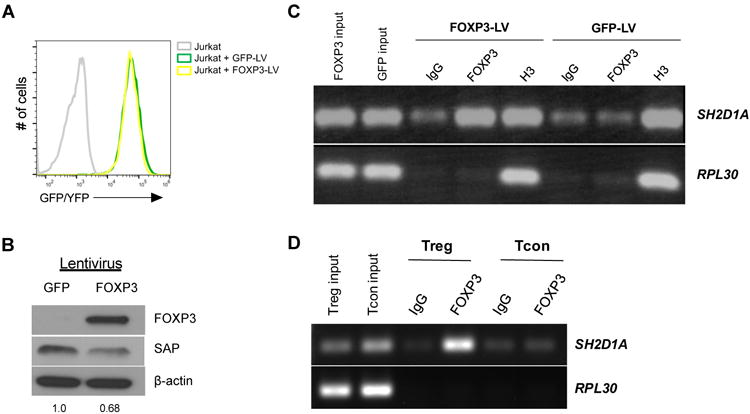
(A) Jurkat T cells were transduced with GFP or YFP-2A-FOXP3 lentiviral (LV) vectors; stable expression was monitored over time by GFP/YFP fluorescence. (B) FOXP3 and SAP expression was measured in LV-transduced Jurkat T cell lysates by Western blot. Numbers represent quantitation of SAP expression by spot densitometry, normalized to LV-GFP transduced cells. (C) LV-transduced Jurkat T cells were lysed for chIP using antibodies specific for FOXP3, histone H3, or an isotype IgG control. Immunoprecipitated DNA was then subjected to PCR using primers specific for the SH2D1A promoter, or the RPL30 housekeeping gene. Results are representative of 3 independent experiments. (D) ChIP analysis was performed on activated human Tregs and Tcons using FOXP3 or IgG isotype antibodies. Immunoprecipitated DNA was then subjected to PCR using primers specific for the SH2D1A promoter, or the RPL30 housekeeping gene. Data are representative of 2 independent experiments.
To provide direct evidence of FOXP3 binding to the SH2D1A promoter, we lysed Jurkat T cells stably transduced with FOXP3 or GFP and performed chromatin immunoprecipitations (chIP) using an anti-FOXP3 or isotype control antibody. Indeed, we detected an enrichment of SH2D1A promoter DNA in FOXP3 IPs relative to an IgG isotype control IP (Figure 5C). Importantly, Jurkat cells expressing GFP alone did not show the same amplification of the SH2D1A promoter region after FOXP3 IP. Immunoprecipitation with an anti-histone H3 antibody reliably detected both SH2D1A and the housekeeping gene RPL30, which confirmed that our ChIP procedure was efficient and selective based on the IP Ab used.
To confirm these findings, lysates derived from activated, primary human Tregs and Tcons were also subjected to chIP with FOXP3 and IgG Abs and analyzed for SH2D1A promoter binding. As expected, activated Tregs showed enrichment of SH2D1A promoter DNA in FOXP3 IPs, whereas Tcons did not (Figure 5D). Taken together, these results demonstrate that FOXP3 can directly bind to the SH2D1A promoter, where it is readily detected in activated human Tregs.
3.4 FOXP3-mediated RICD resistance in Tregs is dependent on repression of SAP expression
To further explore the mechanism of FOXP3-dependent RICD resistance in human Tregs, we next investigated whether FOXP3-mediated repression of SAP is required for RICD resistance. Freshly isolated Tregs were transfected with non-specific (NS), FOXP3-specific, or FOXP3- plus SAP-specific siRNA and stimulated in vitro. Knockdown of FOXP3 expression sensitized Tregs to RICD, confirming previous reports [19]. However, concomitant knockdown of SAP completely blocked the increase in RICD afforded by FOXP3 knockdown (Figure 6A). Reduced target protein expression was confirmed for each siRNA transfection by Western blot analysis, and also revealed a modest but significant increase in SAP expression when FOXP3 was silenced in Tregs (Figure 6B-C).
Figure 6. FOXP3 confers RICD resistance in Tregs by a SAP-dependent mechanism.
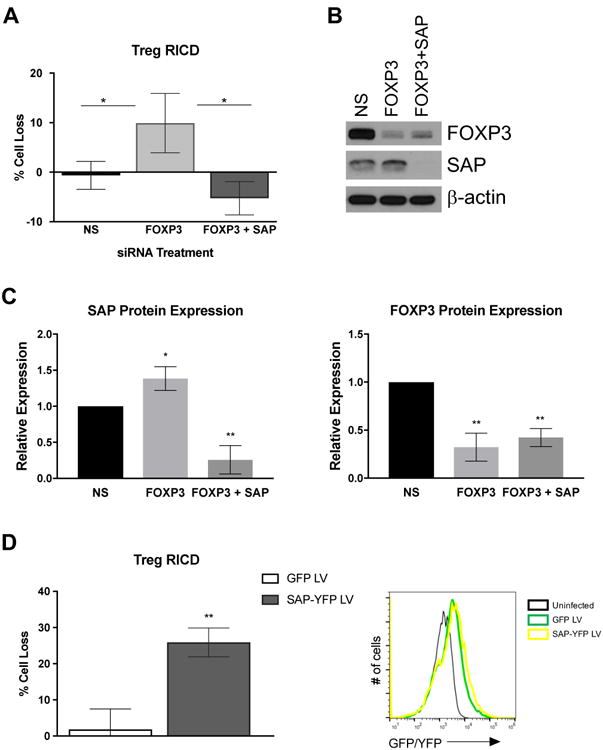
(A) Activated Tregs were transfected with non-specific (NS), FOXP3-specific, or SAP-specific siRNAs. At 4 days post-transfection, cells were restimulated with OKT3 antibody (1 μg/ml) plus Protein A (2 μg/ml) for 24 hours in triplicate. Cell death was measured by flow cytometry using TO-PRO-3 live/dead staining. Data are average ± SD of 3 independent experiments using separate donors. Statistical significance was assessed by one-way ANOVA with Tukey's multiple comparisons test. (B) Changes in FOXP3 and/or SAP expression in siRNA-transfected cells from (A) were assessed by Western blot. β-actin served as a loading control. Data from 4 independent experiments were quantified and assessed for statistical significance by one-way ANOVA with Tukey's multiple comparisons test (right). (C) Activated Tregs were transduced with GFP or YFP-2A-SAP LVs during activation-induced expansion. Cells were then restimulated 24 hours later with OKT3 (1 μg/ml) plus Protein A for an additional 24 hours. Cell loss was determined by TO-PRO-3 live/dead staining within GFP/YFP+ gated cell populations. Data are average ± SD of 2 independent experiments. Transduction efficiency of Tregs was assessed 24 hours post infection by flow cytometry compared to uninfected control cells (right).
If silencing FOXP3 renders Tregs susceptible to RICD by boosting SAP expression, we reasoned that increasing SAP expression alone should also restore RICD sensitivity. To this end, we infected primary activated Tregs with lentiviral vectors expressing YFP-2A-SAP or GFP. At 24 hours post-infection, we restimulated cells with OKT3 and assayed death in the GFP/YFP+ gated population of transduced cells. Ectopic SAP expression dramatically restored RICD sensitivity in YFP+ transduced Tregs, relative to cells transduced with GFP alone (Figure 6D). These results definitively show that RICD resistance in human Tregs is specifically dependent on FOXP3-mediated repression of SAP expression. Furthermore, ectopic expression of SAP alone (using a FOXP3-irrepressible promoter) is sufficient to sensitize activated Tregs to RICD.
4. Discussion
A sufficient CD4+ Treg population must be preserved in order to stave off autoimmunity. A better understanding of apoptosis sensitivity in Tregs is therefore vital to comprehend mechanisms of immune homeostasis and tolerance. Tregs are CD25hi, consume high amounts of IL-2, and experience frequent stimulation from self-antigens [19, 20]. Whereas IL-2 consumption and TCR restimulation would typically induce RICD in conventional effector T cells, Tregs have adopted unique strategies to evade apoptosis and persist under these conditions [30]. In fact, Treg survival and function is dependent on IL-2 consumption for induction of FOXP3 mRNA expression and upregulation of the pro-survival protein MCL-1 to combat pro-apoptotic BIM expression [31]. Surprisingly, Tregs are able to persist despite experiencing even stronger TCR signals in the periphery [32]. We demonstrate here that FOXP3-dependent suppression of SAP renders Tregs resistant to RICD. Although protecting Tregs under these conditions is important for maintaining tolerance, these adaptations to evade RICD could also explain why Tregs are able to persist in the antigen-rich tumor microenvironment, which is likely detrimental to the anti-tumor response [33-35].
Work from our group and others has illuminated a central role for SAP/SLAM receptor signaling in governing RICD sensitivity in T cells [10, 11, 13]. Our results further underscore the importance of SAP-driven signaling in this process, demonstrating that Tregs have evolved to disable this signaling pathway via FOXP3-dependent SAP repression to resist RICD. We posit that other T cell subsets might manipulate SAP-SLAM receptor signaling to elude RICD (e.g. Th17 cells [36]) or adjust RICD sensitivity at distinct phases of the effector T cell response. For example, effector cells that progress into long-lived memory T cell pools could be shaped in part by SAP-SLAM receptor signaling alterations that promote their persistence. After all, dynamic changes in this pathway shape RICD sensitivity at different phases of the conventional effector T cell response; SAP expression is initially kept at low levels after T cell activation, then gradually increases as effector T cells fully differentiate and become more sensitive to RICD [37, 38]. Moreover, transient induction of FOXP3 expression after activation of human Tcons may help preserve these cells from premature RICD during the early clonal expansion phase [39]. However, further work is required to determine whether FOXP3 can repress SAP expression in this context.
Our findings confirm previous work showing FOXP3 is necessary for maintaining RICD resistance in Tregs [19, 20]. Interestingly, ectopic SAP expression was sufficient to sensitize Tregs to RICD, suggesting SAP can override any direct FOXP3-mediated repression of FASL itself to promote apoptosis. On the other hand, ectopic FOXP3 expression alone was not sufficient to significantly reduce SAP expression in Tcons and confer resistance to RICD (data not shown). Indeed, only considerable overexpression of FOXP3 repressed SH2D1A promoter activity in our reporter assays, and reduced endogenous SAP expression by ∼30% in stably-transduced Jurkat T cells. We speculate that additional binding partners may be required to stabilize and/or cooperate with FOXP3 to suppress SAP expression, similar to other FOXP3 target genes [40, 41]. This idea is also consistent with studies that indicate FOXP3 expression alone is not enough to convert Tcons to Tregs with suppressive function, unless expression levels are artificially high [42, 43]. There could also be competition among other transcription factors in Tcons that is not relevant in Tregs. High levels of retrovirally-introduced FOXP3 in Tcons are necessary to efficiently out-compete endogenous AP-1 for NFAT binding and repression of several activation-induced genes [42]. Similarly, a Treg-specific co-factor may cooperate or synergize with FOXP3 to repress SAP expression by interfering with Ets-1/Ets-2 binding or transactivation at the SH2D1A promoter [25]. Finally, additional exogenous factors may also aid in FOXP3-dependent SAP suppression. For example, Bhaskaran and colleagues showed that TGF-β1 promotes RICD resistance in FOXP3+ Tregs, perhaps via increased expression of cFLIP [44].
In conclusion, new mechanistic knowledge regarding apoptosis resistance in Tregs could be utilized to manipulate immune responses by changing Treg apoptosis sensitivity. Tuning T cell apoptosis sensitivity to shape immune responses has great potential in attenuating autoimmunity and/or enhancing eradication of pathogens and tumors [4]. However, this approach also requires a fuller understanding of how T cell subsets could be differentially impacted. In addition, adoptive Treg cell therapies could be improved by maintaining and/or enhancing RICD resistance. Adoptive transfer of infused Tregs has been used in human clinical trials to help prevent graft versus host disease (GVHD) in transplant recipients [45]. Although GVHD rates declined, no infused Tregs were detected in the blood after 14 days, which likely suggests many of the infused Tregs died by apoptosis. Strategies that specifically stabilize FOXP3-dependent repression of the SH2D1A promoter could improve clinical usefulness of Tregs by promoting their longevity in vivo. Stabilizing RICD resistance could also preserve infused FOXP3-transduced “regulatory” T cells from patients with immune-dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, which lack natural Tregs due to debilitating FOXP3 mutations [46]. In conclusion, our findings provide new insight into the puzzling TCR-mediated apoptosis resistance demonstrated by CD4+ Tregs, highlighting the importance of SAP suppression in this phenotype.
Highlights (Katz & Voss et al.).
FOXP3+ regulatory T cells (Tregs) are resistant to restimulation-induced cell death.
Human Tregs express much less SAP compared to conventional T cells.
FOXP3 binds to and suppresses the SH2D1A (SAP) promoter in human Tregs.
RICD resistance in regulatory T cells is dependent on FOXP3-dependent repression of SAP.
Ectopic SAP expression is sufficient to restore RICD sensitivity in regulatory T cells.
Acknowledgments
We thank Dr. Michael Lenardo for providing access to anonymous blood donor samples from the National Institutes of Health Blood Bank, and Kateryna Lund for flow cytometry assistance.
Funding: This work was supported by the National Institute of General Medical Sciences, NIH [1R01GM105821, A.L.S.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Strasser A, Pellegrini M. T-lymphocyte death during shutdown of an immune response. Trends Immunol. 2004;25(11):610–5. doi: 10.1016/j.it.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunological reviews. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 3.Boehme SA, Lenardo MJ. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol. 1993;23(7):1552–60. doi: 10.1002/eji.1830230724. [DOI] [PubMed] [Google Scholar]
- 4.Snow AL, et al. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol Rev. 2010;236:68–82. doi: 10.1111/j.1600-065X.2010.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353(6347):858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 6.Larsen SE, et al. Sensitivity to Restimulation-Induced Cell Death Is Linked to Glycolytic Metabolism in Human T Cells. J Immunol. 2017;198(1):147–155. doi: 10.4049/jimmunol.1601218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruffo E, et al. Inhibition of diacylglycerol kinase alpha restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci Transl Med. 2016;8(321):321ra7. doi: 10.1126/scitranslmed.aad1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol. 2014;34(7):772–9. doi: 10.1007/s10875-014-0083-7. [DOI] [PubMed] [Google Scholar]
- 9.Snow AL, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest. 2009;119(10):2976–89. doi: 10.1172/JCI39518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen G, et al. Increased proliferation of CD8+ T cells in SAP-deficient mice is associated with impaired activation-induced cell death. Eur J Immunol. 2007;37(3):663–74. doi: 10.1002/eji.200636417. [DOI] [PubMed] [Google Scholar]
- 11.Katz G, et al. SAP facilitates recruitment and activation of LCK at NTB-A receptors during restimulation-induced cell death. J Immunol. 2014;192(9):4202–9. doi: 10.4049/jimmunol.1303070. [DOI] [PubMed] [Google Scholar]
- 12.Zhao F, et al. Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity. 2012;36(6):1003–16. doi: 10.1016/j.immuni.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez Del Pino RE, et al. Restimulation-induced T-cell death through NTB-A/SAP signaling pathway is impaired in tuberculosis patients with depressed immune responses. Immunol Cell Biol. 2017;95(8):716–728. doi: 10.1038/icb.2017.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldanzi G, et al. SAP-mediated inhibition of diacylglycerol kinase alpha regulates TCR-induced diacylglycerol signaling. J Immunol. 2011;187(11):5941–51. doi: 10.4049/jimmunol.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res. 2016;4(9):721–5. doi: 10.1158/2326-6066.CIR-16-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XF. Factors regulating apoptosis and homeostasis of CD4+ CD25(high) FOXP3+ regulatory T cells are new therapeutic targets. Front Biosci. 2008;13:1472–99. doi: 10.2741/2775. [DOI] [PubMed] [Google Scholar]
- 17.Pereira LMS, et al. Regulatory T Cell and Forkhead Box Protein 3 as Modulators of Immune Homeostasis. Front Immunol. 2017;8:605. doi: 10.3389/fimmu.2017.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen CR, Yang WC, Chen HW. The fate of regulatory T cells: survival or apoptosis. Cell Mol Immunol. 2014;11(1):11–3. doi: 10.1038/cmi.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss EM, et al. Foxp3-mediated suppression of CD95L expression confers resistance to activation-induced cell death in regulatory T cells. J Immunol. 2011;187(4):1684–91. doi: 10.4049/jimmunol.1002321. [DOI] [PubMed] [Google Scholar]
- 20.Fritzsching B, et al. In contrast to effector T cells, CD4+CD25+FoxP3+ regulatory T cells are highly susceptible to CD95 ligand- but not to TCR-mediated cell death. J Immunol. 2005;175(1):32–6. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- 21.Sadlon TJ, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol. 2010;185(2):1071–81. doi: 10.4049/jimmunol.1000082. [DOI] [PubMed] [Google Scholar]
- 22.Seddiki N, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YC, et al. Oligodeoxynucleotides stabilize Helios-expressing Fox p3+ human T regulatory cells during in vitro expansion. Blood. 2012;119(12):2810–8. doi: 10.1182/blood-2011-09-377895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz G, Snow AL. Fluorescence-activated cell sorting-based quantitation of T cell receptor restimulation-induced cell death in activated, primary human T cells. Methods Mol Biol. 2013;979:15–23. doi: 10.1007/978-1-62703-290-2_2. [DOI] [PubMed] [Google Scholar]
- 25.Okamoto S, et al. Expression of the SH2D1A gene is regulated by a combination of transcriptional and post-transcriptional mechanisms. Eur J Immunol. 2004;34(11):3176–86. doi: 10.1002/eji.200324755. [DOI] [PubMed] [Google Scholar]
- 26.Basu S, et al. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. 2008;180(9):5794–8. doi: 10.4049/jimmunol.180.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee-Basu S, Baxevanis AD. Structural analysis of disease-causing mutations in the P-subfamily of forkhead transcription factors. Proteins. 2004;54(4):639–47. doi: 10.1002/prot.10621. [DOI] [PubMed] [Google Scholar]
- 28.Russell JH, et al. Receptor-stimulated death pathway is opened by antigen in mature T cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(6):2151–2155. doi: 10.1073/pnas.88.6.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wesselborg S, Kabelitz D. Activation-driven death of human T cell clones: time course kinetics of the induction of cell shrinkage, DNA fragmentation, and cell death. Activation-driven death of human T cell clones: time course kinetics of the induction of cell shrinkage, DNA fragmentation, and cell death. 1993 doi: 10.1006/cimm.1993.1106. [DOI] [PubMed] [Google Scholar]
- 30.Yolcu ES, et al. Apoptosis as a mechanism of T-regulatory cell homeostasis and suppression. Immunol Cell Biol. 2008;86(8):650–8. doi: 10.1038/icb.2008.62. [DOI] [PubMed] [Google Scholar]
- 31.Pierson W, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(9):959–65. doi: 10.1038/ni.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran AE, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208(6):1279–89. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Rezende LC, et al. Regulatory T cell as a target for cancer therapy. Arch Immunol Ther Exp (Warsz) 2010;58(3):179–90. doi: 10.1007/s00005-010-0075-0. [DOI] [PubMed] [Google Scholar]
- 34.Mougiakakos D, et al. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127(4):759–67. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 36.Cencioni MT, et al. FAS-ligand regulates differential activation-induced cell death of human T-helper 1 and 17 cells in healthy donors and multiple sclerosis patients. Cell Death Dis. 2015;6:e1785. doi: 10.1038/cddis.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehrle S, et al. SAP and SLAM expression in anti-CD3 activated lymphocytes correlates with cytotoxic activity. Immunol Cell Biol. 2005;83(1):33–9. doi: 10.1111/j.1440-1711.2004.01302.x. [DOI] [PubMed] [Google Scholar]
- 38.Shinozaki K, et al. Activation-dependent T cell expression of the X-linked lymphoproliferative disease gene product SLAM-associated protein and its assessment for patient detection. Int Immunol. 2002;14(10):1215–23. doi: 10.1093/intimm/dxf084. [DOI] [PubMed] [Google Scholar]
- 39.McMurchy AN, et al. A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood. 2013;121(8):1265–75. doi: 10.1182/blood-2012-05-431023. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 41.Rudra D, d P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, Rudensky AY. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13(10):1010–1019. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aarts-Riemens T, et al. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(-) cells. Eur J Immunol. 2008;38(5):1381–90. doi: 10.1002/eji.200737590. [DOI] [PubMed] [Google Scholar]
- 43.Kim HJ, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. 2015;350(6258):334–9. doi: 10.1126/science.aad0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhaskaran N, et al. Transforming growth factor-beta1 sustains the survival of Foxp3(+) regulatory cells during late phase of oropharyngeal candidiasis infection. Mucosal Immunol. 2016;9(4):1015–26. doi: 10.1038/mi.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parmar S, Shpall EJ. Treg adoptive therapy: is more better? Blood. 2016;127(8):962–3. doi: 10.1182/blood-2015-12-682492. [DOI] [PubMed] [Google Scholar]
- 46.Passerini L, Bacchetta R. Forkhead-Box-P3 Gene Transfer in Human CD4+ T Conventional Cells for the Generation of Stable and Efficient Regulatory T Cells, Suitable for Immune Modulatory Therapy. Front Immunol. 2017;8:1282. doi: 10.3389/fimmu.2017.01282. [DOI] [PMC free article] [PubMed] [Google Scholar]