

Abstract
AIM
The impact of mild drinking habit (less than 20 g/d of ethanol) on the clinical course of non-alcoholic fatty liver disease (NAFLD) has not been determined. We examined the influence of a mild drinking habit on liver carcinogenesis from NAFLD.
METHODS
A total of 301 patients who had been diagnosed as having NAFLD by liver biopsy between 2003 and 2016 [median age: 56 years, 45% male, 56% with non-alcoholic steatohepatitis, 26% with advanced fibrosis (F3-4)] were divided into the mild drinking group with ethanol consumption of less than 20 g/d (mild drinking group, n = 93) and the non-drinking group (n = 208). Clinicopathological features at the time of liver biopsy and factors related to hepatocellular carcinoma (HCC) occurrence were compared between the groups.
RESULTS
We observed significant differences in male prevalence (P = 0.01), platelet count (P = 0.04), and gamma-glutamyl transpeptidase (P = 0.02) between the test groups. Over 6 years of observation, the HCC appearance rate was significantly higher in the mild drinking group (6.5% vs 1.4%, P = 0.02). Multivariate survival analysis using Cox’s regression model revealed that hepatic advanced fibrosis (F3-4) (P < 0.01, risk ratio: 11.60), diabetes mellitus (P < 0.01, risk ratio: 89.50), and serum triglyceride (P = 0.04, risk ratio: 0.98) were factors significantly related to HCC in all NAFLD patients, while the effect of a drinking habit was marginal (P = 0.07, risk ratio: 4.43). In patients with advanced fibrosis (F3-4), however, a drinking habit (P = 0.04, risk ratio: 4.83), alpha-fetoprotein (P = 0.01, risk ratio: 1.23), and diabetes mellitus (P = 0.03, risk ratio: 12.00) were identified as significant contributors to HCC occurrence.
CONCLUSION
A mild drinking habit appears to be a risk factor for hepatocarcinogenesis in NAFLD patients, especially those with advanced fibrosis.
Keywords: Non-alcoholic fatty liver disease, Ethanol, Hepatocellular carcinoma, Risk factor
Core tip: This study focused on the impact of a mild drinking habit on liver carcinogenesis in 301 biopsy-proven non-alcoholic fatty liver disease (NAFLD) patients. Multivariate analysis revealed that mild drinking of < 20 g/d might increase the risk of hepatocellular carcinoma in NAFLD patients, particularly those with advanced fibrosis (F3-4). NAFLD patients with severe fibrosis should abstain from even small amounts of regular alcohol consumption.
INTRODUCTION
Over the past several decades, it has become clear that non-alcoholic fatty liver disease (NAFLD) and its advanced form non-alcoholic steatohepatitis (NASH), are major chronic liver diseases worldwide[1,2]. The prevalence rate of NAFLD has doubled during the last 20 years, while those of other chronic liver conditions have remained stable or even decreased[3]. Recent evidence has confirmed that NAFLD and NASH incidence is increasing in Japan as well[4].
The etiology of NAFLD/NASH has not been fully elucidated. The most widely accepted theory involves insulin resistance as an important mechanism leading to liver steatosis and perhaps steatohepatitis[5]. Others have proposed that “multiple hits” of additional oxidative stress and lipotoxicity are necessary for the necro-inflammatory component of steatohepatitis and carcinogenesis[6]. Liver iron, leptin, anti-oxidant deficiency, and intestinal microbiota have also been suggested as potential factors in the progression from steatosis to steatohepatitis[6-9]. However, research on NAFLD/NASH pathogenesis and carcinogenesis is ongoing.
Although it is widely accepted that more than 60 g/d of ethanol consumption may lead to alcoholic liver disease and steatosis, steatohepatitis, and hepatic fibrosis[10], there is in uncertainty on the influence of a mild drinking habit (< 20 g/d of ethanol) on human health. For example, mild habitual drinking improved insulin resistance and hepatic steatosis but either worsened or improved hepatic fibrosis[11-14]. Since NAFLD/NASH is defined as fatty liver disease with average ethanol intake of less than 20 g daily[10], some NAFLD patients may habitually consume small amounts of ethanol while others abstain completely. There are no reports to date investigating the influence of a mild drinking habit on NAFLD/NASH patients despite a growing number of reports on hepatocellular carcinoma (HCC). To investigate the influence of a mild drinking habit on liver carcinogenesis from NAFLD, we compared clinicopathological features and outcomes between NAFLD patients with a mild drinking habit and the non-drinking NAFLD patients.
MATERIALS AND METHODS
Ethics
This study was carried out in accordance with the World Medical Association Helsinki Declaration and was approved by the ethics committee of Shinshu University School of Medicine (approval ID: 2802).
Patients
We enrolled 301 patients who were diagnosed as having NAFLD by liver biopsy between 2003 and 2016 [median age: 56 years, 45% male, 56% with NASH, 26% with advanced fibrosis (F3-4)] at Shinshu University Hospital in Matsumoto, Nagano, Japan. These patients originally referred to our department from local hospitals in Nagano prefecture to confirm the diagnosis by liver biopsy.
The diagnosis of NAFLD was based on the criteria of: (1) the presence of hepato-renal contrast and increased hepatic echogenicity on abdominal ultrasonography (US), (2) an average daily consumption of < 20 g of ethanol, and (3) the absence of other causes of liver dysfunction, such as viral hepatitis, drug-induced liver injury, autoimmune liver diseases, primary sclerosing cholangitis, Wilson’s disease, hereditary hemochromatosis, and citrin deficiency[15,16]. The diagnosis of NAFLD was confirmed based on histological findings of biopsied specimens. Pathology details are described below.
All patients were followed by US or computed tomography with measurements of serum alpha-fetoprotein every 6 mo. HCC was identified radiologically in all affected patients (n = 9). The radiological diagnosis of HCC was based on the American Association for the Study of Liver Diseases practice guidelines on the management of HCC as either: (1) the presence of a hepatic lesion > 2 cm in diameter with typical vascular pattern for HCC on one dynamic imaging technique or alpha-fetoprotein > 200 ng/mL; or (2) the presence of a lesion 1-2 cm in diameter with typical vascular pattern for HCC on two dynamic imaging techniques[17]. Follow-up time was defined as the number of days from biopsy to HCC diagnosis or from biopsy to the last follow-up visit when protocol surveillance confirmed no HCC. Patient drinking habits were confirmed as remaining unchanged during follow-up.
Clinical data collection
All laboratory data in a fasting state on the day of liver biopsy were obtained from our medical database. Past and current drinking habit data were collected by self-reported questionnaires and interviews with doctors performing the liver biopsy. We divided the subjects into two groups: the mild drinking group with ethanol consumption of less than 20 g/d (mild drinking group, n = 93) and the non-drinking group (n = 208). Patients were considered to be hypertensive if their systolic/diastolic pressure was > 140/90 mmHg or if they were taking anti-hypertensive drugs[18]. Patients were judged as having hyperlipidemia if their fasting serum levels of cholesterol or triglyceride were ≥ 220 mg/dL or ≥ 150 mg/dL, respectively, or if they were taking lipid-lowering drugs[19]. Patients were considered to be diabetic if they had a fasting glucose level of ≥ 126 mg/dL or hemoglobin A1c (HbA1c) was ≥ 6.5%, or if they were taking insulin or oral hypoglycemic agents[19].
Histological findings
Liver specimens of at least 1.5 cm in length were obtained from segment 5 or 8 using a 14-gauge needle, as described previously, and immediately fixed in 10% neutral formalin[20]. Sections of 4 μm in thickness were stained by means of the hematoxylin and eosin and Azan-Mallory methods. The histological activity of NAFLD was assessed by an independent expert pathologist in a blinded manner according to the NAFLD scoring system proposed by Kleiner et al[21]. Steatosis grade was scored as 0 to 3 by the fat degeneration rate of hepatocytes (< 5%, 5%-33%, 33%-66%, and > 66%, respectively). Lobular inflammation grade was also scored as 0 to 3 by overall assessment of all inflammatory foci (none, < 2 foci/200 × field, 2-4 foci/200 × field, and > 4 foci/200 × field, respectively). Ballooning grade was determined by the number of degenerating hepatocytes as 0 to 2, corresponding to none, few, and many, respectively. NAFLD activity score was the total of steatosis, lobular inflammation, and ballooning scores. Fibrosis was staged as 0 to 4 depending on the degree of fibrosis (F0, none; F1, perisinusoidal or periportal; F2, perisinusoidal and portal/periportal; F3, bridging fibrosis; and F4, cirrhosis)[21].
Statistical analysis
Clinical and histological data were expressed as a number (percentage) or median (range). Chi-square and Mann-Whitney U tests were used for comparisons between the groups. Kaplan-Meier analysis was performed to estimate HCC cumulative incidence from the time of liver biopsy, and plots of cumulative events vs years of follow-up were constructed. Receiver operating characteristic curves were plotted, and optimal cut-off points were determined as the values showing maximum sensitivity plus specificity. In order to assess which factors were associated with the development of HCC after liver biopsy, univariate and multivariate Cox’s proportional hazard regression analysis was employed. Variables revealed as significant by univariate analysis were further tested by multivariate analysis. P < 0.05 was considered to be statistically significant. Data were analyzed using a statistical software package (SPSS for Windows, SPSS Inc., Chicago, IL, United States).
RESULTS
Overall HCC occurrence rate
HCC appeared in 9 subjects (3%) within a median of 6 years of follow-up from liver biopsy. Kaplan-Mayer analysis revealed the HCC occurrence rate in our cohort to be 0.9/2.6/6.0% in 3/5/10 years, respectively (Figure 1).
Figure 1.

Cumulative incidence rate of hepatocellular carcinoma by Kaplan Meier analysis. The horizontal and vertical axes show days from liver biopsy and cumulative incidence rate of hepatocellular carcinoma, respectively.
Comparison of clinicopathological features at the time of biopsy between the mild drinking and non-drinking groups
Comparisons of clinicopathological features at the time of biopsy between the mild drinking and non-drinking groups revealed significant differences for male prevalence (P = 0.01), platelet count (P = 0.04), and gamma-glutamyl transpeptidase (P = 0.02) (Table 1). No differences were observed between the groups for co-existing disease rate, serum albumin, bilirubin, or alpha-fetoprotein, HbA1c, or pathological features, such as grades for steatosis, lobular inflammation, ballooning, or NAFLD activity score (Table 1). The prevalence of liver cirrhosis (F4) was higher in the mild drinking group compared to non-drinking groups (9 vs 8 cases: 10% vs 4%, P = 0.04) (Table 1), while the rate of hepatic advanced fibrosis (F3-4) was not different between the groups. Interestingly, the HCC appearance rate was higher in the mild drinking group (6 vs 3 cases: 6.5% vs 1.4%, P = 0.02) (Figure 2).
Table 1.
Comparison of clinicopathological features at the time of biopsy between mild drinking and non-drinking groups
| Mild drinking group (n = 93) | Non-drinking group (n = 208) | P value | |
| Age (yr) | 55 (19-77) | 56 (10-84) | 0.50 |
| Male | 52 (56) | 84 (40) | 0.01 |
| Body mass index (kg/m2) | 26.5 (18.3-40.0) | 26.2 (17.8-41.0) | 0.53 |
| Co-existing disease | |||
| Diabetes mellitus | 32 (34) | 78 (38) | 0.57 |
| Hypertension | 41 (44) | 81 (39) | 0.44 |
| Hyperlipidemia | 55 (59) | 135 (66) | 0.29 |
| Laboratory data | |||
| Albumin (mg/dL) | 4.5 (3.2-5.4) | 4.5 (3.0-5.2) | 0.48 |
| Total bilirubin (mg/dL) | 0.91 (0.40-2.20) | 0.86 (0.38-2.64) | 0.12 |
| Aspartate aminotransferase (IU/L) | 44 (20-175) | 47 (13-263) | 0.80 |
| Alanine aminotransferase (IU/L) | 68 (22-237) | 67 (13-522) | 0.74 |
| Gamma-glutamyl transpeptidase (IU/L) | 69 (19-400) | 54 (7-544) | 0.02 |
| Cholinesterase (IU/L) | 363 (171-586) | 384 (189-591) | 0.05 |
| Fasting blood sugar (mg/dL) | 106 (84-215) | 108 (77-221) | 0.63 |
| HOMA-IR | 3.4 (1.0-42.6) | 3.3 (0.3-24.5) | 0.58 |
| HbA1c (%) | 5.9 (5.1-9.8) | 6.0 (4.9-12.3) | 0.21 |
| Total cholesterol (mg/dL) | 205 (138-336) | 208 (70-295) | 0.68 |
| Triglyceride (mg/dL) | 115 (42-404) | 133 (32-801) | 0.14 |
| Platelet (×104/μL) | 21.1 (5.3-45.4) | 22.1 (7.2-40.7) | 0.04 |
| Hyaluronic acid (ng/mL) | 43 (12-320) | 49 (9-1611) | 0.57 |
| Type IV collagen 7s (ng/mL) | 4.3 (2-20) | 4.5 (2-11) | 0.93 |
| Alpha-fetoprotein (ng/mL) | 3.3 (0.7-13.5) | 3.0 (0.7-20.3) | 0.41 |
| Pathology | |||
| Steatosis | 0.86 | ||
| 1 | 33 (36) | 64 (31) | |
| 2 | 36 (39) | 90 (43) | |
| 3 | 24 (26) | 54 (26) | |
| Lobular inflammation | 0.14 | ||
| 0 | 5 (5) | 8 (4) | |
| 1 | 47 (51) | 83 (40) | |
| 2 | 37 (40) | 95 (46) | |
| 3 | 4 (4) | 22 (11) | |
| Ballooning | 0.82 | ||
| 0 | 18 (19) | 41 (20) | |
| 1 | 50 (54) | 118 (57) | |
| 2 | 25 (27) | 49 (24) | |
| NAFLD activity score | 0.79 | ||
| 1 | 4 (4) | 5 (2) | |
| 2 | 8 (9) | 16 (8) | |
| 3 | 16 (17) | 34 (16) | |
| 4 | 17 (18) | 32 (15) | |
| 5 | 25 (27) | 62 (30) | |
| 6 | 14 (15) | 36 (17) | |
| 7 | 9 (10) | 18 (9) | |
| 8 | 0 (0) | 5 (2) | |
| Fibrosis | 0.39 | ||
| 0 | 17 (18) | 40 (19) | |
| 1 | 39 (42) | 93 (45) | |
| 2 | 10 (11) | 25 (12) | |
| 3 | 18 (19) | 42 (20) | |
| 4 | 9 (10) | 8 (4) | |
| Fibrosis 3-4 (Advanced fibrosis) | 27 (29) | 50 (24) | 0.36 |
| Fibrosis 4 (Cirrhosis) | 9 (10) | 8 (4) | 0.04 |
Data are expressed as median (range) or n (%). HbA1c: Hemoglobin A1c; HOMA-IR: Homeostasis model assessment for insulin resistance.
Figure 2.

Comparison of incidence rates of hepatocellular carcinoma between mild drinking and non-drinking groups. The vertical axis shows incidence rate (percentage) of hepatocellular carcinoma during follow-up time.
Comparison of clinicopathological features at the time of biopsy between the HCC and non-HCC groups
In comparisons of clinicopathological features at the time of biopsy between HCC and non-HCC patients (Table 2), those with HCC had significantly higher age (P < 0.01), higher prevalence of a drinking habit (P = 0.02), diabetes mellitus (P < 0.01), and hypertension (P < 0.01), higher HbA1c (P = 0.01), type IV collagen 7S (P = 0.03), and alpha-fetoprotein (P < 0.01), and lower albumin (P < 0.01), cholinesterase (P < 0.01), total cholesterol (P = 0.02), triglyceride (P = 0.02), and platelet count (P < 0.01) (Table 2). In pathological findings, the HCC group had a lower steatosis score (P = 0.01) and significantly higher fibrosis stage (P < 0.01) (Table 2).
Table 2.
Comparison of clinicopathological features at the time of biopsy between hepatocellular carcinoma and non- hepatocellular carcinoma groups
| HCC group (n = 9) | Non-HCC group (n = 292) | P value | |
| Age (yr) | 65 (56-84) | 55 (10-81) | < 0.01 |
| Male | 3 (33) | 133 (46) | 0.52 |
| Body mass index (kg/m2) | 26.0 (18.3-28.7) | 26.2 (17.8-41.0) | 0.23 |
| Drinking habit | 6 (67) | 87 (30) | 0.02 |
| Co-existing disease | |||
| Diabetes mellitus | 8 (89) | 102 (35) | < 0.01 |
| Hypertension | 9 (100) | 113 (39) | < 0.01 |
| Hyperlipidemia | 3 (33) | 187 (65) | 0.06 |
| Laboratory data | |||
| Albumin (mg/dL) | 4.0 (3.7-4.6) | 4.5 (3.0-5.4) | < 0.01 |
| Total bilirubin (mg/dL) | 0.91 (0.44-1.61) | 0.88 (0.38-2.64) | 0.75 |
| Aspartate aminotransferase (IU/L) | 52 (32-95) | 46 (13-263) | 0.71 |
| Alanine aminotransferase (IU/L) | 53 (16-132) | 68 (13-522) | 0.11 |
| Gamma-glutamyl transpeptidase (IU/L) | 65 (28-192) | 56 (7-544) | 0.48 |
| Cholinesterase (IU/L) | 249 (190-434) | 380 (171-591) | < 0.01 |
| Fasting blood sugar (mg/dL) | 133 (84-172) | 106 (77-221) | 0.13 |
| HOMA-IR | 5.8 (1.2-9.8) | 3.3 (0.3-42.6) | 0.18 |
| HbA1c (%) | 6.6 (6.0-7.0) | 5.9 (4.9-12.3) | 0.01 |
| Total cholesterol (mg/dL) | 176 (153-264) | 208 (70-336) | 0.02 |
| Triglyceride (mg/dL) | 85 (64-140) | 130 (32-801) | 0.02 |
| Platelet (×104/μL) | 11.0 (6.4-18.6) | 22.0 (5.3-45.4) | < 0.01 |
| Hyaluronic acid (ng/mL) | 42 (17-263) | 42 (9-1180) | 0.96 |
| Type IV collagen 7s (ng/mL) | 5.6 (4.7-8.4) | 4.4 (2.0-20.0) | 0.03 |
| Alpha-fetoprotein (ng/mL) | 6.0 (3.7-20.3) | 3.0 (0.7-13.2) | < 0.01 |
| Pathology | |||
| Steatosis | 0.01 | ||
| 1 | 7 (78) | 90 (31) | |
| 2 | 2 (22) | 124 (43) | |
| 3 | 0 (0) | 78 (27) | |
| Lobular inflammation | 0.21 | ||
| 0 | 0 (0) | 13 (4) | |
| 1 | 2 (22) | 128 (44) | |
| 2 | 7 (78) | 125 (43) | |
| 3 | 0 (0) | 26 (9) | |
| Ballooning | 0.76 | ||
| 0 | 1 (11) | 58 (20) | |
| 1 | 6 (67) | 162 (56) | |
| 2 | 2 (22) | 72 (25) | |
| NAFLD activity score | 0.53 | ||
| 1 | 0 (0) | 9 (3) | |
| 2 | 0 (0) | 24 (8) | |
| 3 | 3 (33) | 47 (16) | |
| 4 | 2 (22) | 47 (16) | |
| 5 | 4 (44) | 83 (28) | |
| 6 | 0 (0) | 50 (17) | |
| 7 | 0 (0) | 27 (9) | |
| 8 | 0 (0) | 5 (2) | |
| Fibrosis | < 0.01 | ||
| 0 | 0 (0) | 57 (20) | |
| 1 | 0 (0) | 132 (45) | |
| 2 | 0 (0) | 35 (12) | |
| 3 | 4 (44) | 56 (19) | |
| 4 | 5 (56) | 12 (4) |
Data are expressed as median (range) or n (%). HCC: Hepatocellular carcinoma; HOMA-IR: Homeostasis model assessment for insulin resistance; HbA1c: Hemoglobin A1c; NAFLD: Non-alcoholic fatty liver disease.
Kaplan-Meier analysis was carried out for the HCC and non-HCC groups (Figure 3). Factors associated with higher HCC occurrence included age ≥ 63 years at the time of biopsy (P = 0.04, Figure 3A), a mild drinking habit (P < 0.01, Figure 3B), diabetes mellitus (P < 0.01, Figure 3C), hypertension (P < 0.01, Figure 3D), albumin ≤ 4.0 g/dL (P < 0.01, Figure 3E), HbA1c ≥ 6.6% (P = 0.01, Figure 3G), triglyceride < 133 mg/dL (P = 0.02, Figure 3I), platelet count < 13.3 ×104/μL (P < 0.01, Figure 3J), type IV collagen 7S ≥ 5.0 ng/mL (P = 0.01, Figure 3K), alpha-fetoprotein ≥ 6.0 ng/mL (P < 0.01, Figure 3L), steatosis grade 1 (P = 0.01, Figure 3M), and F3-4 (P < 0.01, Figure 3N).
Figure 3.
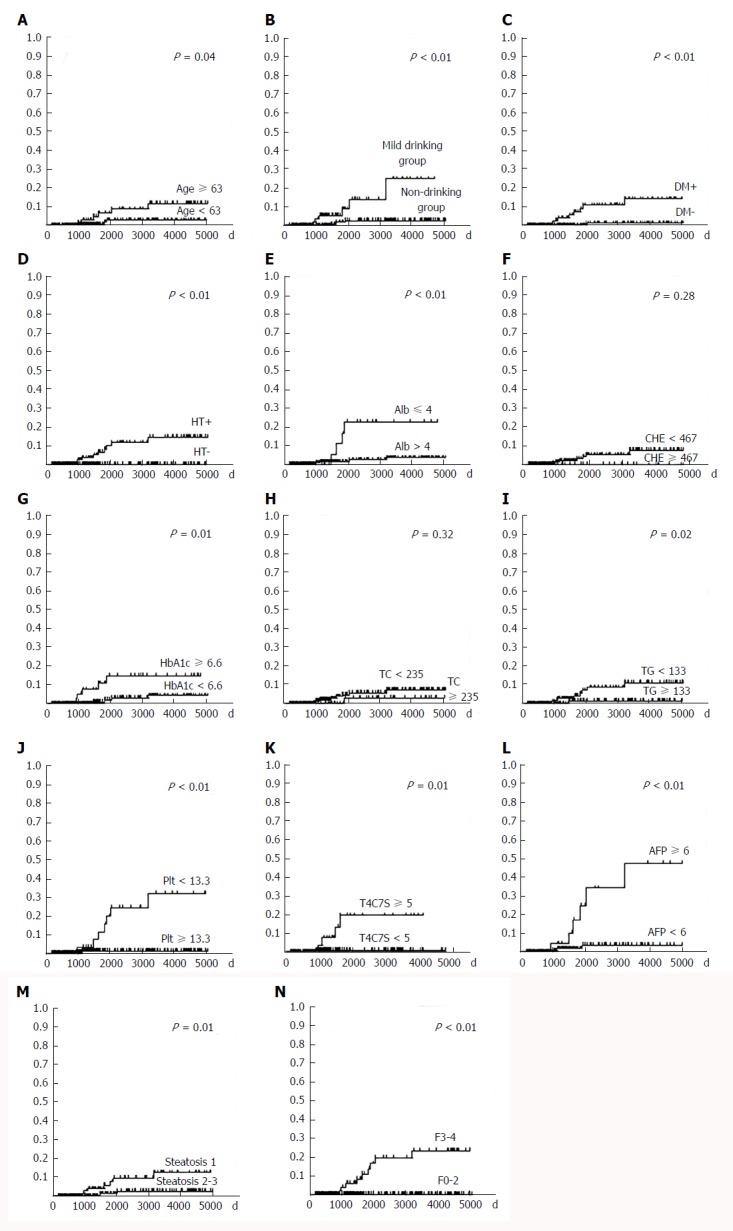
Cumulative incidence rate of hepatocellular carcinoma based on data at the time of liver biopsy. A: Age; B: Drinking habit; C: Diabetes mellitus; D: Hypertension; E: Albumin; F: Cholinesterase; G: HbA1c, H: Total cholesterol; I: Triglyceride; J: Platelet; K: Type IV collagen 7S; L: Alpha-fetoprotein; M: Steatosis; N: Fibrosis. The horizontal and vertical axes show days from liver biopsy and cumulative incidence rate of hepatocellular carcinoma, respectively. DM: Diabetes mellitus; HT: Hypertension; Alb: Albumin; CHE: Cholinesterase; TC: Total cholesterol; TG: Triglyceride; Plt: Platelet; T4C7S: Type IV collagen 7S; AFP: Alpha-fetoprotein; F: Fibrosis.
Multivariate survival analysis using Cox’s regression model revealed that hepatic advanced fibrosis (F3-4) (P < 0.01, risk ratio: 11.60), diabetes mellitus (P < 0.01, risk ratio: 89.50), and serum triglyceride (P = 0.04, risk ratio: 0.98) were factors significantly related to HCC, while a mild drinking habit appeared to be marginally related (P = 0.07, risk ratio: 4.43) (Table 3).
Table 3.
Factors related to hepatic carcinogenesis by multivariate survival analysis using Cox's regression model for all patients
| P value | Relative risk | 95%CI | |
| Fibrosis | < 0.01 | 11.6 | 2.36-56.9 |
| Diabetes mellitus | < 0.01 | 89.5 | 6.01-1331.2 |
| Triglyceride | 0.04 | 0.98 | 0.95-0.99 |
| Drinking habit | 0.07 | 4.43 | 0.88-22.4 |
The result of all HCC patients having advanced hepatic fibrosis (F3-4) at the time of biopsy corroborated the close association between HCC and hepatic fibrosis. To elucidate the additional impact of mild drinking on HCC development in the HCC high-risk group, we evaluated the clinicopathological features of the HCC and non-HCC groups in NAFLD patients with advanced fibrosis (Table 4). Compared with the non-HCC group (n = 68), the HCC group (n = 9) had a significantly higher rate of a drinking habit (P = 0.03), diabetes mellitus (P = 0.02), and hypertension (P = 0.01), higher alpha-fetoprotein (P = 0.04), and lower cholinesterase (P = 0.02), triglyceride (P = 0.02), and platelet count (P < 0.01) (Table 4). There were no differences in pathological findings between the groups (Table 4).
Table 4.
Comparison of clinicopathological features at the time of biopsy between hepatocellular carcinoma and non- hepatocellular carcinoma groups in non-alcoholic fatty liver disease patients with advanced fibrosis (F3-4)
| HCC group (n = 9) | Non-HCC group (n = 68) | P value | |
| Age (yr) | 65 (56-84) | 64 (30-81) | 0.32 |
| Male | 3 (33) | 14 (21) | 0.39 |
| Body mass index (kg/m2) | 26.0 (18.3-28.7) | 27.7 (20.1-41.0) | 0.05 |
| Drinking habit | 6 (67) | 21 (31) | 0.03 |
| Co-existing disease | |||
| Diabetes mellitus | 8 (89) | 32 (47) | 0.02 |
| Hypertension | 9 (100) | 38 (56) | 0.01 |
| Hyperlipidemia | 3 (33) | 35 (52) | 0.31 |
| Laboratory data | |||
| Albumin (mg/dL) | 4.0 (3.7-4.6) | 4.3 (3.2-5.0) | 0.07 |
| Total bilirubin (mg/dL) | 0.91 (0.44-1.61) | 0.91 (0.48-2.15) | 0.99 |
| Aspartate aminotransferase (IU/L) | 52 (32-95) | 63 (20-200) | 0.13 |
| Alanine aminotransferase (IU/L) | 53 (16-132) | 71 (19-298) | 0.09 |
| Gamma-glutamyl transpeptidase (IU/L) | 65 (28-192) | 65 (25-205) | 0.75 |
| Cholinesterase (IU/L) | 249 (190-434) | 345 (171-467) | 0.02 |
| Fasting blood sugar (mg/dL) | 133 (84-172) | 110 (82-215) | 0.30 |
| HOMA-IR | 5.8 (1.2-9.8) | 4.3 (1.1-15.6) | 0.54 |
| HbA1c (%) | 6.6 (6.0-7.0) | 6.1 (5.0-10.9) | 0.14 |
| Total cholesterol (mg/dL) | 176 (153-264) | 201 (131-294) | 0.10 |
| Triglyceride (mg/dL) | 85 (64-140) | 119 (42-351) | 0.03 |
| Platelet (×104/μL) | 11.0 (6.4-18.6) | 16.6 (5.3-32.6) | < 0.01 |
| Hyaluronic acid (ng/mL) | 42 (17-263) | 59 (11-1180) | 0.45 |
| Type IV collagen 7s (ng/mL) | 5.6 (4.7-8.4) | 6.8 (3.5-20.0) | 0.61 |
| Alpha-fetoprotein (ng/mL) | 6.0 (3.7-20.3) | 5.0 (1.1-11.3) | 0.04 |
| Pathology | |||
| Steatosis | 0.07 | ||
| 1 | 7 (78) | 26 (38) | |
| 2 | 2 (22) | 32 (47) | |
| 3 | 0 (0) | 10 (15) | |
| Lobular inflammation | 0.45 | ||
| 0 | 0 (0) | 1 (2) | |
| 1 | 2 (22) | 17 (25) | |
| 2 | 7 (78) | 37 (54) | |
| 3 | 0 (0) | 13 (19) | |
| Ballooning | 0.76 | ||
| 0 | 1 (11) | 1 (2) | |
| 1 | 6 (67) | 34 (50) | |
| 2 | 2 (22) | 33 (49) | |
| NAFLD activity score | 0.25 | ||
| 1 | 0 (0) | 1 (2) | |
| 2 | 0 (0) | 1 (2) | |
| 3 | 3 (33) | 5 (7) | |
| 4 | 2 (22) | 10 (15) | |
| 5 | 4 (44) | 26 (38) | |
| 6 | 0 (0) | 16 (24) | |
| 7 | 0 (0) | 8 (12) | |
| 8 | 0 (0) | 1 (2) |
Data are expressed as median (range) or n (%). HCC: Hepatocellular carcinoma; HOMA-IR: Homeostasis model assessment for insulin resistance; HbA1c: Hemoglobin A1c; NAFLD: Non-alcoholic fatty liver disease.
In NAFLD cases with advanced fibrosis, multivariate survival analysis using Cox’s regression model revealed that a mild drinking habit (P = 0.04, risk ratio: 4.83), alpha-fetoprotein (P = 0.01, risk ratio: 1.23), and diabetes mellitus (P = 0.03, risk ratio: 12.00) were factors significantly associated with HCC (Table 5). Accordingly, a mild drinking habit appeared to be a risk factor for hepatocarcinogenesis in NAFLD patients with advanced fibrosis.
Table 5.
Factors related to hepatic carcinogenesis by multivariate survival analysis using Cox's regression model for patients with advanced fibrosis (F3-4)
| P value | Relative risk | 95%CI | |
| Drinking habit | 0.04 | 4.83 | 1.01-23.00 |
| Alpha-fetoprotein | 0.01 | 1.23 | 1.04-1.44 |
| Diabetes mellitus | 0.03 | 12.00 | 1.20-119.66 |
DISCUSSION
Although continuous and excessive ethanol consumption is harmful to the liver, a mild drinking habit reportedly improves insulin sensitivity and decreases cardiovascular mortality in the general population[22]. One question arises on whether mild drinking is similarly beneficial for NAFLD patients, but there are few studies on NAFLD regarding the impact of light ethanol consumption. This study demonstrated that a mild drinking habit may be associated with HCC occurrence in NAFLD patients with advanced fibrosis. We therefore propose the abstinence of ethanol, even in small amounts, in such individuals.
Mild to moderate alcohol consumption has been shown to decrease insulin resistance and improve components of metabolic syndrome[23]. Dunn et al[12] and Kwon et al[13] reported a positive association between moderate alcohol intake and decreased steatosis/ballooning and fibrosis grades in NAFLD patients, which might explain the protective effects of moderate alcohol intake on preventing histological injury. In our cohort, however, the mild and non-drinking groups did not differ with regard to steatosis/ballooning grade and the rate of liver cirrhosis was higher in the mild drinking group. The reason for these discrepancies is unknown, along with why there were no significant reductions in body mass index or homeostasis model assessment for insulin resistance score between our test groups. We presume that ethnic differences may account for differences in ethanol consumption effects on steatosis, ballooning, and fibrosis.
In 2010, Ascha et al[24] described that a mild drinking habit was associated with an increased risk of carcinogenesis in a NASH-associated cirrhosis cohort. Here, we focused on the impact of a mild drinking habit on liver carcinogenesis originating from NAFLD. All HCC-developing patients (n = 9) had advanced fibrosis (F3-4). Among all NAFLD patients, multivariate analysis revealed that fibrosis, diabetes mellitus, and serum triglyceride were factors significantly related to HCC, while a mild drinking habit appeared to be only marginally related to carcinogenesis. On the other hand, in NAFLD cases with F3-4, multivariate survival analysis showed a mild drinking habit, alpha-fetoprotein, and diabetes mellitus to be factors significantly associated with HCC. Our results indicated that mild drinking may increase the risk of HCC in NAFLD patients with not only cirrhosis (F4), but also advanced fibrosis.
The International Agency for Cancer Research (WHO) has certified that alcohol intake is carcinogenic for humans[25]. Indeed, alcohol consumption has been associated with increased risks of head and neck, oral cavity, pharynx, larynx, esophagus, bowel, breast, and liver cancers[25]. Ethanol is metabolized into acetaldehyde by alcohol dehydrogenase and cytochrome P450 2E1 (CYP2E1) in the liver, which is then oxidized to acetate by aldehyde dehydrogenase (ALDH)[26]. Although the underlying causes of cancers related to ethanol consumption are not yet clear, various factors have been proposed as key contributors of hepatocarcinogenesis, including the direct genotoxicity of ethanol and its metabolite acetaldehyde, malnutrition, chronic inflammation, oxidative stress, interactions with retinoids, methylation level alterations, immunological surveillance, and angiogenesis[27]. Ethanol also reduces the levels of glutathione S-transferase, a detoxifier of oxidative stress, and increases the expression of CYP2E1, a generator of oxidative stress[28-32]. The net increases in oxidative stress by long-term ethanol consumption may lead to hepatocarcinogenesis in the presence of steatosis, while it is undetermined which factor is most affecting this oncogenic process[33-36]. The impact of ethanol per hepatocyte might be greater in cirrhotic patients because of the decreases in the number and function of hepatocytes. Actually, Vidal et al[37] reported that ALDH activity was significantly reduced in patients with advanced liver fibrosis compared with those having mild fibrosis. Therefore, we presume that increased acetaldehyde and resultant DNA damage may induce pro-carcinogenic gene mutations and/or epigenetic changes, even with mild drinking, in NAFLD patients with advanced fibrosis.
Based on the results of the present study, mild ethanol consumption should be abandoned for NAFLD patients, especially those with advanced fibrosis, due to the possible risk of liver tumorigenesis. The main limitation of this study was its retrospective design; there remains a need for future large-scale longitudinal studies that evaluate the outcomes of NAFLD patients with mild ethanol intake. Prospective studies investigating the effect of ethanol cession in NAFLD patients with a mild drinking habit are also required to confirm the impact of mild drinking on the clinical course of NAFLD.
In conclusion, in NAFLD patients, especially those with advanced fibrosis, a mild drinking habit is a risk factor for hepatocarcinogenesis that should be discouraged.
ARTICLE HIGHLIGHTS
Research background
The prevalence rate of non-alcoholic fatty liver disease (NAFLD) has doubled during the last 20 years.
Research motivation
The impact of mild drinking habit (less than 20 g/d of ethanol) on the clinical course of NAFLD has not been determined. We examined the influence of a mild drinking habit on liver carcinogenesis from NAFLD.
Research objectives
A total of 301 patients who had been diagnosed as having NAFLD by liver biopsy between 2003 and 2016 (median age: 56 years, 45% male, 56% with non-alcoholic steatohepatitis, 26% with advanced fibrosis (F3-4) were divided into the mild drinking group with ethanol consumption of less than 20 g/d (mild drinking group, n = 93) and the non-drinking group (n = 208).
Research methods
Clinicopathological features at the time of liver biopsy and factors related to hepatocellular carcinoma (HCC) occurrence were compared between the groups.
Research results
We observed significant differences in male prevalence (P = 0.01), platelet count (P = 0.04), and gamma-glutamyl transpeptidase (P = 0.02) between the test groups. Over 6 years of observation, the HCC appearance rate was significantly higher in the mild drinking group (6.5% vs 1.4%, P = 0.02). Multivariate survival analysis using Cox’s regression model revealed that hepatic advanced fibrosis (F3-4) (P < 0.01, risk ratio: 11.60), diabetes mellitus (P < 0.01, risk ratio: 89.50), and serum triglyceride (P = 0.04, risk ratio: 0.98) were factors significantly related to HCC in all NAFLD patients, while the effect of a drinking habit was marginal (P = 0.07, risk ratio: 4.43). In patients with advanced fibrosis (F3-4), however, a drinking habit (P = 0.04, risk ratio: 4.83), alpha-fetoprotein (P = 0.01, risk ratio: 1.23), and diabetes mellitus (P = 0.03, risk ratio: 12.00) were identified as significant contributors to HCC occurrence.
Research conclusions
A mild drinking habit appears to be a risk factor for hepatocarcinogenesis in NAFLD patients, especially those with advanced fibrosis.
Research perspectives
Prospective studies investigating the effect of ethanol cession in NAFLD patients with a mild drinking habit are also required to confirm the impact of mild drinking on the clinical course of NAFLD.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Committee for Medical Ethics of Shinshu University School of Medicine Institutional Review Board.
Informed consent statement: Informed written consent was obtained from all patients.
Conflict-of-interest statement: The authors declare that no conflict of interest exists.
Data sharing statement: No additional data are available.
Peer-review started: January 31, 2018
First decision: February 26, 2015
Article in press: March 10, 2018
P- Reviewer: Peltec A, Sharafi H, Sanal MG S- Editor: Wang XJ L- Editor: A E- Editor: Huang Y
Contributor Information
Takefumi Kimura, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Naoki Tanaka, Department of Metabolic Regulation, Shinshu University Graduate School of Medicine, Matsumoto 390-8621, Japan; Research Center for Agricultural Food Industry, Shinshu University, Matsumoto 390-8621, Japan. naopi@shinshu-u.ac.jp.
Naoyuki Fujimori, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Ayumi Sugiura, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Tomoo Yamazaki, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Satoru Joshita, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Michiharu Komatsu, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Takeji Umemura, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Akihiro Matsumoto, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
Eiji Tanaka, Department of Internal Medicine, Division of Gastroenterology, Shinshu University School of Medicine, Matsumoto 390-8621, Japan.
References
- 1.NCD Risk Factor Collaboration (NCD-RisC) Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet. 2016;387:1377–1396. doi: 10.1016/S0140-6736(16)30054-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 3.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 4.Eguchi Y, Hyogo H, Ono M, Mizuta T, Ono N, Fujimoto K, Chayama K, Saibara T; JSG-NAFLD. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: a multicenter large retrospective study. J Gastroenterol. 2012;47:586–595. doi: 10.1007/s00535-012-0533-z. [DOI] [PubMed] [Google Scholar]
- 5.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 6.Duvnjak M, Lerotić I, Barsić N, Tomasić V, Virović Jukić L, Velagić V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–4550. doi: 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. 2017;179:142–157. doi: 10.1016/j.pharmthera.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka N, Takahashi S, Hu X, Lu Y, Fujimori N, Golla S, Fang ZZ, Aoyama T, Krausz KW, Gonzalez FJ. Growth arrest and DNA damage-inducible 45α protects against nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet. Biochim Biophys Acta. 2017;1863:3170–3182. doi: 10.1016/j.bbadis.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitabatake H, Tanaka N, Fujimori N, Komatsu M, Okubo A, Kakegawa K, Kimura T, Sugiura A, Yamazaki T, Shibata S, et al. Association between endotoxemia and histological features of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23:712–722. doi: 10.3748/wjg.v23.i4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura T, Kobayashi A, Tanaka N, Sano K, Komatsu M, Fujimori N, Yamazaki T, Shibata S, Ichikawa Y, Joshita S, et al. Clinicopathological characteristics of non-B non-C hepatocellular carcinoma without past hepatitis B virus infection. Hepatol Res. 2017;47:405–418. doi: 10.1111/hepr.12762. [DOI] [PubMed] [Google Scholar]
- 11.Bell RA, Mayer-Davis EJ, Martin MA, D’Agostino RB Jr, Haffner SM. Associations between alcohol consumption and insulin sensitivity and cardiovascular disease risk factors: the Insulin Resistance and Atherosclerosis Study. Diabetes Care. 2000;23:1630–1636. doi: 10.2337/diacare.23.11.1630. [DOI] [PubMed] [Google Scholar]
- 12.Dunn W, Sanyal AJ, Brunt EM, Unalp-Arida A, Donohue M, McCullough AJ, Schwimmer JB. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD) J Hepatol. 2012;57:384–391. doi: 10.1016/j.jhep.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon HK, Greenson JK, Conjeevaram HS. Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int. 2014;34:129–135. doi: 10.1111/liv.12230. [DOI] [PubMed] [Google Scholar]
- 14.Ajmera VH, Terrault NA, Harrison SA. Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology. 2017;65:2090–2099. doi: 10.1002/hep.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimori N, Tanaka N, Shibata S, Sano K, Yamazaki T, Sekiguchi T, Kitabatake H, Ichikawa Y, Kimura T, Komatsu M, et al. Controlled attenuation parameter is correlated with actual hepatic fat content in patients with non-alcoholic fatty liver disease with none-to-mild obesity and liver fibrosis. Hepatol Res. 2016;46:1019–1027. doi: 10.1111/hepr.12649. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu M, Kimura T, Yazaki M, Tanaka N, Yang Y, Nakajima T, Horiuchi A, Fang ZZ, Joshita S, Matsumoto A, et al. Steatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARα. Biochim Biophys Acta. 2015;1852:473–481. doi: 10.1016/j.bbadis.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura T, Shinji A, Tanaka N, Koinuma M, Yamaura M, Nagaya T, Joshita S, Komatsu M, Umemura T, Horiuchi A, et al. Association between lower air pressure and the onset of ischemic colitis: a case-control study. Eur J Gastroenterol Hepatol. 2017;29:1071–1078. doi: 10.1097/MEG.0000000000000913. [DOI] [PubMed] [Google Scholar]
- 19.Kimura T, Shinji A, Horiuchi A, Tanaka N, Nagaya T, Shigeno T, Nakamura N, Komatsu M, Umemura T, Arakura N, et al. Clinical characteristics of young-onset ischemic colitis. Dig Dis Sci. 2012;57:1652–1659. doi: 10.1007/s10620-012-2088-5. [DOI] [PubMed] [Google Scholar]
- 20.Umemura T, Joshita S, Sekiguchi T, Usami Y, Shibata S, Kimura T, Komatsu M, Matsumoto A, Ota M, Tanaka E. Serum Wisteria floribunda Agglutinin-Positive Mac-2-Binding Protein Level Predicts Liver Fibrosis and Prognosis in Primary Biliary Cirrhosis. Am J Gastroenterol. 2015;110:857–864. doi: 10.1038/ajg.2015.118. [DOI] [PubMed] [Google Scholar]
- 21.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 22.Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW Jr, Doll R. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. 1997;337:1705–1714. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- 23.Alkerwi A, Boutsen M, Vaillant M, Barre J, Lair ML, Albert A, Guillaume M, Dramaix M. Alcohol consumption and the prevalence of metabolic syndrome: a meta-analysis of observational studies. Atherosclerosis. 2009;204:624–635. doi: 10.1016/j.atherosclerosis.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 24.Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- 25.Boffetta P, Hashibe M. Alcohol and cancer. Lancet Oncol. 2006;7:149–156. doi: 10.1016/S1470-2045(06)70577-0. [DOI] [PubMed] [Google Scholar]
- 26.Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
- 27.Testino G, Leone S, Borro P. Alcohol and hepatocellular carcinoma: a review and a point of view. World J Gastroenterol. 2014;20:15943–15954. doi: 10.3748/wjg.v20.i43.15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsumi T, Suzuki T, Moriya K, Shintani Y, Fujie H, Miyoshi H, Matsuura Y, Koike K, Miyamura T. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology. 2003;38:820–828. doi: 10.1053/jhep.2003.50399. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanbe H, Kamijo Y, Nakajima T, Tanaka N, Sugiyama E, Wang L, Fang ZZ, Hara A, Gonzalez FJ, Aoyama T. Chronic ethanol consumption decreases serum sulfatide levels by suppressing hepatic cerebroside sulfotransferase expression in mice. Arch Toxicol. 2014;88:367–379. doi: 10.1007/s00204-013-1132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okiyama W, Tanaka N, Nakajima T, Tanaka E, Kiyosawa K, Gonzalez FJ, Aoyama T. Polyenephosphatidylcholine prevents alcoholic liver disease in PPARalpha-null mice through attenuation of increases in oxidative stress. J Hepatol. 2009;50:1236–1246. doi: 10.1016/j.jhep.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, Fukushima Y, Peters JM, Gonzalez FJ, Aoyama T. Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology. 2004;40:972–980. doi: 10.1002/hep.20399. [DOI] [PubMed] [Google Scholar]
- 33.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–4370. [PubMed] [Google Scholar]
- 35.Ramadori P, Cubero FJ, Liedtke C, Trautwein C, Nevzorova YA. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers (Basel) 2017;9 doi: 10.3390/cancers9100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambade A, Satishchandran A, Gyongyosi B, Lowe P, Szabo G. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J Gastroenterol. 2016;22:4091–4108. doi: 10.3748/wjg.v22.i16.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vidal F, Toda R, Gutiérrez C, Broch M, Fernández-Muixí F, Lorenzo A, Richart C. Influence of chronic alcohol abuse and liver disease on hepatic aldehyde dehydrogenase activity. Alcohol. 1998;15:3–8. doi: 10.1016/s0741-8329(97)00073-6. [DOI] [PubMed] [Google Scholar]