

Abstract
AIM
To analyze the alterations of fecal microbiota in Chinese patients with inflammatory bowel disease (IBD).
METHODS
Fecal samples from 15 patients with Crohn’s disease (CD) (11 active CD, 4 inactive CD), 14 patients with active ulcerative colitis (UC) and 13 healthy individuals were collected and subjected to 16S ribosomal DNA (rDNA) gene sequencing. The V4 hypervariable regions of 16S rDNA gene were amplified from all samples and sequenced by the Illumina MiSeq platform. Quality control and operational taxonomic units classification of reads were calculated with QIIME software. Alpha diversity and beta diversity were displayed with R software.
RESULTS
Community richness (chao) and microbial structure in both CD and UC were significantly different from those in normal controls. At the phyla level, analysis of the microbial compositions revealed a significantly greater abundance of Proteobacteria in IBD as compared to that in controls. At the genera level, 8 genera in CD and 23 genera in UC (in particular, the Escherichia genus) showed significantly greater abundance as compared to that in normal controls. The relative abundance of Bacteroidetes in the active CD group was markedly lower than that in the inactive CD group. The abundance of Proteobacteria in patients with active CD was nominally higher than that in patients with inactive CD; however, the difference was not statistically significant after correction. Furthermore, the relative abundance of Bacteroidetes showed a negative correlation with the CD activity index scores.
CONCLUSION
Our study profiles specific characteristics and microbial dysbiosis in the gut of Chinese patients with IBD. Bacteroidetes may have a negative impact on inflammatory development.
Keywords: Crohn’s disease, Ulcerative colitis, Chinese, Microbial dysbiosis, 16S ribosomal DNA
Core tip: Intestinal microbiota plays an important role in the pathogenesis of inflammatory bowel disease. However, there are few data on global alteration of microbiota in Chinese patients. In this study, fecal samples were subjected to 16S ribosomal DNA sequencing. Community richness and microbial structure in inflammatory bowel disease were significantly different from those in normal controls. The relative abundance of Bacteroidetes in the active Crohn’s disease group was significantly lower than that in the inactive Crohn’s disease group, and it showed a negative correlation with Crohn’s disease activity index, which indicates that Bacteroidetes may have a negative impact on inflammatory development.
INTRODUCTION
Inflammatory bowel disease (IBD) is characterized by chronic relapsing inflammation of the gastrointestinal tract and includes two main clinical phenotypes: Crohn’s disease (CD) and ulcerative colitis (UC). The etiopathogenesis of IBD is not completely understood. Several disease susceptibility genes, such as NOD2, ATG16L1 and IRGM, have been implicated in its pathogenesis[1]. However, the rapid increase in the incidence of IBD cannot be explained by genetic factors alone; an accumulating body of evidence indicates that environmental factors play a key role in the development of IBD by triggering intestinal microbiota dysbiosis[2].
Currently available data from experimental models and clinical studies suggest that intestinal microbiota plays an important role in the pathogenesis of IBD[3]. The alterations in intestinal microbiota related to IBD include decrease in Bacteroides, Firmicutes, Clostridia, Ruminococcaceae, Bifidobacterium, Lactobacillus, and Faecalibacterium prausnitzii, but increase in Gamma Proteobacteria and presence of Fusobacterium and Escherichia coli, especially adherent-invasive E. coli (AIEC). In addition, IBD is also associated with alterations in the microbial metabolic functions, including decrease of short-chain fatty acids (SCFAs) and amino acid biosynthesis, and increase of auxotrophy, amino acid and sulfate transport, oxidative stress, and type II secretion system[4-7].
With respect to changes (increase or decrease) in intestinal microbiota in IBD patients, some conflicting findings have been reported for several bacteria, including Bifidobacterium, Clostridiales, Clostridium difficile, Campylobacter, Helicobacter and Faecalibacterium prausnitzii[8]. For example, the levels of F. prausnitzii in IBD patients were found to be reduced in several studies[9-11]. However, one study of de novo pediatric IBD revealed an increase in F. prausnitzii in CD, but not in UC[12]. Another study of twins showed an increase in F. prausnitzii in patients with colonic CD, but a decrease of F. prausnitzii in patients with ileal CD[13].
The intestinal microbiota of Western IBD patients has been extensively studied. However, the intestinal microbial profiles of Chinese IBD patients are not well characterized[14]. In the present study, we profiled and compared the fecal microbial community of IBD patients at different disease stages and healthy controls by using 16S rDNA amplicon-based analysis.
MATERIALS AND METHODS
Study population
Twenty-nine IBD patients (11 active CD, 4 inactive CD and 14 active UC patients) who regularly visited the First Affiliated Hospital of Nanjing Medical University (Jiangsu, China) from 2014 to 2016 were recruited to the study. The diagnosis of IBD was based on standard clinical, endoscopic, radiological and histological criteria[15]. The control group consisted of sex- and age-matched healthy subjects. Patients with IBD who met any of the following criteria were excluded: (1) use of antibiotics, probiotics or prebiotics in the 3-mo period immediately preceding the sampling time point; (2) current infectious diarrhea; and (3) malignancy. UC activity was evaluated using the Mayo score[16]; active UC was defined as UC disease activity index > 2. Activity of CD was scored by Crohn’s disease activity index (CDAI)[17]; active CD was defined as a CDAI > 150. Written informed consent was obtained from all subjects prior to their enrollment and the study was approved by the Ethics Committee at the First Affiliated Hospital of Nanjing Medical University, Jiangsu, China.
Fecal sample collection and extraction of genomic DNA
Fecal samples were collected from all subjects and subsequently stored at -80 °C within 2 h to prevent exposure of anaerobic bacteria to oxygen and to avoid bacterial overgrowth prior to DNA extraction. Genomic DNA was extracted from fecal samples using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Feces (200 mg) was added to a 2-mL screw cap vial containing 300 mg of 0.1-mm glass beads (Sigma, St. Louis, MO, United States) which was maintained on ice. The samples were added of 1.4 mL ASL buffer and then subjected to bead beating (45 s, speed 6.5) twice using a FastPrep-24 machine (MP Biomedicals, Solon, OH, United States) before the initial incubation for heat and chemical lysis at 95 °C for 5 min. Subsequent DNA extraction was performed following the QIAamp kit protocol for pathogen detection.
Sequencing
16S rDNA genes of V4 regions were amplified using specific primer with the barcode. All PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA, United States). The same volume of 1 × loading buffer (containing SYB green) was mixed with PCR products and electrophoresis on 2% agarose gel was carried out for detection. Samples with the bright main band between 400-450 bp were chosen for further experiments. PCR products were mixed in equidensity ratios. The mixture of PCR products was subsequently purified with Qiagen Gel Extraction Kit. Sequencing libraries were generated using TruSeq®DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, United States) following manufacturer’s recommendations, and index codes were added. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific, Waltham, MA, United States) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina MiSeq platform and 250 bp paired-end reads were generated.
Data analysis
Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end reads were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/)[18], which was designed to merge paired-end reads when at least some of the reads overlapped the read generated from the opposite end of the same DNA fragment, and the splicing sequences were called raw tags. Quality filtering of the raw tags was performed under specific filtering conditions to obtain high-quality clean tags[19] according to the QIIME (V1.7.0, http://qiime.org/index.html)[20] quality controlled process. The tags were compared with the reference database (Gold database, http://drive5.com/uchime/uchime_download.html) using UCHIME algorithm (UCHIME Algorithm, http://www.drive5.com/usearch/manual/uchime_algo.html)[21] to detect chimera sequences, and then the chimera sequences were removed[22]. Finally, the effective tags were obtained. Analysis of sequences was performed with Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/)[23]. Sequences with ≥ 97% similarity were assigned to the same operational taxonomic units (OTUs). Representative sequence for each OTU was screened for further annotation. For each representative sequence, the GreenGene Database (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi)[24] was used based on the RDP classifier (version 2.2, http://sourceforge.net/projects/rdp-classifier/)[25] algorithm to annotate taxonomic information.
In order to study the phylogenetic relationship of different OTUs, and the difference of the dominant species in different samples (groups), multiple sequence alignment was conducted using the MUSCLE software (version 3.8.31, http://www.drive5.com/muscle/)[26]. OTUs’ abundance information was normalized using a standard sequence number corresponding to the sample with the least sequences. Subsequent analysis of alpha diversity and beta diversity were all performed based on this output normalized data. Alpha diversity and beta diversity were calculated with QIIME (version 1.7.0) and displayed with R software (version 2.15.3).
Statistical analysis was performed using Statistical Package for Social Sciences version 19.0 (SPSS Inc., Chicago, IL, United States). The microbiota data and community estimates were analyzed by Kruskal-Wallis one-way analysis of variance to compare median values of microbiota data between CD, UC and controls. Spearman correlation analysis was used to analyze the correlation between intestinal bacterial abundance and intestinal inflammatory status. P values were corrected for multiple comparisons using false discovery rate (FDR); P < 0.05 was considered statistically significant.
RESULTS
Patients’ characteristics and sequencing data
Fecal samples from patients with active CD (n = 11), inactive CD (n = 4), active UC (n = 14), and 13 healthy individuals were analyzed in the current study. The median disease duration in patients with CD and UC was 10 mo (range: 3-48 mo) and 30 mo (range: 2-93 mo), respectively. Detailed clinical characteristics of the study subjects are presented in Table 1.
Table 1.
Clinical characteristics of enrolled patients
| CD | UC | Control | |
| n | 15 | 14 | 13 |
| Age, mean ± SD, yr | 37.7 ± 13.0 | 37.5 ± 17.1 | 39.8 ± 14.3 |
| Sex, male/female | 11/4 | 7/7 | 10/3 |
| Disease duration in months, median (range) | 10 (3-48) | 30 (2-93) | - |
| Smoking habits | 4 (26.7) | 1 (7.1) | 2 (15.4) |
| Abdominal surgery | 4 (26.7) | 0 | 0 |
| Montreal A (age of onset) | |||
| A1 (< 17) | 1 (6.7) | - | - |
| A2 (17-40) | 7 (46.7) | - | - |
| A3 (> 40) | 7 (46.7) | - | - |
| Montreal L (location) | |||
| L1 (ileal) | 8 (53.3) | - | - |
| L2 (colonic) | 1 (6.7) | - | - |
| L3 (ileocolonic) | 6 (40) | - | - |
| L4 (upper gastrointestinal tract) | 0 | - | - |
| Montreal B (behavior) | |||
| B1(nonstricturing, nonpenetrating) | 8 (53.3) | - | - |
| B2 (stricturing) | 6 (40) | - | - |
| B3 (penetrating) | 1 (6.7) | - | - |
| p (perianal disease) | 4 (26.7) | - | - |
| Montreal | |||
| E1 ulcerative proctitis | - | 4 (28.6) | - |
| E2 left sided ulcerative colitis | - | 5 (35.7) | - |
| E3 extensive ulcerative colitis | - | 5 (35.7) | - |
| CDAI score | |||
| < 150 | 4 (26.7) | - | - |
| 150-220 | 5 (33.3) | - | - |
| 221-450 | 6 (40) | - | - |
| > 450 | 0 | - | - |
| Mayo score | |||
| 0-2 | - | 0 | - |
| 3-5 | - | 7 (50.0) | - |
| 6-10 | - | 5 (35.7) | - |
| 11-12 | - | 2 (14.3) | - |
| Therapy | |||
| 5-ASA | 14 (93.3) | 14 (100) | - |
| Azathioprine | 2 (13.3) | 0 | - |
| Steroids | 1 (6.7) | 6 (42.9) | - |
| Infliximab | 0 | 0 | - |
Data are presented as n (%). 5-ASA: 5-aminosalicylic acid; CD: Crohn’s disease; CDAI: CD activity index; SD: Standard deviation; UC: Ulcerative colitis.
Paired-end reads were generated with the Illumina MiSeq platform. The reads with sequencing adapters, N base, poly base, and low quality were filtered out with default parameters. High quality paired-end reads were combined to tags based on overlaps. A total of 1747775 tags were obtained with an average of 41613 tags per sample; the average length was 252 bp. Filtered tags were clustered into OTUs at 97% similarity and a total of 878 OTUs were generated from 42 samples (see Supplementary File 1).
Characteristics of the microbial community in IBD patients and controls
When comparing bacterial alpha diversity, including community richness (observed species, chao, and ace) and diversity (Shannon and Simpson) between CD, UC and control groups, we found overall differences with respect to each diversity index (Figure 1). Significant differences (P < 0.05) with respect to community richness (chao) were observed both between CD and controls and between UC and controls. The observed species and ace indices of CD patients were lower than those of controls; however, the differences were not statistically significant (P > 0.05). Moreover, the pattern of richness was found to be similar in CD and UC. When considering the species diversity of microbiota (Shannon and Simpson), the differences between each group were not statistically significant.
Figure 1.
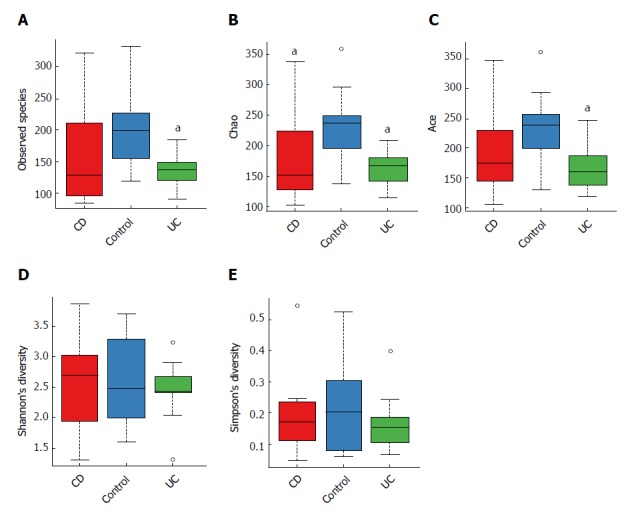
Alpha diversity indices boxplot, including community richness (observed species, chao, ace) and diversity (Shannon, Simpson) varied among each group. A: Observed species; B: Chao; C: Ace; D: Shannon; E: Simpson. aP < 0.05 vs control. CD: Crohn’s disease; UC: Ulcerative colitis.
We subsequently surveyed the alpha diversity in IBD patients at different disease stages (see Supplementary Figure 1). Generally, the richness indices in IBD patients showed a decreasing trend (controls > inactive CD > active CD), but the between-group differences were not statistically significant. However, the diversity indices in IBD patients were not significantly different from those in controls.
Microbial community structures in IBD are distinct from those in normal controls
We used principal component analysis (PCA) to investigate the community structure of microbiota in CD, UC and controls. We found that samples tended to cluster together based on disease; however, to a certain extent, there was an overlap between all groups. IBD samples were mostly distinct from those of normal controls, which indicated differences with respect to community structure of the microbiota between IBD and controls (Anosim: CD vs control, P = 0.02; UC vs control, P = 0.001). However, samples of CD and UC were located closely, which suggested a similar bacterial community structure in the context of both CD and UC (Anosim: P = 0.133) (Figure 2A).
Figure 2.
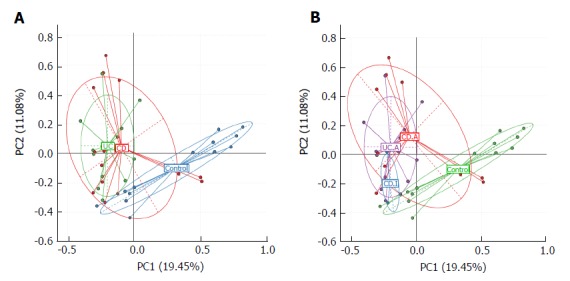
Principal component analysis based on the overall structure of the fecal microbiota in the entire study population. Each data point represents an individual sample. A: Disease phenotype group; B: Stages of disease group. CD: Crohn’s disease; CD.A: Active CD; CD.I: Inactive CD; UC: Ulcerative colitis; UC.A: Active UC.
Next, we visualized the PCA to compare the microbial structure in patients at different disease stages (Figure 2B). The results showed that samples could be well separated between active CD and controls (Anosim: P = 0.016) as well as between active CD and active UC (Anosim: P = 0.01). However, there were no distinct microbiota structural patterns apparent between active CD and inactive CD groups, although the samples seemed to be clearly separated (Anosim: P = 0.719). There was also no separation between inactive CD and controls (Anosim: P = 0.564) based on the PCA. Our results indicated that the bacterial community structure in active CD was different from that in active UC; however, there was no difference with respect to the alterations of bacterial community structure in fecal samples of the total UC and CD patients.
Overall taxonomic analysis of IBD patients and controls
Taxonomic composition distribution histograms of each sample were summarized at the phyla level (Figure 3A). The dominant sequences belonged to four bacterial phyla (Bacteroidetes, Firmicutes, Proteobacteria and Fusobacteria), which accounted for over 97% of taxonomy generally (Figure 3B). Among all the relatively abundant dominant strains in IBD and normal controls, Bacteroidetes was, as a rule, the most abundant bacterial phylum.
Figure 3.
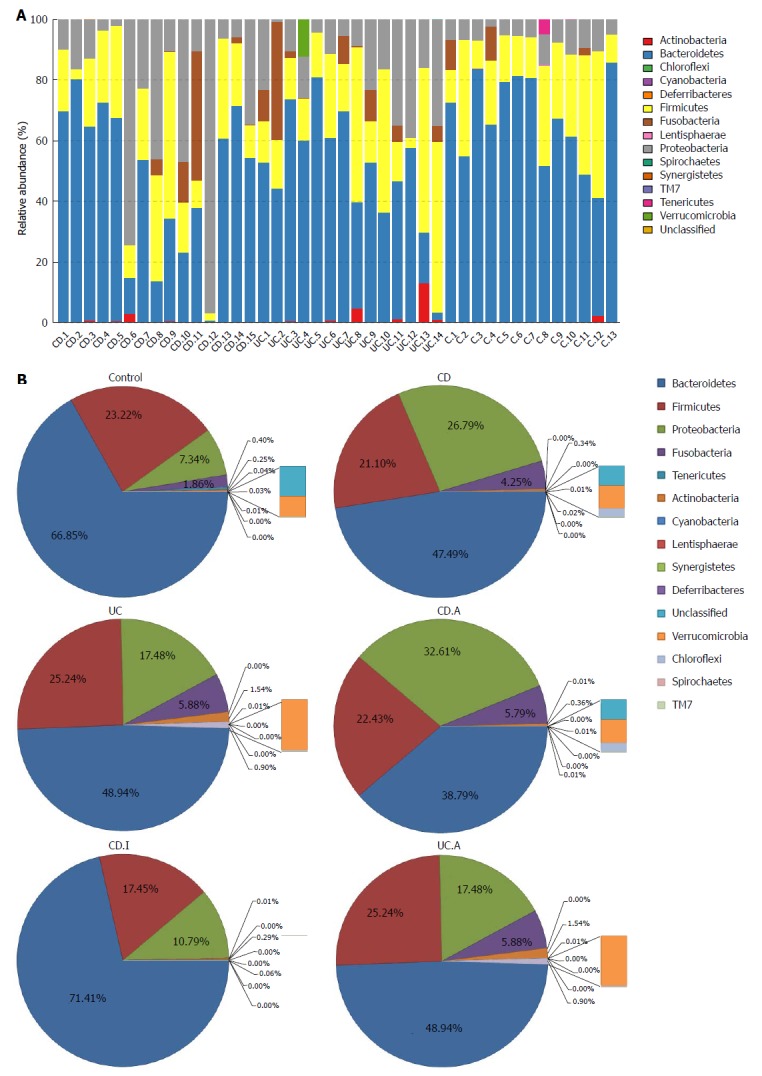
Taxonomic composition distribution in samples of phylum level. A: Individually; B: Integrally. CD: Crohn’s disease; CD.A: Active CD; CD.I: Inactive CD; UC: Ulcerative colitis; UC.A: Active UC.
Phylum-level analysis (Figure 3B, Table 2) revealed a nominal decrease in the relative abundance of Bacteroidetes in both CD and UC patients (CD vs control, 47.49% vs 66.85%, P = 0.015; UC vs control, 48.94% vs 66.85%, P = 0.019); however, these differences were not significant after adopting the FDR. On the contrary, Proteobacteria was significantly increased in both CD and UC, as compared to that in controls (CD vs control, 26.79% vs 7.34%, P = 0.002; UC vs control, 17.48% vs 7.34%, P = 0.005). In addition, no Spirochaetes phylum was detected in CD and controls but it was observed in UC (0.015%). Similarly, Lentisphaerae phylum was found in the control group (accounting for 0.031%), but almost none was found in patients with IBD.
Table 2.
Significant differences in microbial distribution of taxa (phylum and genus) in patients with inflammatory bowel disease
| CD | UC | CD/UC | CD.A/CD.I | CD.A/UC.A | |
| Firmicutes | |||||
| Abiotrophia1 | ↑c | ||||
| Butyricicoccus | ↓c | c3 | |||
| RFN201 | ↑c | c2 | |||
| Pseudoramibacter_Eubacterium1 | ↑b | c2 | |||
| Holdemania1 | ↓c | c2 | |||
| 02d06 | ↓c | c2 | c2 | ||
| Lachnobacterium | ↓c | ||||
| Megamonas | ↓c | ||||
| Mitsuokella | ↓c | ↓c | |||
| Granulicatella | ↑b | ||||
| Peptostreptococcus | ↑b | ||||
| Schwartzia1 | ↑b | ||||
| Moryella1 | c3 | ||||
| Staphylococcus1 | c3 | c3 | |||
| Epulopiscium | c2 | ||||
| Sarcina | c2 | ||||
| Bacteroidetes | b3 | ||||
| Alistipes | ↓c | ||||
| Butyricimonas | ↓c | ||||
| Capnocytophaga1 | ↑c | c3 | c3 | ||
| Prevotella | ↓c | ||||
| Proteobacteria | ↑b | ↑b | |||
| Escherichia | ↑c | ↑b | |||
| Haemophilus | ↓c | b3 | b3 | ||
| Desulfovibrio | ↓c | b2 | c2 | ||
| Oxalobacte1 | ↓c | ||||
| Janthinobacterium1 | ↑b | b3 | |||
| Campylobacter | ↑b | ||||
| Cardiobacterium1 | c3 | ||||
| Lautropia1 | c3 | ||||
| Lupinus1 | c3 | ||||
| Shewanella1 | b3 | ||||
| Actinobacteria | |||||
| Actinomyces | ↑c | ||||
| Eggerthella1 | ↑b | ||||
| Corynebacterium1 | ↑b | c3 | b3 | ||
| Slackia1 | b2 | c2 | |||
| Synergistetes | |||||
| Pyramidobacter1 | ↓c | ||||
| Synergistes1 | ↓c | ||||
| TG51 | c3 | ||||
| Spirochaetes | ↑c | c3 | |||
| Lentisphaerae | ↓c | c2 | |||
| Victivallis1 | ↓c | ↓c |
↑ and ↓ relative to controls;
Relative abundance of genera < 0.01%;
Increase in value;
Decrease in value. aP < 0.05; bP < 0.01; cP < 0.001. CD: Crohn’s disease; CD.A: Active CD; CD.I: Inactive CD; IBD: Inflammatory bowel disease; UC: Ulcerative colitis; UC.A: Active UC.
At the genus level, the relative abundance of all genera varied between different samples (Figure 4A). The top 10 abundant genera in UC, CD and controls were Bacteroides, Escherichia, Faecalibacterium, Fusobacterium, Haemophilus, Lachnospira, Prevotella, Roseburia, Streptococcus, and Sutterella (Figure 4B). Among these, the relative abundance of Escherichia in CD and UC was significantly higher than that in controls. In addition, abundance of Haemophilus in CD and Prevotella in UC patients were both markedly lower than that in normal controls. Moreover, the abundance of Haemophilus in CD was dramatically lower than that in UC. Besides the top 10 abundant genera, the relative abundance of remaining genera was comparable between IBD patients and normal controls (Table 2). The abundance of 12 genera, Butyricicoccus, Mitsuokella, 02d06, Actinomyces, Alistipes, Butyricimonas, Campylobacter, Desulfovibrio, Granulicatella, Lachnobacterium, Megamonas and Peptostreptococcus, was significantly different after correction among each group within the community; the sequence percentages for each of these 12 genera were more than 0.01%.
Figure 4.
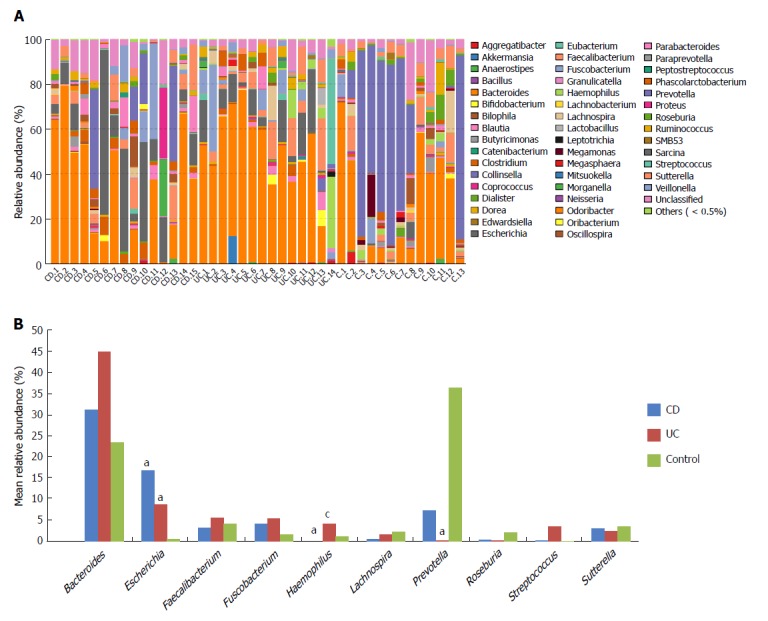
A: The taxonomic composition distribution in samples of genus level; B: Genera shown represent the 10 most abundant genera of CD, UC and control. aP < 0.05 vs control, cP < 0.05 vs CD. CD: Crohn’s disease; UC: Ulcerative colitis.
Taxonomic comparisons in IBD patients at different disease stages
On analysis of the alterations at the phyla level between active CD and inactive CD, we found that the dominant bacterial phyla were the same as described earlier (accounting for over 99% of taxonomy), with the exception that Fusobacteria was replaced by Actinobacteria in inactive CD (Figure 3B, Table 2). However, the abundance of Bacteroidetes was dramatically decreased in active CD group, as compared to that in the inactive CD group (CD.A vs CD.I, 38.79% vs 71.41%, P = 0.001). The abundance of Proteobacteria was just nominally increased in active CD, as compared to that in inactive CD (P = 0.023), which did not hold significance after correction. Similarly, no differences were detected with respect to the remaining dominant bacteria between active CD and inactive CD. Microbiota in active CD and active UC were found to be similar at the phyla level.
We then investigated the genera with percentages of sequences > 0.01% of community in different phases of IBD and found that the abundance of Bacteroides and Prevotella in active CD were only nominally different from that in inactive CD. However, Desulfovibrio, 02d06, Epulopiscium, and Sarcina detected in active CD were markedly higher than that in active UC, while Haemophilus was markedly lower than that in active UC (Table 2).
Association between the inflammatory index of CD patients and microbiome
We assessed the correlation between the relative abundance of Bacteroidetes and CDAI scores of each CD patient; surprisingly, we found a negative correlation between the two (r = -0.538, P = 0.039) (Figure 5A). On the contrary, there was a trend of positive correlation between the abundance of Proteobacteria and CDAI (r = 0.250, P = 0.369); however, the correlation was not statistically significant.
Figure 5.
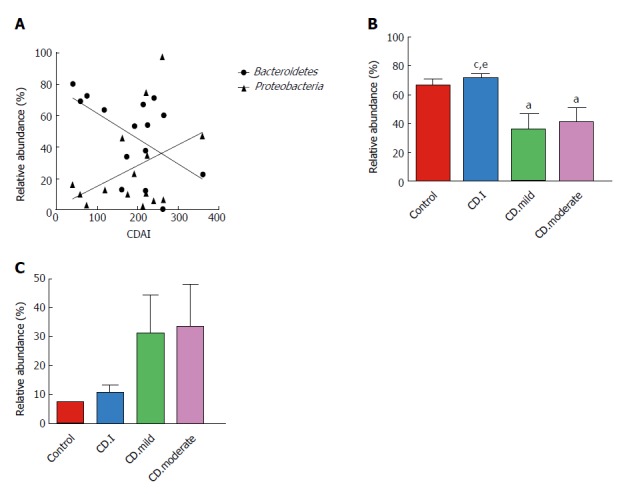
Correlation of the relative abundance of Bacteroidetes and Proteobacteria with Crohn’s disease activity index scores (A). Bacteroidetes (r = -0.538, P = 0.039); Proteobacteria (r = 0.250, P = 0.369); B: Microbial composition of Bacteroidetes in patients with inactive/mild/moderate CD and in control; C: Microbial composition of Proteobacteria in patients with inactive/mild/moderate CD and in controls. aP < 0.05 vs control; cP < 0.05 vs CD.mild; eP < 0.05 vs CD.moderate. CDAI: CD activity index; CD.I: Inactive CD; CD.mild: Mild CD; CD.moderate: Moderate CD.
Next, we analyzed the correlation between microbial composition and disease severity. Patients with mild and moderate CD had notably decreased levels of Bacteroidetes as compared to that in patients with inactive CD; however, no significant difference in this respect was noted between patients with mild and moderate CD (Figure 5B). Interestingly, Proteobacteria exhibited a noteworthy trend (controls < inactive CD < mild CD < moderate CD); however, the trend did not attain statistical significance (Figure 5C).
Effect of age and sex on intestinal microbial compositions
Although IBD mostly occurs in young adults (20- to 30-years-old), it can happen at any age. In the present research, no correlation was observed between microbial composition and age (see Supplementary Figure 2). Considering that most participants in our study (with the exception of one patient aged 14 years with UC) were adults, we divided the participants into two groups: age < 40 years and age > 40 years. However, no significant difference in microbial compositions was observed between the two groups (see Supplementary Table 1). On subgroup analysis based on sex, no notable differences were observed between male and female patients in either subject subgroup (see Supplementary Table 2).
DISCUSSION
IBD is one of the most frequently studied human diseases linked to the gut microbiota. Distinctive microbial composition and its interaction with the host immunological response are believed to play a critical role in the pathogenesis of IBD[27,28]; however, several aspects of the relationship are not well-characterized. In this study, we demonstrated differences with respect to fecal microbiota between Chinese IBD patients and healthy controls based on 16S rDNA sequencing analysis.
The dominant dysbiosis pattern unraveled by the present study was the decrease in community abundance of fecal microbiota both in CD and UC patients; while microbial diversity in CD patients was lower than that in controls, the difference was not statistically significant. Previous studies have shown reduced diversity of fecal microbiota in both Western[29,30] and Chinese patients with IBD[14], as compared to that in healthy controls. These inconsistencies are likely attributable to differences with respect to study design, stage of disease, or technique employed to survey the gut microbiota. The reasons for the changes of diversity in these conditions are still not known. Indeed, despite general trends such as a reduction in diversity, the response to IBD may, to some extent, be subject-specific.
We analyzed the bacterial community structure of microbiota in IBD patients and healthy individuals. The results showed distinct differences both in CD and UC, as compared to controls; however, the microbiota were similar within CD and UC groups or within active CD and inactive CD groups, which were not structurally distinguishable according to PCA. These data were also consistent with the previous studies conducted in Chinese and Western populations[14,31]. However, Forbes et al[32] found a difference in the structure of microbiota between CD and UC. This result differed from those of other studies, as this study involved analysis of intestinal mucosa, while other studies were based on fecal analysis.
Detailed compositional alterations in fecal microbiota in IBD patients were detected at distinct taxonomic levels. The principle finding in our study was that the phylum Proteobacteria was significantly increased in IBD patients, which was in agreement with a consistent finding across published literature[33,34]. The genus Escherichia, especially Escherichia coli (data not shown), was also found to be notably higher in IBD patients, as compared to that in normal controls. Escherichia coli, particularly AIEC, as an important pathobiont that may play a role in IBD development, has been isolated from ileal CD biopsy specimens[35]. The initial lesions in the colon mucosa can be aggravated by alpha-hemolysin secreted by Escherichia coli, which can damage host cell membranes and epithelial barrier[36].
Moreover, both E. coli and Campylobacter (affiliated with Proteobacteria) are known to release cytolethal distending toxins, which leads to cell cycle arrest, chromatin fragmentation and apoptosis, all of which are involved in the pathogenesis of IBD[37].
In the present study, patients with IBD exhibited relatively less number of Bacteroidetes compared to that in controls. The lower proportion of Bacteroidetes was mainly attributable to notably reduced abundance of Prevotella genus. The results were largely similar to those of another study which employed 16S rDNA sequencing analysis[38]. Actually, alterations in Bacteroidetes in CD still remain controversial. Rehman et al[39] reported increased Bacteroidetes in CD patients and even demonstrated a notable increase in transcriptional activity, as compared to that in controls. Further studies are needed to clarify this issue. To minimize potential confounding factors, future studies should define gut dysbiosis in detail. Moreover, prospective cohort studies on newly diagnosed treatment-naïve patients will provide more definitive evidence in this respect.
In the present study, we documented increased abundance of Haemophilus and decreased Desulfovibrio (affiliated with Proteobacteria) in patients with UC. These findings were not observed in a previous study on fecal microbiota dysbiosis conducted by Chen et al[14] in Chinese patients with IBD. Recently, Haemophilus has been reported to contribute to oral dysbiosis in patients with IBD[40] and Haemophilus spp., like the Enterobacteriaceae, are well adapted to survive under conditions of increased oxidative stress[41]. To our knowledge, Rowan et al[42] demonstrated an increase of Desulfovibrio (sulfate-reducing bacteria) in patients with UC. In vitro studies have shown that 5-aminosalicylic acid (5-ASA) inhibits fecal sulfide production and fecal samples from patients not treated with this drug revealed higher levels of sulfide[43]. It is conceivable that all participants in the present study were treated with 5-ASA, which may have contributed to the opposite phenomenon.
In addition, the study found an abundance of Butyricicoccus, Mitsuokella, 02d06, Lachnobacterium and Megamonas (all affiliated with Clostridia class, Firmicutes phylum), which are obligate anaerobes. These were found significantly decreased in IBD patients in the current study. Dysanaerobiosis in patients with UC was observed recently[44] and there seems to be a shift from anaerobiosis in healthy state to dysanaerobiosis in IBD, with an elevated oxygen level in the gut[45]. Furthermore, studies conducted on experimental colitis models showed decrease in obligate anaerobes of Firmicutes and increase in facultative anaerobes of Proteobacteria, which indicates a role of oxygen in gut dysbiosis[46]. In fact, both Butyricicoccus (affiliated with Ruminococcaceae family) and Lachnobacterium (affiliated with Lachnospiraceae family) produce SCFAs, which are known as the primary energy source for colonic epithelial cells[47] and were shown to induce the expansion of colonic regulatory T cells[48]. These alterations in microbial composition suggested that reduction in beneficial microbiota (Clostridia class and SCFA-producing bacteria) is more associated with IBD patients compared to the increment of pathobionts (Escherichia and Campylobacter).
When analyzing the fecal microbiota at different disease stages of IBD, only the abundance of Bacteroidetes was dramatically decreased in active CD, as compared to that in inactive CD. About the relationship between microbiome and disease activity, we also found a negative correlation between the relative abundance of Bacteroidetes and CDAI in the present study. The relative abundance of Bacteroidetes in active CD patients was lower than that in inactive CD or controls, but the relative abundance of Bacteroidetes was similar between mild and moderate CD. All these findings suggest that Bacteroidetes may have a negative impact on inflammatory development.
Potential links between age or sex and microbial compositions have been suggested recently[49]. Gut microbiota vary in different age groups: infants, adults or the elderly. The microbiota in infants is often affected by the birth route, feeding patterns and illness history[50]. Not until adulthood does the microbiota become stable, complex and shows improved resilience against perturbations[51]. Then, the stability decreases in the elderly (≥ 65 years of age)[52]. However, we did not find the effect of age and sex on microbiota in the current study. So, a different role for the microbiota in disease initiation and progression should be researched.
Our study faces several limitations. First of all, due to the small sample number and relatively high variability of microbial composition in each group, some of the relative abundances of specific bacteria between groups could not reach statistical significance after adopting the FDR. Secondly, 16S rDNA sequencing mainly focuses on the taxonomic profiling rather than providing greater insight into the function of the intestinal microbiota in disease[53,54]. Thirdly, the nature and extent of difference between the fecal microbiota and mucosa-associated microbiota in IBD remains unclear. Controversy still exists between them because of different techniques used in separate studies[55]. Several studies indicated that the fecal microbiota and mucosa-associated microbiota were similar[13,56,57]. However, some studies have found a significant difference between them[14,58,59]. It seems that the fecal microbiota represents a combination of a separate nonadherent luminal population and shed mucosal bacteria[59]. Further study with a large population is required to confirm our data and mucosa-associated microbiota needs to be researched in Chinese patients with IBD.
In conclusion, we presented a comprehensive analysis of fecal microbiota in Chinese patients with IBD. Significant differences in microbial composition of patients with IBD and controls were observed. Additionally, the negative correlation between Bacteroidetes and CDAI suggested that Bacteroidetes might have a negative impact on development of inflammation.
ARTICLE HIGHLIGHTS
Research background
Inflammatory bowel disease (IBD) is generally defined by two nonspecific inflammatory disorders, Crohn’s disease (CD) and ulcerative colitis (UC), which are characterized by chronic persistent inflammation of the intestinal mucosa lining the intestinal tract. Recently, distinctive microbial composition and its interaction with the host immunological response are believed to play critical roles in the pathogenesis of IBD. Although the intestinal microbial composition of Western IBD patients has been extensively studied, there are conflicting reports about changes of the bacterial abundance. What’s more, the intestinal microbial profiles of Chinese IBD patients are not well characterized. In the present study, we use 16S rDNA amplicon-based analysis to analyze the alterations of fecal microbiota in Chinese patients with IBD.
Research motivation
Although the microbial community is gaining increasing attention for its influence on IBD, there is a lack of data on global alteration of microbiota in Chinese patients and the relationship is poorly understood. This study would characterize the important differences of fecal microbiota between Chinese IBD patients and healthy controls based on a 16S rDNA sequencing analysis, hoping to explore which kinds of the microbiota could be involved in the pathogenesis of IBD or providing important references for diagnosis or treatment of IBD.
Research objectives
The research aimed to investigate the differences in quantity, diversity and similarity of the fecal bacterial population taken from Chinese IBD patients at different stages of disease and healthy individuals.
Research methods
Twenty-nine IBD patients (11 active CD, 4 inactive CD and 14 active UC patients) from the First Affiliated Hospital of Nanjing Medical University (Jiangsu, China) and 13 sex and age well-matched healthy individuals were enrolled in the study. 16S rDNA amplicon-based sequencing was used to analyze the fecal microbiota of each sample.
Research results
In this study, community richness (chao) and microbial structure in IBD were significantly different from those in normal controls. The relative abundance of Bacteroidetes in the active CD group was significantly lower than that in the inactive CD group, and it showed a negative correlation with Crohn’s disease activity index (CDAI). At the phyla level, the abundance of Proteobacteria was significantly higher in IBD than in controls. At the genera level, 8 genera in CD and 23 genera in UC (in particular, the Escherichia genus) showed significantly greater abundance as compared to that in normal controls.
Research conclusions
Our study presented a comprehensive analysis of fecal microbiota in the gut of Chinese patients with IBD. Significant differences in microbial composition of patients with IBD and controls were observed. Additionally, the negative correlation between Bacteroidetes and CDAI suggested that Bacteroidetes might have a negative impact on development of inflammation.
Research perspectives
Fecal microbial examination is noninvasive and easily collected compared with the mucosal biopsy, which may increase the risk of unexpected bleeding.However, the mucosa-associated microbiota is believed to directly affect epithelial and mucosal function. In the future, both the fecal and mucosa-associated microbiota should be investigated together to better understand the role of the intestinal microbiota in health and disease.
ACKNOWLEDGMENTS
The authors appreciate technical and statistical supports of BGI Tech Solutions Co., Ltd (Shenzhen, China) and would like to express thanks.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Supported by the National Natural Science Foundation of China, No. 81470827.
Institutional review board statement: This study was reviewed and approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University.
Informed consent statement: All study participants provided informed written consent prior to study enrollment.
Conflict-of-interest statement: All authors declare no conflicts of interest related to this article.
Data sharing statement: No additional data are available.
Peer-review started: January 10, 2018
First decision: February 5, 2018
Article in press: March 7, 2018
P- Reviewer: Naito Y, Zouiten-Mekki L S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Hai-Qin Ma, Department of Gastroenterology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Province, China.
Ting-Ting Yu, Department of Gastroenterology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Province, China.
Xiao-Jing Zhao, Department of Gastroenterology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Province, China.
Yi Zhang, Department of Gastroenterology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Province, China.
Hong-Jie Zhang, Department of Gastroenterology, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Province, China. hjzhang06@163.com.
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42; quiz e30. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krause DO, Little AC, Dowd SE, Bernstein CN. Complete genome sequence of adherent invasive Escherichia coli UM146 isolated from Ileal Crohn’s disease biopsy tissue. J Bacteriol. 2011;193:583. doi: 10.1128/JB.01290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 7.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Butcher J, Mack D, Stintzi A. Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:139–153. doi: 10.1097/MIB.0000000000000215. [DOI] [PubMed] [Google Scholar]
- 9.Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, Tysk C, Jansson JK. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 10.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 11.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, Mukhopadhya I, Bisset WM, Barclay AR, Bishop J, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am J Gastroenterol. 2012;107:1913–1922. doi: 10.1038/ajg.2012.335. [DOI] [PubMed] [Google Scholar]
- 13.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Wang W, Zhou R, Ng SC, Li J, Huang M, Zhou F, Wang X, Shen B, A Kamm M, et al. Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine (Baltimore) 2014;93:e51. doi: 10.1097/MD.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouyang Q, Tandon R, Goh KL, Pan GZ, Fock KM, Fiocchi C, Lam SK, Xiao SD. Management consensus of inflammatory bowel disease for the Asia-Pacific region. J Gastroenterol Hepatol. 2006;21:1772–1782. doi: 10.1111/j.1440-1746.2006.04674.x. [DOI] [PubMed] [Google Scholar]
- 16.D’Haens G, Sandborn WJ, Feagan BG, Geboes K, Hanauer SB, Irvine EJ, Lémann M, Marteau P, Rutgeerts P, Schölmerich J, et al. A review of activity indices and efficacy end points for clinical trials of medical therapy in adults with ulcerative colitis. Gastroenterology. 2007;132:763–786. doi: 10.1053/j.gastro.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 17.Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology. 1976;70:439–444. [PubMed] [Google Scholar]
- 18.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 24.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 28.Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 29.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andoh A, Imaeda H, Aomatsu T, Inatomi O, Bamba S, Sasaki M, Saito Y, Tsujikawa T, Fujiyama Y. Comparison of the fecal microbiota profiles between ulcerative colitis and Crohn’s disease using terminal restriction fragment length polymorphism analysis. J Gastroenterol. 2011;46:479–486. doi: 10.1007/s00535-010-0368-4. [DOI] [PubMed] [Google Scholar]
- 32.Forbes JD, Van Domselaar G, Bernstein CN. Microbiome Survey of the Inflamed and Noninflamed Gut at Different Compartments Within the Gastrointestinal Tract of Inflammatory Bowel Disease Patients. Inflamm Bowel Dis. 2016;22:817–825. doi: 10.1097/MIB.0000000000000684. [DOI] [PubMed] [Google Scholar]
- 33.Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2011;8:152–168. doi: 10.1038/nrgastro.2011.3. [DOI] [PubMed] [Google Scholar]
- 34.Sokol H, Lepage P, Seksik P, Doré J, Marteau P. Temperature gradient gel electrophoresis of fecal 16S rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J Clin Microbiol. 2006;44:3172–3177. doi: 10.1128/JCM.02600-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 36.Bücker R, Schulz E, Günzel D, Bojarski C, Lee IF, John LJ, Wiegand S, Janßen T, Wieler LH, Dobrindt U, et al. α-Haemolysin of Escherichia coli in IBD: a potentiator of inflammatory activity in the colon. Gut. 2014;63:1893–1901. doi: 10.1136/gutjnl-2013-306099. [DOI] [PubMed] [Google Scholar]
- 37.Smith JL, Bayles DO. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit Rev Microbiol. 2006;32:227–248. doi: 10.1080/10408410601023557. [DOI] [PubMed] [Google Scholar]
- 38.Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Doré J, Raedler A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141:227–236. doi: 10.1053/j.gastro.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol. 2010;59:1114–1122. doi: 10.1099/jmm.0.021170-0. [DOI] [PubMed] [Google Scholar]
- 40.Said HS, Suda W, Nakagome S, Chinen H, Oshima K, Kim S, Kimura R, Iraha A, Ishida H, Fujita J, et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014;21:15–25. doi: 10.1093/dnares/dst037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrison A, Bakaletz LO, Munson RS Jr. Haemophilus influenzae and oxidative stress. Front Cell Infect Microbiol. 2012;2:40. doi: 10.3389/fcimb.2012.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowan F, Docherty NG, Murphy M, Murphy B, Calvin Coffey J, O’Connell PR. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum. 2010;53:1530–1536. doi: 10.1007/DCR.0b013e3181f1e620. [DOI] [PubMed] [Google Scholar]
- 43.Pitcher MC, Beatty ER, Cummings JH. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut. 2000;46:64–72. doi: 10.1136/gut.46.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walujkar SA, Dhotre DP, Marathe NP, Lawate PS, Bharadwaj RS, Shouche YS. Characterization of bacterial community shift in human Ulcerative Colitis patients revealed by Illumina based 16S rRNA gene amplicon sequencing. Gut Pathog. 2014;6:22. doi: 10.1186/1757-4749-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 2013;7:1256–1261. doi: 10.1038/ismej.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Podolsky DK. Inflammatory bowel disease (1) N Engl J Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. [DOI] [PubMed] [Google Scholar]
- 47.Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46:493–499. doi: 10.1136/gut.46.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer F, Trimble WL, Chang EB, Handley KM. Functional predictions from inference and observation in sequence-based inflammatory bowel disease research. Genome Biol. 2012;13:169. doi: 10.1186/gb-2012-13-9-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Presley LL, Ye J, Li X, Leblanc J, Zhang Z, Ruegger PM, Allard J, McGovern D, Ippoliti A, Roth B, et al. Host-microbe relationships in inflammatory bowel disease detected by bacterial and metaproteomic analysis of the mucosal-luminal interface. Inflamm Bowel Dis. 2012;18:409–417. doi: 10.1002/ibd.21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Cruz P, Prideaux L, Wagner J, Ng SC, McSweeney C, Kirkwood C, Morrison M, Kamm MA. Characterization of the gastrointestinal microbiota in health and inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:372–390. doi: 10.1002/ibd.21751. [DOI] [PubMed] [Google Scholar]
- 56.van der Waaij LA, Harmsen HJ, Madjipour M, Kroese FG, Zwiers M, van Dullemen HM, de Boer NK, Welling GW, Jansen PL. Bacterial population analysis of human colon and terminal ileum biopsies with 16S rRNA-based fluorescent probes: commensal bacteria live in suspension and have no direct contact with epithelial cells. Inflamm Bowel Dis. 2005;11:865–871. doi: 10.1097/01.mib.0000179212.80778.d3. [DOI] [PubMed] [Google Scholar]
- 57.Bibiloni R, Tandon P, Vargas-Voracka F, Barreto-Zuniga R, Lupian-Sanchez A, Rico-Hinojosa MA, Guban J, Fedorak R, Tannock GW. Differential clustering of bowel biopsy-associated bacterial profiles of specimens collected in Mexico and Canada: what do these profiles represent? J Med Microbiol. 2008;57:111–117. doi: 10.1099/jmm.0.47321-0. [DOI] [PubMed] [Google Scholar]
- 58.Durbán A, Abellán JJ, Jiménez-Hernández N, Ponce M, Ponce J, Sala T, D’Auria G, Latorre A, Moya A. Assessing gut microbial diversity from feces and rectal mucosa. Microb Ecol. 2011;61:123–133. doi: 10.1007/s00248-010-9738-y. [DOI] [PubMed] [Google Scholar]
- 59.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]