

Abstract
Chronic obstructive pulmonary disease (COPD) is a complex and heterogenous syndrome that represents a major global health burden. COPD phenotypes have recently emerged based on large cohort studies addressing the need to better characterize the syndrome. Though comprehensive phenotyping is still at an early stage, factors such as ethnicity and radiographic, serum, and exhaled breath biomarkers have shown promise. COPD is also an immunological disease where innate and adaptive immune responses to the environment and tobacco smoke are altered. The frequent overlap between COPD and other systemic diseases, such as cardiovascular disease, have influenced COPD therapy, and treatments for both conditions may lead to improved patient outcomes. Here we discuss current paradigms that center on improving the definition of COPD, understanding the immunological overlap between COPD and vascular inflammation, and the treatment of COPD—with a focus on comorbid cardiovascular disease.
Keywords: Chronic Obstructive Pulmonary Disease (COPD), Phenotypes, Comorbidities, Immunobiology
Introduction
Chronic obstructive pulmonary disease (COPD) represents a major global health burden that is widely recognized as a complex, heterogenous syndrome rather than a single disease. Epidemiologic data reveal three major themes: First, COPD consists of several clinical phenotypes, most of which need further refinement in definition. By understanding these COPD phenotypes, it may be possible to improve treatment. Second, COPD is often recognized as a chronic inflammatory lung disorder with important immunological mechanisms and systemic manifestations. Appreciating the immunobiology of COPD may facilitate better treatment paradigms and shed light on common mechanisms shared between COPD and cardiovascular disease. And, third, COPD often exists with and may potentiate cardiovascular disease independent of tobacco smoking. How COPD treatment affects cardiovascular disease, and vice versa, is also unclear.
In this review, we aim to: 1) discuss current COPD phenotypes based on relevant epidemiologic biomarker studies; 2) review COPD immunobiology with a focus on the overlap with cardiovascular disease; and 3) discuss recent advances in COPD treatment, including treatments that can affect both COPD and cardiovascular disease.
COPD Phenotypes
Decades ago medical schools taught the concept that COPD existed as two basic clinical phenotypes: chronic bronchitis versus pulmonary emphysema. We currently understand that COPD is far more heterogeneous. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) took steps to categorize COPD with greater sophistication beginning in 20011. GOLD has subsequently taken the established staging of COPD by spirometry, primarily forced expiratory volume in one second as a percent of forced vital capacity (FEV1/FVC or FEV1%), and created patient groups that include evaluation of the burden of symptoms and exacerbation frequency in a more comprehensive assessment of the impact of COPD on patient lives. These groups include patients with good lung function and minimal symptoms (GOLD A) to patients with advanced lung disease and a high degree of symptoms (GOLD D). As expected it also includes a group of COPD patients with advanced decline in lung function but with relatively few symptoms (GOLD C), and a group with preserved lung function but a high degree of symptoms (GOLD B). Groups B and Group C patients were not a surprise to clinicians, and having an empiric approach to categorizing these complex phenotypes of COPD was welcome.
Large prospective clinical cohort studies have improved our understanding of the heterogeneity of COPD. We review and highlight major discoveries that have emerged from these studies with particular emphasis on phenotyping schemes, contribution of CT scans, and the relationship of COPD with comorbid conditions, including cardiovascular disease.
Cohort Studies
COPDGene®
COPDGene® was originally designed to be an observational study to identify genetic factors associated with COPD2, but it was refined to be a prospective cohort study enrolling 4,500 smoker controls between 2008 and 2011 at 21 different clinical centers: 1,500 GOLD stage 1, and 4,500 GOLD stage 2 to 4 (total 10,500 subjects). Patients that were classified as “smoker controls” had an FEV1/FVC of >0.70 and a forced expiratory volume in one second (FEV1) >80%, all post-bronchodilator. A small group of non-smoker controls were also included as a comparison for the quantitative CT scan data. Additionally, an interesting sub-cohort emerged labelled GOLD–U, which was an unclassified COPD cohort of smokers with a decrease in FEV1 but a preserved FEV1/FVC ratio2. The study goals were to characterize each of these groups with respect to symptoms, medications, and spirometry; inspiratory and expiratory CT scans; exercise capacity; and genome-wide association patterns to compare within each of these cohorts. At final enrollment two thirds of the subjects were non-Hispanic whites while one third was African American. The defining contribution of COPDGene® is taking existing clinical staging (GOLD) and defining novel phenotypes within these stages.
ECLIPSE
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) was the first large prospective cohort designed to characterize COPD with the goal of discovering novel biomarkers3,4. ECLIPSE enrolled patients over a three-year period of time including 2,164 COPD patients and 582 control subjects (of which 337 were smokers). Patients were assessed at eight different time -points with the following studies; PFTs (including body plethysmography, spirometry and forced oscillometry, but not carbon monoxide diffusing capacity), biomarkers (including exhaled breath condensate), clinical health outcomes (e.g., death and disability), CT scans, body impedance, oxygen saturation, and six-minute walk distance. The advantage of the approach in ECLIPSE, as compared to COPDGene, is that it took the basic definition of COPD and sought to define new phenotypes from that starting point over a 3-year period, e. g., the “frequent exacerbator.”
MESA-LUNG
The Multi-Ethnic Study of Atherosclerosis (MESA) is prospective cohort study that was designed to study the prevalence and progression of subclinical cardiovascular disease5. The MESA cohort enrolled a total of 6,814 subjects between the ages of 45 to 84 from 6 separate clinical sites across the United States. One of the recruitment emphases was to include a highly multi-ethnic cohort, and the participants included non-Hispanic whites, Hispanics, African Americans, and Asians. MESA-LUNG is a nested study that utilizes the data from MESA to test a specific hypothesis: that endothelial dysfunction plays a specific role in the pathogenesis of COPD, and more specifically, emphysema6. Initially MESA-LUNG recruited 3,965 randomly sampled participants from MESA that included 24% African American, 23% Hispanics, and 18% Chinese-Americans. These patients had spirometry, quantitative CT scan data, as well as a wide range of genetic and biometric data. MESA-LUNG defined cardiovascular outcomes seeking correlations with COPD within the same cohort.
UPLIFT and TIOSPIR
Although not technically a cohort study, the Understanding Potential Long-Term Impact on Function with Tiotropium (UPLIFT) was a large interventional trial that included 5993 subjects with moderate to severe COPD7. Patients worldwide were randomized to either the long-acting antimuscarinic agent (LAMA) tiotropium or placebo, in addition to their usual respiratory medications. The primary endpoint was rate of FEV1 decline. Secondary endpoints included overall- and respiratory-specific death. The addition of tiotropium conferred an improvement in FEV1 decline, but, surprisingly, the rate of cardiac-specific death was also reduced in the tiotropium group8 (HR 0.86, 95% CI 0.75–0.99), despite similar smoking rates of ~30%. UPLIFT identified a subgroup of patients with COPD and cardiovascular disease (occult or known) that benefitted from COPD-specific therapy, though pre-existing cardiovascular disease was not among the inclusion/exclusion criteria.
A similar study, Tiotropium Safety and Performance in Respimat® (TIOSPIR), randomized COPD subjects to inhaled tiotropium in different doses and different inhaler delivery devices on top of their usual non-anticholinergic medications9. TIOSPIR included >17,000 subjects with GOLD 2–4 disease, and patients with stable cardiovascular disease were included. In addition to showing that tiotropium inhaled as a dry powder using the Handihaler® or as a soft mist using the Respimat® were equally effective in standard COPD outcomes, TIOSPIR found overall low rates of cardiac events (0.1–0.2% myocardial infarction, 1.2%–1.4% cardiac death). The authors found no evidence that one delivery device for tiotropium is safer than the other or was associated with a greater risk of major adverse cardiovascular events.
It is known that a substantial proportion of patients with COPD die from cardiovascular disease10, an “overlap group”, and the UPLIFT and TIOSPIR trials suggest that this group benefits from COPD treatment. It is noteworthy, however, that some studies have not supported the concept that an “overlap group” may derive cardiovascular benefit from COPD treatment11–15. An early meta-analysis by Singh, et al. suggested as much as a 52% increased risk of mortality associated with tiotropium mist inhaler use in COPD patients12, with another meta-analysis supporting a similar conclusion13. However, the weight of the evidence, including the large randomized TIOSPIR trial that featured a pre-specified subgroup analysis involving patients with underlying cardiovascular disease, supports that tiotropium is safe as the overall hazard of major adverse cardiovascular events, including death, was not increased9,16. Post-marketing surveillance focused on cardiovascular events was recommended by the authors to validate their study findings.
Phenotyping and Biomarkers
Thoracic CT Scanning
One common feature in each of the three population studies COPDGene, ECLIPSE, and MESA-LUNG is the incorporation of computed tomography (CT) scans to identify novel radiography-based “biomarkers.” Washko et al. validated that CT scan-based measurements of airway wall attenuation are reproducible and correlate to the FEV1/FVC ratio17. This study suggests that airway measurements by CT could be complementary to spirometry. A related study determined that the total number of small airways inversely correlated with the percent of emphysema, and that total airway count was predictive of BODE score (the prognostication metric calculated by assessing Body mass index, degree of airflow Obstruction, degree of Dyspnea, and Exercise capacity)18.
Building further on the relationship between airway size, caliber, and parenchymal changes, researchers established that the distensibility of medium- to large-sized airways is reduced in individuals with a predominantly emphysema phenotype versus an airway inflammatory phenotype on CT19. When Martinez et al. assessed the correlation between radiologic features of COPD, quality of life, and symptom measures, they discovered that patients with airway-limited disease had worse St. George Respiratory Questionaire (SGRQ) scores while those with more emphysema had increased (worse) BODE scores20. Measures of air-trapping, defined as low attenuation areas of <856 Hounsfield units, were additive to the value of airway measures alone in correlating with FEV1 and FEV1/FVC ratio21. The presence of emphysema, separate from evidence of airflow limitation, was found to be associated with a lower total FEV1 and worse functional status22. ECLIPSE showed a higher risk of emphysema progression in women and active smokers. A similar risk related to gender and African American ethnicity was identified by the COPDGene group23. The biomarkers surfactant protein D (SP-D) and soluble receptor for advanced glycation endproduct (sRAGE) were more common in the progressive emphysema cohort24. However, correlation between COPD and cardiovascular disease outcomes was not the primary purpose of these studies.
Using MRI and CT scans, the MESA-LUNG and MESA-COPD investigators were the first to report that pulmonary microvascular changes are present in patients with mild, moderate, and severe COPD (defined by reduced FEV1)25. This study identified a decrease in the microvascular blood flow that was separate from the degree of emphysema present in those areas. The MESA-LUNG group also reported that CT evidence of pulmonary emphysema occurred in smokers with and without COPD, and this emphysema was associated with symptoms if it was anatomically centrilobular or panlobular but not paraseptal26. MESA-LUNG investigators also commented on the relationship with between emphysema and impaired left ventricular filling, concluding that pulmonary vein dimensions are reduced in patients with emphysema and COPD27.
Serum Biomarkers
Chronic persistent inflammation is generally thought to be a central feature of COPD, despite very little evidence that systemic anti-inflammatory therapy improves markers of inflammation. Assessment and discovery of distinct inflammatory patterns in COPD was a goal of all of the prospective cohort studies. Interestingly, Bowler et al. discovered that decreased levels of IL-16 were associated with emphysema28 and may be related to the development of autoimmunity.
The hypothesis of systemic inflammation was most comprehensively explored by the ECLIPSE investigators. They found inflammation is not present in all patients with COPD, but when present appeared to be associated with poorer outcomes29. Additionally, they found that combining biomarkers as a composite score of inflammation, including C-reactive protein (CRP), fibrinogen, and white blood count (WBC), was associated with more frequent exacerbations and co-morbidities30. Fibrinogen was found to be elevated in 36% of patients with COPD as compared to 5% of control patients, and this has been identified as a candidate biomarker to identify patients at higher risk of frequent exacerbations, hospitalization, or mortality31. Interestingly, fibrinogen is also known biomarker of cardiac disease32.
Breath Biomarkers
Breath biomarkers are an attractive and novel way to study COPD phenotypes as they are largely noninvasive and may complement existing biomarkers of disease. To date, there have been efforts to utilize breath metabolites as a diagnostic matrix from patients with developing COPD33–35 and smokers at risk of COPD36. Studies of exhaled breath condensate (EBC)—the liquid formed from breath passed through a cold tube—identified lower fluid pH and higher hydrogen peroxide levels correlating with COPD37–39. Other efforts have looked at EBC conductivity in emphysema40, EBC alpha-1-antitrypsin levels in acute COPD exacerbations41, and fractional exhalation of nitric oxide (FeNO) in COPD subjects42. Although these studies show promise, differing biomarker collection techniques and analytic methods make standardization problematic, and larger-scale studies and standardized procedures will surely advance the field. There are limited studies of exhaled biomarkers in patients with cardiovascular disease. Still, noninvasive and low-risk assessment tools that may add an important dimension to phenotyping COPD make breath analysis an exciting area of research.
COPD Associated Comorbidities
Possibly as a consequence of systemic inflammation, COPD patients are at higher risk of developing associated diseases independent of smoking-induced airway disease. COPDGene researchers reported a relationship between COPD and cardiovascular disease. Matsuoka et al. showed that the cross-section of small pulmonary arteries correlates with the degree of aortic calcification43. Another study reported that distal pruning of the pulmonary vasculature is a characteristic signature of smoking-related lung disease and associated with accelerated loss of lung tissue44. Researchers have established that seven common co-morbid conditions are associated with COPD, including sleep apnea, stroke, coronary disease, peripheral vascular disease, osteoporosis, gastroesophageal reflux, and congestive heart failure45–47. These associations are more pronounced amongst African Americans47. Additionally, cardiovascular disease was independently associated with COPD48. The prevalence of veno-thromboembolic disease was higher in patients with COPD and co-morbid conditions, and the overlap leads to worse exercise performance49. Finally two separate investigations reported an increased association between COPD and diabetes mellitus50,51. Similar findings were noted in the ECLIPSE cohort, where comorbid COPD and cardiovascular disease was associated with more symptoms31. Additionally, diabetes was identified as increasing the risk of poor clinical outcomes when associated with COPD. Depression was also identified as being more prevalent in COPD52.
COPD Immunobiology with a Focus on Vascular Disease
COPD leads to anatomic distortion of normal airway architecture, resulting in a critical reduction in airway diameter and airflow limitation53. The major mechanisms thought responsible for airflow limitation include accumulated debris and mucus in the airway lumen, chronic bronchoconstriction, airway wall thickening, and increased external airway compression from a loss of elastic tissue. However, the rate of development of airflow limitation, i.e., lung function loss, varies widely between COPD patients. Factors such as quantity and quality of toxicant exposure (e.g., tobacco smoke), innate and adaptive immune responses, and genetic and epigenetic elements that regulate airway inflammation and remodeling all contribute to the clinical progression in any single person. Clearly, the interplay between immune cells, toxicant exposure, and host background is complex and may evolve over the life of the COPD patient.
As COPD stems from abnormal lung and systemic inflammation, and advanced COPD is associated with comorbid vascular disease, there is considerable interest in understanding the immunologic links between lung and vascular inflammation. It is known that COPD and coronary arterial disease (CAD) are connected54–56, and the dominant theory is that shared risk factors (e.g., smoking) elicit a chronic inflammatory response that affects both the lungs and vasculature56–58. In fact, COPD patients with elevated levels of systemic inflammatory markers such as C-reactive protein, fibrinogen, and leukocytes have increased rates of myocardial infarction and congestive heart failure (MI and CHF) based on large cohort studies59. Efforts to unravel genetic links by comparing COPD-specific single nucleotide polymorphisms to carotid thickness and CAD are underway60. While at present a clear connection is not well established, it is imperative to understand concepts of shared cellular and molecular pathways such as oxidative stress, cell death, airway structural changes and impaired tissue repair underlying both chronic vascular conditions and COPD. The following sections discuss pulmonary structural and inflammatory cells in COPD with a focus on how these may relate to vascular inflammation (see Fig. 2).
Figure 2.
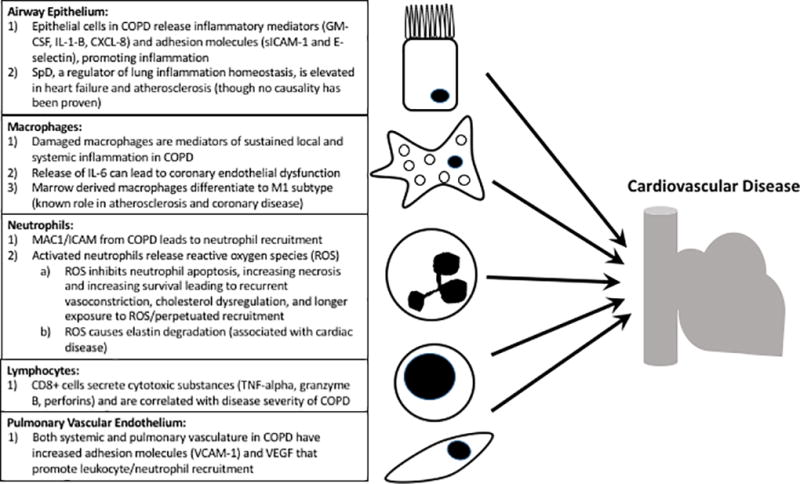
Schematic showing the overlap between dysfunctional lung structural cells and inflammatory calls in COPD that may have a connection to cardiovascular disease.
Immune cells, Inflammation, and the Lung-Vascular Connection
The gross insult by tobacco smoke to the respiratory tract is the result of repeated and prolonged exposure to a range of toxicants through inflammation and oxidative stress, or, to individual toxicants through specific mechanisms61. In COPD, damaged epithelial cells express high levels of inflammatory mediators (CXCL-8, IL-1-β, and GM-CSF)62 and adhesion molecules (sICAM-162 and E-selectin63). This inflammatory response facilitates a continuous recruitment and activation of inflammatory cells from the blood. In addition, damaged lung epithelial cells have an altered ability to regulate normal immune functions such as pathogen binding64, antigen presentation, and TNF-alpha expression65–68. In addition to its pro-inflammatory function, the airway epithelium is also responsible for maintaining immune homeostasis and protection against chronic inflammatory changes in the lung and the pulmonary vasculature. The protective function of airway epithelial cells has been attributed to the constitutive production of lung immune modulators called collectins: SP-A and SP-D. Although we currently do not have any direct evidence of a shared mechanism, SP-D-related immune regulatory pathways can be impaired in the development of atherosclerotic plaques69 and increased levels of SP-D have been observed in heart failure70 and carotid artery atherosclerosis71. These data suggest that SP-D may be a biomarker or may play a putative role in coexistent lung and vascular disease.
Alveolar macrophages are the most abundant immune cell type in the lungs and airways. They function to clear inhaled particles, identify and destroy pathogens, and remove dead or dying cells in the distal air spaces. In COPD, however, the function of these cells is severely impaired72 despite increased numbers of macrophages in COPD patients73,74. In fact, macrophages, activated locally or recruited during inflammation, can account for many of the known features of COPD74,75. Macrophages isolated from the lungs of patients with COPD exhibit reduced apoptosis and increased survival compared to those found in patients with normal lungs. Though this increased survival may be anti-inflammatory in the lung, damaged lung macrophages can produce IL-656,76, which, in turn, can potentiate coronary endothelial dysfunction77. Indeed bone marrow-derived macrophages in the COPD lung differentiate into the highly proinflammatory M1 subtype and the anti-inflammatory M2 subtype; M1 macrophages have a well-accepted pathogenic role in atherosclerosis and CAD.
Normal, healthy lung parenchyma contains few if any neutrophils. In COPD, damaged epithelial cells, activated macrophages, and T-cells (via CXCL8, CXCL1, and leukotriene B4) cause direct migration of neutrophils towards the airways. Adhesion molecules expressed on endothelial and epithelial cells mediate neutrophil migration with the MAC1/ICAM1 interactions being the most crucial, and COPD patients who smoke have increased surface expression of MAC1 on their neutrophils78. Neutrophils play a major role in COPD exacerbations elicited by air pollution, viral, and/or bacterial infections79–81. Recruited neutrophils secrete a number of pro-inflammatory cytokines that elicit reactive oxygen species (ROS) formation, which further perpetuates neutrophil recruitment82. Oxidative stress also causes elevated levels of cytokine and growth factor expression responsible for activating and preventing apoptosis of neutrophils. This effect can lead to either increased survival or necrotic death of these cells. An important feature of the COPD lung is an increased number of dead neutrophils due to necrotic cell death and a reduced ability of alveolar macrophages to perform their scavenger function. As with chronically-activated macrophages, chronic neutrophil activity can lead to repeated endothelial exposure to cytotoxic agents (i.e. ROS such as myeloperoxidase) and likely potentiate inflammatory changes, recurrent vasoconstriction, and cholesterol dysregulation83. In particular, neutrophil-derived ROS can potentiate elastin degradation which has been associated with significant comorbid cardiac disease in COPD patients84. Although the exact association between lung neutrophil activity and cardiovascular disease is not entirely clear, clinical evidence links circulating myeloperoxidase levels with adverse cardiac outcomes85–87.
Lymphocyte accumulation in the pulmonary interstitium and peribronchial areas correlate with the severity of the symptoms of COPD and are considered to be part of the mechanism leading to exacerbation of symptoms brought on by air pollution or infections79. Lymphocytes organized in follicular structures with B lymphocyte-containing germinal centers surrounded by CD4+Th1-cells have been observed in clinically advanced cases of chronic bronchitis, while increases in the numbers of CD8+ cytotoxic Tc1 lymphocytes in the alveolar wall appears to be proportional with the severity of emphysema88. Th1 cells are CD4+T-cells that lead to interferon-γ (IFN-γ) secretion, and this, in turn, helps activate CD8+ cytotoxic Tc1 cells89. CD8+ T-cells synthesize, store, and release cytokines and cytotoxic substances like tumor necrosis factor-α (TNF-α), granzyme B, and perforins, and their numbers inversely correlate with the FEV1 of patients suffering of COPD90.
Pulmonary Endothelial Cells, COPD, and Vascular Effects
Often described as the silent player in COPD pathogenesis, the pulmonary vasculature has been increasingly recognized as a major contributor to disease. Beyond their physiological function, endothelial cells also secrete a variety of pro-inflammatory molecules including cytokines, chemokines, growth factors, and lipids relevant to COPD91. Because COPD itself is a systemic inflammatory condition, both the systemic and pulmonary vasculature have enhanced expression of adhesion molecules (e.g. VCAM-1) which further promote adherence of activated leukocytes to endothelial surfaces76. The pulmonary and airway vasculature also express VEGF and various adhesion molecules important in the immune response that mediates the transmigration of neutrophils to the airways. As described above, the inflammatory milieu in COPD likely correlates with cardiovascular disease through, in part, endothelial dysfunction. However, a recent study by Chandra et al. challenges this notion as the authors did not find a significant correlation between endothelial dysfunction and reduced lung function (FEV1) in cohorts of patients with atherosclerotic disease92. This study underscores the need to better define COPD patients based on biologic parameters other than lung function in order to truly understand the link between COPD and cardiovascular disease.
Alterations in the structure of the pulmonary vasculature in COPD contributes to the development of pulmonary arterial hypertension (PAH) which is associated with reduced survival in COPD and has higher prevalence in more advanced disease93. The underlying dysfunction of the endothelial compartment in COPD leads to an imbalance between vasoconstrictive and vasodilatory mediators further contributing to the development of PAH. This imbalance is in part driven by cigarette smoke which also damages pulmonary endothelial cells via protease activity, dysregulated apoptosis, and oxidative stress94. The development of alveolar destruction and emphysema is in part also due to this vasculopathy. Pulmonary capillary septal endothelial cell apoptosis and reduced local alveolar production of VEGF and its receptor VEGFRII also contribute to the development of emphysema. Interestingly, in healthy smokers who quit smoking, pulmonary capillary apoptosis is reversible. However, in patients with COPD this mechanism of endothelial cell apoptosis continues to be active despite smoking cessation further contributing to the development of progressive airflow obstruction95. This may explain in part the continued decline in lung function over many years in COPD despite smoking cessation.
COPD Treatment: Focusing on Comorbid Cardiac Disease
Current COPD Therapies
Several excellent reviews of the pharmacological treatment of COPD have been written96–98. The GOLD guidelines (2017) use patient grouping (Groups A–D) based on spirometry (FEV1), frequency of exacerbations, and burden symptoms as assessed by symptom scores to guide treatment considerations. In addition to smoking cessation and vaccines, GOLD treatment guidelines use a step-up approach based on Groups A–D with the goals to reduce symptoms with combination bronchodilators and to reduce risks, particularly acute exacerbations with anticholinergic bronchodilators, and, if indicated, inhaled corticosteroids or roflumilast. (see Fig. 1). Table 1 summarizes the currently available combination inhalers for maintenance therapy. No current therapies have been demonstrated to change the natural course of COPD except for smoking cessation.
Figure 1. Recommended therapy for stable COPD by GOLD category.
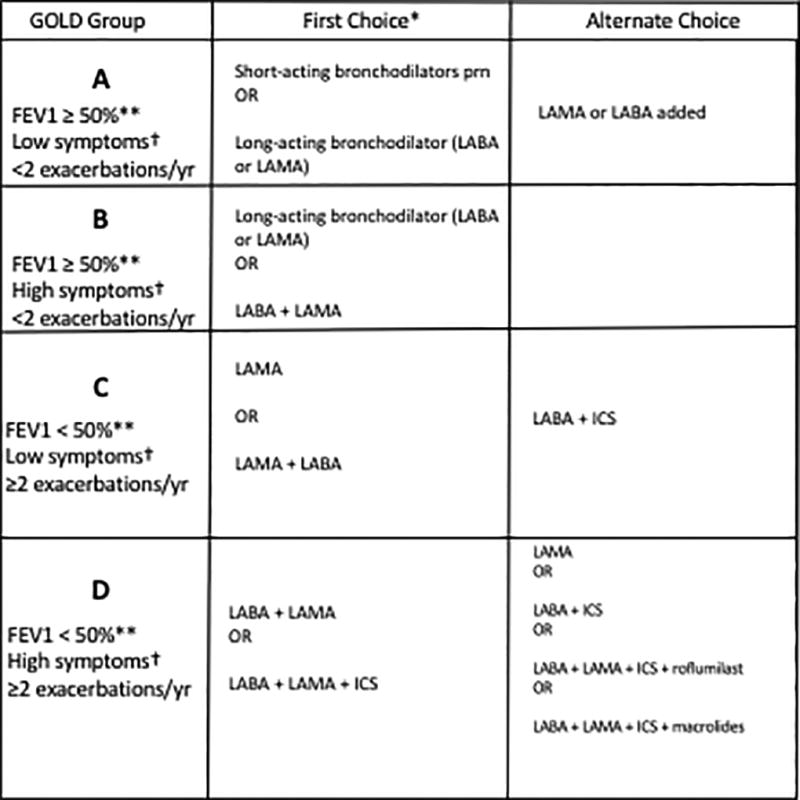
Figure adapted from the recommendations of the Global Initiative for Chronic Obstructive Lung Disease1 (www.goldcopd.org, accessed Jan 2017). LABA, long-acting beta-2-agonist; LAMA, long-acting anti-muscarinic; ICS, inhaled corticosteroid.
*First choice therapy includes short-acting beta2-agonists or short-acting anticholinergic medications as needed for all categories. First choice therapy also includes the first entry followed by a clinical evaluation. If the patient still has symptoms, then moving to the second entry is advised.
**FEV1 impairment is FEV1≥50% predicted for GOLD Categories A and B, and FEV1<50% predicted for Categories C and D.
†Symptoms based on the modified Medical Research Council (mMRC) scale: 0-shortness of breath (SOB) with strenuous exertion, 1-SOB with hurrying on level ground or inclines, 2-SOB with normal walking on level ground >100m, 3-SOB within 100m, and 4-SOB with daily activities; mMRC <2 = low symptoms and mMRC ≥2 = high symptoms.
Table 1.
Combination Drug Inhalers Used for Maintenance Treatment of COPD
| Drug 1 + Drug 2 | DI(mcg) | DF | Type DD | DD Name | Name | |
|---|---|---|---|---|---|---|
| LABA + LAMA | ||||||
| Indacterol | glycopyrronium | 110/50 | qd | DP | Ultibro Breezhaler® | QVQ149 |
| Vilanterol | umeclidinium | 25/62.5 | qd | DP | Ellipta® | Anoro® |
| Olodaterol | tiotropium | 3.5/2.5 | qd | SDM | Respimat® | Stiolto® |
| Formoterol | aclidinium | 12/400 | bid | DP | Genuair® | Duaklir® |
| Formoterol | glycopyrrolate | 4.8/9 | bid | MDI | Bevespi Aerosphere | |
|
| ||||||
| SABA + SAMA | ||||||
| Albuterol | ipratropium | 2.5/0.5 (mg) | q6h | Neb | DuoNeb® | |
| Albuterol | ipratropium | 0.1/0.33 (mg) | q6h | SDM | Respimat® | Combivent® |
|
| ||||||
| LABA + ICS | ||||||
| Vilanterol | fluticasone F | 25/100 | qd | DP | Ellipta® | Breo® |
| Formoterol | budesonide | 4.5/160 or 80 | bid | MDI | Symbicort® | |
| Formoterol | budesonide | 6,6,12/100,200,400 | bid | DP | Turbuhaler® | Symbicort® |
| Formoterol | mometasone | 5/100 or 200 | bid | MDI | Dulera®A | |
| Salmeterol | fluticasone P | 50/100,250,500 | bid | DP | Diskus® | Advair® |
| Salmeterol | fluticasone P | 21/45,115,230 | bid | MDI | Advair® HFAA | |
Abbrv. DI = dose per inhalation; DF = dose frequency; Type DD = type of delivery device; qd = once a day; q6h = every six hours; bid = twice a day; DP = dry powder; Neb = nebulization; SDM = spring driven mist; F = furoate; P = propionate; A = approved for asthma indication only, all others approved for COPD or COPD + asthma; LABA = long-acting beta2 agonist; LAMA = long-acting muscarinic antagonist; SABA = short-acting beta2 agonist; SAMA = short-acting muscarinic antagonist; ICS = inhaled corticosteroid
Novel and Investigative COPD Therapies
Several approaches to new drug therapy for COPD are ongoing. These include novel agents that are dual phosphodiesterase (PDE3 and PDE4) inhibitors and other agents that are more specific PDE4 inhibitors. Some of these can potentially be delivered by inhalation98. Novel macrolide/fluoroketolide compounds appear to have better anti-inflammatory profiles than current marcolides and may be useful in treating COPD. Agents that are antagonists of the human CXCR2 receptor modulating neutrophil trafficking have potential in the treatment and prevention of COPD. The p38 mitogen-activated protein kinase (p38 MAPK) inhibitors also have potential in COPD98.
Agents that antagonize matrix metalloproteinases (MMP) have the potential to inhibit the development of emphysema and small airway fibrosis in animal models but none have been effective in humans. Many new anti-cytokine therapies that have potential use in the treatment of COPD including humanized monoclonal antibodies directed at interleukin (IL-5 and IL-17) receptors. Phosphoinositide 3-kinase inhibitors, soluble epoxide hydrolase inhibitors and orally active, gamma-selective retinoid agonists are new potential approaches to treating COPD98. Exciting new approaches to the treatment and prevention of COPD are on the horizon.
The statin drugs (‘statins’) have garnered much interest as a potential therapy for COPD. Despite several large retrospective studies that suggest statins have a benefit in preserving lung function and reducing mortality and morbidity in patients with COPD99, prospective studies have failed to show an advantage in COPD patients without significant cardiovascular risk factor100. The STATCOPE clinical trial did not show that simvastatin reduced exacerbations in patients with moderate-to-severe COPD101. However, smaller clinical trials with pravastatin did show benefit in patients with COPD. In two randomized clinical trials, pravastatin was associated with increased exercise time and reduced systemic inflammation in COPD102, and in COPD patients with pulmonary hypertension treatment with pravastatin increased functional capacity and exercise time, reduced systolic pulmonary pressures, and improved the BORG dyspnea score103. Based on these data in sum, statins cannot be recommended for the treatment of COPD especially with the results of the STATCOPE trial. However, one limitation in the STATCOPE study is it did not include COPD patients with overt cardiovascular disease or those with significant cardiac risk factors. STATCOPE excluded the very group of COPD patients who benefited from statin use in multiple observational studies104.
Cardiac Treatment in Patients with COPD
The association of tobacco use and COPD is unequivocal and puts patients with COPD at higher risk for cardiovascular comorbidities105. Patients with COPD are more likely to have cardiovascular disease than matched non-COPD populations (odds ratio (OR) = 2.46 (95% CI 2.02–3.00, p<0.00001)106. This includes a 2 to 5 time increased risk for MI, cardiac dysrhythmia, CHF, disease of the pulmonary vasculature, and peripheral vascular diseases. Hypertension is also more common in COPD patients (OR = 1.33, 95% CI 1.13–1.56, p=0.0007)106. Medications used to treat these cardiovascular comorbidities such as diuretics and beta-blockers can have potential detrimental drug-disease interactions and effects in COPD patients.
The treatment of hypertension in patients with COPD has been reviewed elsewhere107. Thiazide (hydrochlorothiazide, chlorthalidone) and loop (furosemide, bumetanide, torsemide) diuretics used in the treatment of hypertension and CHF can cause serious toxicity through urinary potassium losses. This can be exacerbated when diuretics are used with inhaled beta-2-receptor agonists, which cause the movement of potassium into the cell. This combination can lead to severe hypokalemia. These drugs can also generate a volume-contraction metabolic alkalosis leading to a further suppression in ventilatory drive, thus resulting in worsening hypoxemia and hypercapnia. Alkalemia also increase risk for cardiac arrhythmias further exacerbating cardiac disease.
Angiotensin-converting enzyme (ACE) inhibitors are effective in the control of hypertension and the treatment of CHF in COPD patients. However, because 5 to 20% of the patients on ACE inhibitors can develop cough, they must be used with caution in COPD. Prior use of ACE inhibitors has been shown to reduce mortality in COPD patients admitted with exacerbations107. Although ACE inhibitors have been suggested to improve skeletal muscle function in COPD patients, a recent randomized controlled 3-month trial of the ACE inhibitor fosinopril in COPD patients failed to show improvement in strength of the quadriceps or exercise performance108.
Amiodarone is a class III antiarrhythmic drug used to treat complex and life threatening cardiac arrhythmias. The use of amiodarone is associated with significant pulmonary toxicity. In a large study of patients with atrial fibrillation, amiodarone use was associated with a nearly 40% increase in pulmonary toxicity in males compared to females (HR = 1.37, 95% CI 1.19–1.57, p<0.0001) and more than a doubled risk in pulmonary toxicity was seen in patients with COPD (HR = 2.53, 95% CI 2.21–2.89, p<0.0001)109. Approximately 3.1% of atrial fibrillation patients without pre-existing pulmonary disease were found to have pulmonary toxicity after four years of taking amiodarone compared to 5.9% (p=0.015) of those patients with pre-existing pulmonary disease in another study110. Patients with CHF and COPD who were treated with amiodarone and survived at least 1 year had a significantly greater decrease in lung diffusion capacity (DLCO) compared to patients treated with placebo (2.05 vs. 0.09 ml/min per mm Hg, p = 0.008) but had no difference in survival-free of cardiac deaths111. Taken together, these limited data suggest that the risk-benefit ratio must be considered before treating patients that have significant COPD with amiodarone and they need to be carefully monitored with chest imaging and DLCO measurements while on amiodarone.
As noted above, the risk of cardiovascular disease is increased in COPD patients106. Beta-blockers are used widely in the treatment of CHF, hypertension, atrial fibrillation, and MI. Non-selective beta-blockers such as propranolol have been shown to have a negative effect on lung function (FEV1, FVC, and FEV1% predicted) as compared to beta-1 selective receptor blockers like atenolol. This effect holds true both at baseline and after albuterol inhalation in patients with COPD or asthma112,113. Non-selective beta-blocking agents should therefore be avoided in COPD patients in favor of the more selective beta1-receptor blocker agents.
Use of labetalol, a non-selective beta-blocker that also blocks alpha1-receptor, did not affect FEV1 or the mid-expiratory flow volumes in patients with COPD and hypertension two hours after the administration of the maximum labetalol dose114. Another non-selective beta-blocker/alpha1-receptor blocker, carvedilol, was studied in patients with CHF and COPD and compared to the selective beta1-blockers metoprolol and bisoprolol. Six-minute walk and left ventricular ejection fraction did not change with the three drugs. However, FEV1 was lowest with carvedilol, better in metoprolol, and best in the patients treated with bisoprolol115. In CHF patients with COPD (n=31) or asthma (n=12), 3.2% of COPD patients and 25% of asthma patients developed wheezing after starting carvedilol116. In contrast, actual improvement in peak expiratory flow rate (PEFR) of 17% (p = 0.04) was seen in COPD patients and 4% (p=NS) in asthma patients two hours after starting carvedilol. Beta- and alpha-adrenergic blocking agents should be used with caution in patients with COPD until more information is available.
In COPD patients who had an MI, those discharged on beta-blockers compared to those who did not had a lower all-cause mortality after adjusting for confounders (HR = 0.87, 95% CI 0.64–0.95) during a follow-up period that was as long as 7.2 years117. More impressive was the survival advantage seen in those COPD patients discharged on a beta-blocker after a MI and who also had CHF (HR=0.77, 95% CI 0.63–0.95).
Meta-analysis of the use of selective beta1-receptor blockers for hypertension, CHF, coronary artery disease and during the perioperative period in COPD patients concluded that they did not produce adverse respiratory effects118. However, a large prospective cohort observational trial showed that both cardio-selective and non-cardio-selective beta blockers in patients without lung disease were associated with significant reductions in FEV1 measures over a mean of 6.1+0.5 years. The use of selective beta1-blockers resulted in less reduction in FEV1 (−118 ml, 95% CI −157 to −78, p <0.001) than the reduction seen with the use of non-cardio-selective beta blockers (−198 ml, 95% CI −301 to −96, p <0.001)119. When patients with COPD, asthma, and CHF were included, the same trends held.
In a clinical trial where patients with COPD and CHF were randomized to either selective the beta1-blocker bisoprolol or the non-selective beta-blocker/alpha1-blocker carvedilol, both agents reduced heart rate and had no effect on N-terminal pro brain natriuretic peptide (BNP). Bisoprolol, but not carvedilol, significantly increased FEV1 by 127 ml120. Another randomized triple-crossover trial evaluated carvedilol, metoprolol, and bisoprolol in CHF patients and found that in those patients with COPD, bisoprolol had the highest and carvedilol the lowest FEV1 measurements115. However, bisoprolol use is also associated with worsening dynamic hyperinflation compared to placebo in moderate-to-severe COPD patients without reducing the duration of exercise121. Conversely, the rate of CHF and/or COPD exacerbations were higher in the those patients treated with carvedilol as compared to bisoprolol122.
Beyond lung function, beta-blockers have been associated with important hard outcomes such as mortality. A mortality advantage was seen with the use of bisoprolol, but not carvedilol or metoprolol, in patients with COPD and CHF123. Another study demonstrated reduced mortality rates in COPD patients with CHF on bisoprolol or carvedilol (HR = 0.41, 95% CI 0.17–0.99, P=0.047). In a large Scottish retrospective cohort study of beta-blockers with a mean follow-up of 4.35 years, there was a 22% reduction in overall mortality in COPD patients taking beta-blockers124.
Two large trials have demonstrated significant reductions in COPD exacerbations regardless of the severity of airflow obstruction when the patients are on beta-blockers125,126. A trial of 520 COPD patients undergoing lung resection found that the use of perioperative beta-blockers compared to not using them did not change the rate of postoperative COPD exacerbations (5.4% versus 6.3%)127. Selective beta1-receptor blockers appear to have an advantage over non-selective beta-blockers in COPD patients with CHF, hypertension and MI’s but the advantages have been small and not always consistent.
Summary
COPD is the third most common cause of death worldwide. The definition of COPD is evolving, due to complex disease mechanisms, clinical heterogeneity, and variable immune response to inhaled toxicants and environmental pollutants. Large cohort studies are important to help define COPD phenotypes and identify useful biomarkers, and these studies give rise to important and testable clinical questions such as how patients with certain radiologic features respond to therapeutic interventions. As our understanding of COPD immunobiology improves, we may better identify specific and effective immune-modulating therapies at various stages of COPD, including monoclonal antibodies in the current age of biologics and precision medicine. The recognition that COPD often coexists with cardiovascular disease underscores the link between these disorders. Therapies directed at both COPD and heart disease seem to confer benefit beyond treating each separately, and the future of COPD research and treatment approaches needs to bear this in mind.
Acknowledgments
The authors graciously acknowledge the following funding agencies for this work: NIH-NHLBI K23HL127185 [MS]; NIH-NIAID R2111612 [AH]; NIH-NHLBI K08HL114882 [AZ]
Footnotes
Conflict of Interest
No conflicts of interest are reported by the authors for this manuscript
References
- 1.2017 GIfCOLDG. The Global Strategy for the Diagnosis, Management, and Prevention of COPD. 2017 http://goldcopd.org/
- 2.Wan ES, Cho MH, Boutaoui N, et al. Genome-wide association analysis of body mass in chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology. 2011;45:304–10. doi: 10.1165/rcmb.2010-0294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rennard SI, Locantore N, Delafont B, et al. Identification of five chronic obstructive pulmonary disease subgroups with different prognoses in the ECLIPSE cohort using cluster analysis. Annals of the American Thoracic Society. 2015;12:303–12. doi: 10.1513/AnnalsATS.201403-125OC. [DOI] [PubMed] [Google Scholar]
- 4.Rennard SI. The Promise of Observational Studies (ECLIPSE, SPIROMICS, and COPDGene) in Achieving the Goal of Personalized Treatment of Chronic Obstructive Pulmonary Disease. Seminars in respiratory and critical care medicine. 2015;36:478–90. doi: 10.1055/s-0035-1555609. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama K, Donekal S, Venkatesh BA, et al. Natural History of Myocardial Function in an Adult Human Population: Serial Longitudinal Observations From MESA. JACC Cardiovascular imaging. 2016;9:1164–73. doi: 10.1016/j.jcmg.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 6.Barr RG, Ahmed FS, Carr JJ, et al. Subclinical atherosclerosis, airflow obstruction and emphysema: the MESA Lung Study. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2012;39:846–54. doi: 10.1183/09031936.00165410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. The New England journal of medicine. 2008;359:1543–54. doi: 10.1056/NEJMoa0805800. [DOI] [PubMed] [Google Scholar]
- 8.Celli B, Decramer M, Kesten S, et al. Mortality in the 4-year trial of tiotropium (UPLIFT) in patients with chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 2009;180:948–55. doi: 10.1164/rccm.200906-0876OC. [DOI] [PubMed] [Google Scholar]
- 9.Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat inhaler and the risk of death in COPD. The New England journal of medicine. 2013;369:1491–501. doi: 10.1056/NEJMoa1303342. [DOI] [PubMed] [Google Scholar]
- 10.Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. American journal of respiratory and critical care medicine. 2012;186:975–81. doi: 10.1164/rccm.201207-1299OC. [DOI] [PubMed] [Google Scholar]
- 11.Gershon A, Croxford R, Calzavara A, et al. Cardiovascular safety of inhaled long-acting bronchodilators in individuals with chronic obstructive pulmonary disease. JAMA Intern Med. 2013;173:1175–85. doi: 10.1001/jamainternmed.2013.1016. [DOI] [PubMed] [Google Scholar]
- 12.Singh S, Loke YK, Enright PL, Furberg CD. Mortality associated with tiotropium mist inhaler in patients with chronic obstructive pulmonary disease: systematic review and meta-analysis of randomised controlled trials. Bmj. 2011;342:d3215. doi: 10.1136/bmj.d3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong YH, Lin HH, Shau WY, Wu YC, Chang CH, Lai MS. Comparative safety of inhaled medications in patients with chronic obstructive pulmonary disease: systematic review and mixed treatment comparison meta-analysis of randomised controlled trials. Thorax. 2013;68:48–56. doi: 10.1136/thoraxjnl-2012-201926. [DOI] [PubMed] [Google Scholar]
- 14.Karner C, Chong J, Poole P. Tiotropium versus placebo for chronic obstructive pulmonary disease. The Cochrane database of systematic reviews. 2014:CD009285. doi: 10.1002/14651858.CD009285.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verhamme KM, Sturkenboom MC, Brusselle GG. Use of tiotropium Respimat versus HandiHaler and mortality in patients with COPD. The European respiratory journal. 2014;43:1818–9. doi: 10.1183/09031936.00036314. [DOI] [PubMed] [Google Scholar]
- 16.Tan CK, Say GQ, Geake JB. Long-term safety of tiotropium delivered by Respimat(R) SoftMist Inhaler: patient selection and special considerations. Ther Clin Risk Manag. 2016;12:1433–44. doi: 10.2147/TCRM.S109011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Washko GR, Dransfield MT, Estepar RS, et al. Airway wall attenuation: a biomarker of airway disease in subjects with COPD. J Appl Physiol (1985) 2009;107:185–91. doi: 10.1152/japplphysiol.00216.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz AA, Valim C, Yamashiro T, et al. Airway count and emphysema assessed by chest CT imaging predicts clinical outcome in smokers. Chest. 2010;138:880–7. doi: 10.1378/chest.10-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz AA, Come CE, Ross JC, et al. Association between airway caliber changes with lung inflation and emphysema assessed by volumetric CT scan in subjects with COPD. Chest. 2012;141:736–44. doi: 10.1378/chest.11-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez CH, Chen YH, Westgate PM, et al. Relationship between quantitative CT metrics and health status and BODE in chronic obstructive pulmonary disease. Thorax. 2012;67:399–406. doi: 10.1136/thoraxjnl-2011-201185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder JD, McKenzie AS, Zach JA, et al. Relationships between airflow obstruction and quantitative CT measurements of emphysema, air trapping, and airways in subjects with and without chronic obstructive pulmonary disease. AJR American journal of roentgenology. 2013;201:W460–70. doi: 10.2214/AJR.12.10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castaldi PJ, San Jose Estepar R, Mendoza CS, et al. Distinct quantitative computed tomography emphysema patterns are associated with physiology and function in smokers. American journal of respiratory and critical care medicine. 2013;188:1083–90. doi: 10.1164/rccm.201305-0873OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foreman MG, Zhang L, Murphy J, et al. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPDGene Study. American journal of respiratory and critical care medicine. 2011;184:414–20. doi: 10.1164/rccm.201011-1928OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coxson HO, Dirksen A, Edwards LD, et al. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: a prospective analysis from the ECLIPSE study. The lancet Respiratory medicine. 2013;1:129–36. doi: 10.1016/S2213-2600(13)70006-7. [DOI] [PubMed] [Google Scholar]
- 25.Hueper K, Vogel-Claussen J, Parikh MA, et al. Pulmonary Microvascular Blood Flow in Mild Chronic Obstructive Pulmonary Disease and Emphysema. The MESA COPD Study. American journal of respiratory and critical care medicine. 2015;192:570–80. doi: 10.1164/rccm.201411-2120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith BM, Austin JH, Newell JD, Jr, et al. Pulmonary emphysema subtypes on computed tomography: the MESA COPD study. The American journal of medicine. 2014;127:94, e7–23. doi: 10.1016/j.amjmed.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith BM, Prince MR, Hoffman EA, et al. Impaired left ventricular filling in COPD and emphysema: is it the heart or the lungs? The Multi-Ethnic Study of Atherosclerosis COPD Study. Chest. 2013;144:1143–51. doi: 10.1378/chest.13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowler RP, Bahr TM, Hughes G, et al. Integrative omics approach identifies interleukin-16 as a biomarker of emphysema. Omics : a journal of integrative biology. 2013;17:619–26. doi: 10.1089/omi.2013.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PloS one. 2012;7:e37483. doi: 10.1371/journal.pone.0037483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA : the journal of the American Medical Association. 2013;309:2353–61. doi: 10.1001/jama.2013.5732. [DOI] [PubMed] [Google Scholar]
- 31.Miller J, Edwards LD, Agusti A, et al. Comorbidity, systemic inflammation and outcomes in the ECLIPSE cohort. Respir Med. 2013;107:1376–84. doi: 10.1016/j.rmed.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Emerging Risk Factors C, Kaptoge S, Di Angelantonio E, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. The New England journal of medicine. 2012;367:1310–20. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Besa V, Teschler H, Kurth I, et al. Exhaled volatile organic compounds discriminate patients with chronic obstructive pulmonary disease from healthy subjects. International journal of chronic obstructive pulmonary disease. 2015;10:399–406. doi: 10.2147/COPD.S76212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basanta M, Ibrahim B, Dockry R, et al. Exhaled volatile organic compounds for phenotyping chronic obstructive pulmonary disease: a cross-sectional study. Respiratory research. 2012;13:72. doi: 10.1186/1465-9921-13-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schivo M, Seichter F, Aksenov AA, et al. A mobile instrumentation platform to distinguish airway disorders. Journal of breath research. 2013;7:017113. doi: 10.1088/1752-7155/7/1/017113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malerba M, Montuschi P. Non-invasive biomarkers of lung inflammation in smoking subjects. Current medicinal chemistry. 2012;19:187–96. doi: 10.2174/092986712803414204. [DOI] [PubMed] [Google Scholar]
- 37.Murata K, Fujimoto K, Kitaguchi Y, Horiuchi T, Kubo K, Honda T. Hydrogen peroxide content and pH of expired breath condensate from patients with asthma and COPD. Copd. 2014;11:81–7. doi: 10.3109/15412555.2013.830094. [DOI] [PubMed] [Google Scholar]
- 38.Inonu H, Doruk S, Sahin S, et al. Oxidative stress levels in exhaled breath condensate associated with COPD and smoking. Respiratory care. 2012;57:413–9. doi: 10.4187/respcare.01302. [DOI] [PubMed] [Google Scholar]
- 39.MacNee W, Rennard SI, Hunt JF, et al. Evaluation of exhaled breath condensate pH as a biomarker for COPD. Respiratory medicine. 2011;105:1037–45. doi: 10.1016/j.rmed.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Stolk J, Fumagalli M, Viglio S, Iadarola P. Conductivity in Exhaled Breath Condensate from Subjects with Emphysema and Type ZZ alpha-1-Antitrypsin Deficiency. Copd. 2015;12(Suppl 1):32–5. doi: 10.3109/15412555.2015.1021910. [DOI] [PubMed] [Google Scholar]
- 41.Koczulla AR, Noeske S, Herr C, et al. Alpha-1 antitrypsin is elevated in exhaled breath condensate and serum in exacerbated COPD patients. Respiratory medicine. 2012;106:120–6. doi: 10.1016/j.rmed.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 42.Malerba M, Radaeli A, Olivini A, et al. Exhaled nitric oxide as a biomarker in COPD and related comorbidities. BioMed research international. 2014;2014:271918. doi: 10.1155/2014/271918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuoka S, Yamashiro T, Diaz A, et al. The relationship between small pulmonary vascular alteration and aortic atherosclerosis in chronic obstructive pulmonary disease: quantitative CT analysis. Acad Radiol. 2011;18:40–6. doi: 10.1016/j.acra.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Estepar RS, Kinney GL, Black-Shinn JL, et al. Computed tomographic measures of pulmonary vascular morphology in smokers and their clinical implications. Am J Respir Crit Care Med. 2013;188:231–9. doi: 10.1164/rccm.201301-0162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laforest L, Roche N, Devouassoux G, et al. Frequency of comorbidities in chronic obstructive pulmonary disease, and impact on all-cause mortality: A population-based cohort study. Respir Med. 2016;117:33–9. doi: 10.1016/j.rmed.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 46.Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1:73–83. doi: 10.1016/S2213-2600(12)70060-7. [DOI] [PubMed] [Google Scholar]
- 47.Putcha N, Han MK, Martinez CH, et al. Comorbidities of COPD have a major impact on clinical outcomes, particularly in African Americans. Chronic Obstr Pulm Dis. 2014;1:105–14. doi: 10.15326/jcopdf.1.1.2014.0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Black-Shinn JL, Kinney GL, Wise AL, et al. Cardiovascular disease is associated with COPD severity and reduced functional status and quality of life. COPD. 2014;11:546–51. doi: 10.3109/15412555.2014.898029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim V, Goel N, Gangar J, et al. Risk Factors for Venous Thromboembolism in Chronic Obstructive Pulmonary Disease. Chronic Obstr Pulm Dis. 2014;1:239–49. doi: 10.15326/jcopdf.1.2.2014.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinney GL, Black-Shinn JL, Wan ES, et al. Pulmonary function reduction in diabetes with and without chronic obstructive pulmonary disease. Diabetes Care. 2014;37:389–95. doi: 10.2337/dc13-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hersh CP, Make BJ, Lynch DA, et al. Non-emphysematous chronic obstructive pulmonary disease is associated with diabetes mellitus. BMC Pulm Med. 2014;14:164. doi: 10.1186/1471-2466-14-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanania NA, Mullerova H, Locantore NW, et al. Determinants of depression in the ECLIPSE chronic obstructive pulmonary disease cohort. Am J Respir Crit Care Med. 2011;183:604–11. doi: 10.1164/rccm.201003-0472OC. [DOI] [PubMed] [Google Scholar]
- 53.Ge MQ, Kokalari B, Flayer CH, et al. Cutting Edge: Role of NK Cells and Surfactant Protein D in Dendritic Cell Lymph Node Homing: Effects of Ozone Exposure. J Immunol. 2016;196:553–7. doi: 10.4049/jimmunol.1403042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Man SF, Leipsic JA, Man JP, Sin DD. Is atherosclerotic heart disease in COPD a distinct phenotype? Chest. 2011;140:569–71. doi: 10.1378/chest.11-0928. [DOI] [PubMed] [Google Scholar]
- 55.Williams MC, Murchison JT, Edwards LD, et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax. 2014;69:718–23. doi: 10.1136/thoraxjnl-2012-203151. [DOI] [PubMed] [Google Scholar]
- 56.Roversi S, Roversi P, Spadafora G, Rossi R, Fabbri LM. Coronary artery disease concomitant with chronic obstructive pulmonary disease. Eur J Clin Invest. 2014;44:93–102. doi: 10.1111/eci.12181. [DOI] [PubMed] [Google Scholar]
- 57.Man SF, Van Eeden S, Sin DD. Vascular risk in chronic obstructive pulmonary disease: role of inflammation and other mediators. Can J Cardiol. 2012;28:653–61. doi: 10.1016/j.cjca.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 58.Boschetto P, Beghe B, Fabbri LM, Ceconi C. Link between chronic obstructive pulmonary disease and coronary artery disease: implication for clinical practice. Respirology. 2012;17:422–31. doi: 10.1111/j.1440-1843.2011.02118.x. [DOI] [PubMed] [Google Scholar]
- 59.Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG. Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:982–8. doi: 10.1164/rccm.201206-1113OC. [DOI] [PubMed] [Google Scholar]
- 60.Sabater-Lleal M, Malarstig A, Folkersen L, et al. Common genetic determinants of lung function, subclinical atherosclerosis and risk of coronary artery disease. PLoS One. 2014;9:e104082. doi: 10.1371/journal.pone.0104082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shepperd CJ, Newland N, Eldridge A, et al. Changes in levels of biomarkers of exposure and biological effect in a controlled study of smokers switched from conventional cigarettes to reduced-toxicant-prototype cigarettes. Regul Toxicol Pharmacol. 2015;72:273–91. doi: 10.1016/j.yrtph.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 62.Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir Res. 2002;3:22. doi: 10.1186/rr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Stefano A, Maestrelli P, Roggeri A, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med. 1994;149:803–10. doi: 10.1164/ajrccm.149.3.7509705. [DOI] [PubMed] [Google Scholar]
- 64.Rosseau S, Guenther A, Seeger W, Lohmeyer J. Phagocytosis of viable Candida albicans by alveolar macrophages: lack of opsonin function of surfactant protein A. J Infect Dis. 1997;175:421–8. doi: 10.1093/infdis/175.2.421. [DOI] [PubMed] [Google Scholar]
- 65.Brinker KG, Martin E, Borron P, et al. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1453–63. doi: 10.1152/ajplung.2001.281.6.L1453. [DOI] [PubMed] [Google Scholar]
- 66.Brinker KG, Garner H, Wright JR. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L232–41. doi: 10.1152/ajplung.00187.2002. [DOI] [PubMed] [Google Scholar]
- 67.Hansen S, Lo B, Evans K, Neophytou P, Holmskov U, Wright JR. Surfactant protein D augments bacterial association but attenuates major histocompatibility complex class II presentation of bacterial antigens. Am J Respir Cell Mol Biol. 2007;36:94–102. doi: 10.1165/rcmb.2006-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hortobagyi L, Kierstein S, Krytska K, et al. Surfactant protein D inhibits TNF-alpha production by macrophages and dendritic cells in mice. J Allergy Clin Immunol. 2008;122:521–8. doi: 10.1016/j.jaci.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrow AD, Palarasah Y, Bugatti M, et al. OSCAR is a receptor for surfactant protein D that activates TNF-alpha release from human CCR2+ inflammatory monocytes. J Immunol. 2015;194:3317–26. doi: 10.4049/jimmunol.1402289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gargiulo P, Banfi C, Ghilardi S, et al. Surfactant-derived proteins as markers of alveolar membrane damage in heart failure. PLoS One. 2014;9:e115030. doi: 10.1371/journal.pone.0115030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu F, Zhong Q, Gong J, Qin Y, Cui L, Yuan H. Serum Surfactant Protein D is Associated with Atherosclerosis of the Carotid Artery in Patients on Maintenance Hemodialysis. Clin Lab. 2016;62:97–104. doi: 10.7754/clin.lab.2015.150536. [DOI] [PubMed] [Google Scholar]
- 72.Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–96. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- 73.Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152:1666–72. doi: 10.1164/ajrccm.152.5.7582312. [DOI] [PubMed] [Google Scholar]
- 74.Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- 75.Barnes PJ. Alveolar macrophages in chronic obstructive pulmonary disease (COPD) Cell Mol Biol (Noisy-le-grand) 2004;50 Online Pub:OL627-37. [PubMed] [Google Scholar]
- 76.van Eeden SF, Sin DD. Chronic obstructive pulmonary disease: a chronic systemic inflammatory disease. Respiration; international review of thoracic diseases. 2008;75:224–38. doi: 10.1159/000111820. [DOI] [PubMed] [Google Scholar]
- 77.Zhang J, Alcaide P, Liu L, et al. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS One. 2011;6:e14525. doi: 10.1371/journal.pone.0014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domagala-Kulawik J. Effects of cigarette smoke on the lung and systemic immunity. J Physiol Pharmacol. 2008;59(Suppl 6):19–34. [PubMed] [Google Scholar]
- 79.Papi A, Luppi F, Franco F, Fabbri LM. Pathophysiology of exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:245–51. doi: 10.1513/pats.200512-125SF. [DOI] [PubMed] [Google Scholar]
- 80.Tsoumakidou M, Siafakas NM. Novel insights into the aetiology and pathophysiology of increased airway inflammation during COPD exacerbations. Respir Res. 2006;7:80. doi: 10.1186/1465-9921-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Celli BR, Barnes PJ. Exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 2007;29:1224–38. doi: 10.1183/09031936.00109906. [DOI] [PubMed] [Google Scholar]
- 82.Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys. 2005;43:167–88. doi: 10.1385/CBB:43:1:167. [DOI] [PubMed] [Google Scholar]
- 83.King PT. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin Transl Med. 2015;4:68. doi: 10.1186/s40169-015-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rabinovich RA, Miller BE, Wrobel K, et al. Circulating desmosine levels do not predict emphysema progression but are associated with cardiovascular risk and mortality in COPD. Eur Respir J. 2016;47:1365–73. doi: 10.1183/13993003.01824-2015. [DOI] [PubMed] [Google Scholar]
- 85.Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem. 2011;57:33–9. doi: 10.1373/clinchem.2010.152827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meuwese MC, Stroes ES, Hazen SL, et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. Journal of the American College of Cardiology. 2007;50:159–65. doi: 10.1016/j.jacc.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 87.Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. The New England journal of medicine. 2003;349:1595–604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 88.Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–53. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 89.Barnes PJ, Rennard SI. Pathophysiology of COPD. In: Barnes PJ, Drazen JM, Rennard SI, Thomson NC, editors. Asthma and COPD: Basic Mechanisms and Clinical Management. 2. Elsevier; 2009. [Google Scholar]
- 90.Freeman CM, Han MK, Martinez FJ, et al. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. 2010;184:6504–13. doi: 10.4049/jimmunol.1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138:16–27. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 92.Chandra D, Gupta A, Strollo PJ, Jr, et al. Airflow Limitation and Endothelial Dysfunction. Unrelated and Independent Predictors of Atherosclerosis. Am J Respir Crit Care Med. 2016;194:38–47. doi: 10.1164/rccm.201510-2093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blanco I, Piccari L, Barbera JA. Pulmonary vasculature in COPD: The silent component. Respirology. 2016;21:984–94. doi: 10.1111/resp.12772. [DOI] [PubMed] [Google Scholar]
- 94.Voelkel NF, Cool CD. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:28s–32s. doi: 10.1183/09031936.03.00000503. [DOI] [PubMed] [Google Scholar]
- 95.Strulovici-Barel Y, Staudt MR, Krause A, et al. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax. 2016 doi: 10.1136/thoraxjnl-2015-208274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Albertson TE, Schivo M, Zeki AA, et al. The pharmacological approach to the elderly COPD patient. Drugs Aging. 2013;30:479–502. doi: 10.1007/s40266-013-0080-1. [DOI] [PubMed] [Google Scholar]
- 97.Bateman ED, Mahler DA, Vogelmeier CF, Wedzicha JA, Patalano F, Banerji D. Recent advances in COPD disease management with fixed-dose long-acting combination therapies. Expert Rev Respir Med. 2014;8:357–79. doi: 10.1586/17476348.2014.910457. [DOI] [PubMed] [Google Scholar]
- 98.Barjaktarevic IZ, Arredondo AF, Cooper CB. Positioning new pharmacotherapies for COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:1427–42. doi: 10.2147/COPD.S83758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dobler CC, Wong KK, Marks GB. Associations between statins and COPD: a systematic review. BMC Pulm Med. 2009;9:32. doi: 10.1186/1471-2466-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Howard ML, Vincent AH. Statin Effects on Exacerbation Rates, Mortality, and Inflammatory Markers in Patients with Chronic Obstructive Pulmonary Disease: A Review of Prospective Studies. Pharmacotherapy. 2016 doi: 10.1002/phar.1740. [DOI] [PubMed] [Google Scholar]
- 101.Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. 2014;370:2201–10. doi: 10.1056/NEJMoa1403086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee TM, Lin MS, Chang NC. Usefulness of C-reactive protein and interleukin-6 as predictors of outcomes in patients with chronic obstructive pulmonary disease receiving pravastatin. Am J Cardiol. 2008;101:530–5. doi: 10.1016/j.amjcard.2007.09.102. [DOI] [PubMed] [Google Scholar]
- 103.Lee TM, Chen CC, Shen HN, Chang NC. Effects of pravastatin on functional capacity in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Clin Sci (Lond) 2009;116:497–505. doi: 10.1042/CS20080241. [DOI] [PubMed] [Google Scholar]
- 104.Young RP, Hopkins RJ, Agusti A. Statins as adjunct therapy in COPD: how do we cope after STATCOPE? Thorax. 2014;69:891–4. doi: 10.1136/thoraxjnl-2014-205814. [DOI] [PubMed] [Google Scholar]
- 105.Brown JP, Martinez CH. Chronic obstructive pulmonary disease comorbidities. Current opinion in pulmonary medicine. 2016;22:113–8. doi: 10.1097/MCP.0000000000000241. [DOI] [PubMed] [Google Scholar]
- 106.Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. The Lancet Respiratory medicine. 2015;3:631–9. doi: 10.1016/S2213-2600(15)00241-6. [DOI] [PubMed] [Google Scholar]
- 107.Chandy D, Aronow WS, Banach M. Current perspectives on treatment of hypertensive patients with chronic obstructive pulmonary disease. Integrated blood pressure control. 2013;6:101–9. doi: 10.2147/IBPC.S33982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shrikrishna D, Tanner RJ, Lee JY, et al. A randomized controlled trial of angiotensin-converting enzyme inhibition for skeletal muscle dysfunction in COPD. Chest. 2014;146:932–40. doi: 10.1378/chest.13-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jackevicius CA, Tom A, Essebag V, et al. Population-level incidence and risk factors for pulmonary toxicity associated with amiodarone. Am J Cardiol. 2011;108:705–10. doi: 10.1016/j.amjcard.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 110.Olshansky B, Sami M, Rubin A, et al. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol. 2005;95:404–5. doi: 10.1016/j.amjcard.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 111.Singh SN, Fisher SG, Deedwania PC, Rohatgi P, Singh BN, Fletcher RD. Pulmonary effect of amiodarone in patients with heart failure. The Congestive Heart Failure-Survival Trial of Antiarrhythmic Therapy (CHF-STAT) Investigators (Veterans Affairs Cooperative Study No. 320) J Am Coll Cardiol. 1997;30:514–7. doi: 10.1016/s0735-1097(97)00157-5. [DOI] [PubMed] [Google Scholar]
- 112.Doshan HD, Rosenthal RR, Brown R, Slutsky A, Applin WJ, Caruso FS. Celiprolol, atenolol and propranolol: a comparison of pulmonary effects in asthmatic patients. J Cardiovasc Pharmacol. 1986;8(Suppl 4):S105–8. [PubMed] [Google Scholar]
- 113.Fogari R, Zoppi A, Tettamanti F, Poletti L, Rizzardi G, Fiocchi G. Comparative effects of celiprolol, propranolol, oxprenolol, and atenolol on respiratory function in hypertensive patients with chronic obstructive lung disease. Cardiovasc Drugs Ther. 1990;4:1145–9. doi: 10.1007/BF01856511. [DOI] [PubMed] [Google Scholar]
- 114.George RB, Manocha K, Burford JG, Conrad SA, Kinasewitz GT. Effects of labetalol in hypertensive patients with chronic obstructive pulmonary disease. Chest. 1983;83:457–60. doi: 10.1378/chest.83.3.457. [DOI] [PubMed] [Google Scholar]
- 115.Jabbour A, Macdonald PS, Keogh AM, et al. Differences between beta-blockers in patients with chronic heart failure and chronic obstructive pulmonary disease: a randomized crossover trial. Journal of the American College of Cardiology. 2010;55:1780–7. doi: 10.1016/j.jacc.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 116.Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2002;21:1290–5. doi: 10.1016/s1053-2498(02)00459-x. [DOI] [PubMed] [Google Scholar]
- 117.Andell P, Erlinge D, Smith JG, et al. beta-blocker use and mortality in COPD patients after myocardial infarction: a Swedish nationwide observational study. Journal of the American Heart Association. 2015;4 doi: 10.1161/JAHA.114.001611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salpeter S, Ormiston T, Salpeter E. Cardioselective beta-blockers for chronic obstructive pulmonary disease. The Cochrane database of systematic reviews. 2005:CD003566. doi: 10.1002/14651858.CD003566.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Loth DW, Brusselle GG, Lahousse L, Hofman A, Leufkens HGM, Stricker BH. beta-adrenoceptor blockers and pulmonary function in the general population: the Rotterdam Study. Brit J Clin Pharmaco. 2014;77:190–200. doi: 10.1111/bcp.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lainscak M, Podbregar M, Kovacic D, Rozman J, von Haehling S. Differences between bisoprolol and carvedilol in patients with chronic heart failure and chronic obstructive pulmonary disease: a randomized trial. Resp Med. 2011;105:S44–S9. doi: 10.1016/S0954-6111(11)70010-5. [DOI] [PubMed] [Google Scholar]
- 121.Mainguy V, Girard D, Maltais F, et al. Effect of Bisoprolol on Respiratory Function and Exercise Capacity in Chronic Obstructive Pulmonary Disease. American Journal of Cardiology. 2012;110:258–63. doi: 10.1016/j.amjcard.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 122.Kubota Y, Asai K, Furuse E, et al. Impact of beta-blocker selectivity on long-term outcomes in congestive heart failure patients with chronic obstructive pulmonary disease. International journal of chronic obstructive pulmonary disease. 2015;10:515–23. doi: 10.2147/COPD.S79942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Su VY, Chang YS, Hu YW, et al. Carvedilol, Bisoprolol, and Metoprolol Use in Patients With Coexistent Heart Failure and Chronic Obstructive Pulmonary Disease. Medicine. 2016;95:e2427. doi: 10.1097/MD.0000000000002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Short PM, Lipworth SI, Elder DH, Schembri S, Lipworth BJ. Effect of beta blockers in treatment of chronic obstructive pulmonary disease: a retrospective cohort study. Bmj. 2011;342:d2549. doi: 10.1136/bmj.d2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhatt SP, Wells JM, Kinney GL, et al. beta-Blockers are associated with a reduction in COPD exacerbations. Thorax. 2016;71:8–14. doi: 10.1136/thoraxjnl-2015-207251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Farland MZ, Peters CJ, Williams JD, Bielak KM, Heidel RE, Ray SM. beta-Blocker use and incidence of chronic obstructive pulmonary disease exacerbations. The Annals of pharmacotherapy. 2013;47:651–6. doi: 10.1345/aph.1R600. [DOI] [PubMed] [Google Scholar]
- 127.Kamath A, Stover DE, Hemdan A, et al. Effect of Perioperative beta-Blockers on Pulmonary Complications among Patients with Chronic Obstructive Pulmonary Disease Undergoing Lung Resection Surgery. Lung cancer international. 2015;2015:204826. doi: 10.1155/2015/204826. [DOI] [PMC free article] [PubMed] [Google Scholar]