

Abstract
We present an insert-based approach to fabricate scalable and multiplexable microfluidic devices for 3D cell culture and integration with downstream detection modules. Laser cut inserts with a layer of electrospun fibers are used as a scaffold for 3D cell culture, with the inserts being easily assembled in a 3D-printed fluidic device for flow-based studies. With this approach, the number and types of cells (on the inserts) in one fluidic device can be customized. Moreover, after an investigation (i.e., stimulation) under flowing conditions, the cell-laden inserts can be removed easily for subsequent studies including imaging and cell lysis. In this paper, we first discuss the fabrication of the device and characterization of the fibrous inserts. Two device designs containing two (channel width = 260 μm) and four inserts (channel width = 180 μm), respectively were used for different experiments in this study. Cell adhesion on the inserts with flowing media through the device was tested by culturing endothelial cells. Macrophages were cultured and stimulated under different conditions, the results of which indicate that the fibrous scaffolds under flow conditions result in dramatic effects on the amount and kinetics of TNF-α production (after LPS stimulation). Finally, we show that the cell module can be integrated with a downstream absorbance detection scheme. Overall, this technology represents a new and versatile way to culture cells in a more in vivo fashion for in vitro studies with on-line detection modules.
Graphical abstract
This paper describes the an insert-based microfluidic device for 3D cell culture that can be easily scaled, multiplexed, and integrated with downstream analytical modules
Introduction
Microfluidics has emerged as a powerful tool for in vitro cell studies due to some advantages over conventional 2D culture on petri dishes and well plates [1–3]. In addition to reducing the amount of required media and reagents, a microfluidic device can continuously provide a nutrient supply to cultured cells and remove metabolism waste products [4, 5]. Such microfluidic devices can also be integrated with downstream analytical schemes such as electrochemistry and spectroscopy [6, 7]. Importantly, microfluidics enables precise gradient control of chemicals that may affect the biology of cells [8]. For example, Bennett and colleagues have reported a microfluidic device that allows dynamic gradients of glucose and galactose over S. cerevisiae, with which a correlation between gene expression and the gradients was found [9]. Furthermore, the flow feature of a microfluidic device allows introduction of shear stress to cells, which is known to affect the functions of many cell types [10]. A classic example is the study of endothelial cells under physiologically relevant shear stress [11]. However, most of the reported cells-on-a-chip models used cells cultured on a two-dimensional (2D) surface (a monolayer in a reservoir or a channel), which in many cases does not represent the cellular microenvironment in vivo, where many cells grow in a 3D fashion within the network of extracellular matrix (ECM) [12–14]. Composed of macromolecules such as collagen and polysaccharides, the ECM is a mesh-like structure that plays a role in cell adhesion and cell-to-cell interactions [15]. It has proven that the ECM can affect many important cellular properties and processes such as morphology, growth, migration and differentiation [16–19].
Therefore, to increase biological relevance of cells-on-a-chip devices, 3D cell culture techniques are needed to mimic the ECM environment in vivo. Also, on-chip circulation has been recently studied as an important factor that can affect the biology of cells [10, 20–22]. Compared with flow-through models, circulation in a microfluidic device can accumulate signal molecules [23] and apply physical factors such as shear stress [10] and blood pressure mimic [21] to the cells, which is especially useful for cross talk investigations between different cell types [20, 22]. 3D cell culture protocols in microfluidic devices have been reported by several groups, most of which rely on embedding cells in a structure of hydrogels [24]. For example, Beebe’s group reported a device of parallel channels, where different cells can be immobilized in hydrogels. Cell-free hydrogel structures were designed connecting the channels so that cellular cross talk can occur via molecular diffusion and be detected [25]. However, most of the hydrogel devices are diffusion based due to the low to mid-nanometer pore size of a hydrogel structure [26], which makes it difficult to circulate through the gel.
Numerous studies have shown that electrospun micro/nanofibers can be a good ECM mimic for in vitro cell studies under static conditions (i.e., in a petri dish) [27–30]. Our group adopted this technology to design microfluidic devices for 3D cell culture and subsequent investigation. For example, we recently reported a 3D-printed fluidic device with a layer of electrospun fibers directly deposited on the inner wall of a channel, on which, macrophages showed increased immune response to flowing lipopolysaccharide (LPS) stimulation compared with those cultured on a flat surface [31]. In this paper, we are presenting a new approach to make a scalable and versatile 3D cell culture microfluidic device. In this setup, fibers are electrospun onto polystyrene sheets, after which, inserts are laser cut to desired dimensions. Cells cultured on the inserts can be simply placed into a 3D-printed device with locking slots. The advantage of this approach is that inserts with desired cell types can be inserted and removed easily from the device, which provides a new way for carrying out cell-to-cell interaction studies (by integrating inserts of different cell types in one device). In addition, one can place multiple inserts of the same type for time point studies. It also enables the control of the amount of cells by increasing the amount of inserts for analytical methods of moderate sensitivity. Specifically, we measured the attachment strength of the fibrous scaffolds on the insert and used a downstream optical module to show integration of detection. Endothelial cells were immobilized on inserts and cultured with flowing media (420 μL/min) for 24 hours, the results of which indicate no significant cell loss. Furthermore, we investigated the activation of macrophages on the device with flowing stimulant (LPS). We show that the 3D fibrous scaffolds and the flow component have dramatic effects on the amount and kinetics of TNF-α production.
Experimental
Fabrication of the fluidic devices via 3D-printing
A high-resolution 3D-printer (Objet Eden 260 V, Stratasys, Ltd, Edina, MN, USA) was used to create the devices used in this study. The material used in this work was called Full Cure 720 (Stratasys Ltd., MN, USA), the composition of which is propriety, but approximately containing 10–30% isobornyl acrylate, 10–30% acrylic monomer, 15– 30% acrylate oligomer, 0.1–1% photo initiator) [32]. All devices were designed by Autodesk Inventor and saved in an .stl format for the printer to read. The design sketches of the devices used in this work can be found in Figs. S1-S4 in the Electronic Supplementary Material (ESM).
Fabrication of fiber-coated inserts
To make silk fibroin fibers, silk fibroin was first extracted from crude silk as described elsewhere [33], which was then dissolved in 1,1,1,3,3,3-Hexafluoro-2-propanol (HFP) at room temperature at 5% (w/v). After the polymer solution was homogenized on a shaker, it was loaded in a 3 mL syringe fitted with an 18 gauge blunt tip syringe needle (Cole-Parmer, IL, USA). An alligator clip was attached to the needle and connected to a 25 kV supply. A piece of 12 cm × 10 cm polystyrene sheet (300 μm thick; ShrinkyDinks) was backed by aluminum foil, which can be grounded as the fiber collector. Once started, the grounded collector was placed ~ 25 cm away from the syringe needle and the silk fibroin solution was driven by a syringe pump at a flow rate of 16 μL/min. Fibers can then be produced by the high voltage and be collected on the PS sheet. After 2 hours of electrospinning, a layer of silk fibroin fibers of ~ 100 μm thick can be collected. The fibrous sheet was then placed in the chamber of a laser cutter (Epilog Laser, CO, USA) to obtain the rectangular inserts (15 mm × 3.5 mm). The speed, power and frequency parameters of the laser cutting process are 100%, 9%, and 5000, respectively. The inserts were stored in a 10 mm petri dish to avoid dust contamination.
To make PS fibers on inserts, PS particles (192 kDa; Sigma-Aldrich, MO, US) were dissolved in a mixture of 60% dimethylformamide and 40% tetrahydrofuran to make a 15% (w/v) solution, which needs to be homogenized on a shaker overnight. Then, a similar protocol was used to prepare PS fibrous inserts as described above. To make polycaprolactone (PCL) fibers on inserts, PCL particles (80 kDa; Sigma-Aldrich, MO, US) were dissolved in 1,1,1,3,3,3-Hexafluoro-2-propanol (HFP) to make a 12% (w/v) solution. After the solution was homogenized on a shaker, the same electrospinning and laser cutting protocols were followed to obtain PCL fibrous inserts.
Fiber Attachment Studies
Adhesion of the fibers was tested by exposing the inserts to N2 flow. Each row of 30 inserts was held under 10 psi N2 pressure and the weight of the inserts and remaining fibers was recorded at 10 second increments. After a total of 60 seconds, the remaining fibers were removed using a tissue. Compressed air was used to remove any dust that remained from the fibers or wipes before the empty inserts were massed. No water or ethanol was used on the inserts that would affect the weight. The difference between the masses of the bare PS sheets and the fiber-coated sheets taken at each time point was used to calculate fiber loss over time. To visualize the sintering of fibers, the edges of the fibers were imaged (SEM). Inserts were sputter coated with gold for 30 seconds and SEM images were obtained.
Endothelial Cell Immobilization Studies
PS inserts coated with electrospun PS were sprayed with 70% ethanol and placed under UV light for at least 24 hours for sterilization. A piece of double sided tape was placed in the bottom of a 40 mm petri dish and placed under UV. The inserts were then secured to the bottom. A confluent 100 mm diameter petri dish of bovine pulmonary artery endothelial cells (Millipore-Sigma) was detached by trypsin, which was then centrifuged and re-suspended in 600 μL of fresh warm media (DMEM with 7.5% FBS, 2.5% ABS and 1% pen-strep). The cell suspension (~8×106 cells in 600 μL) was dropped onto each insert and incubated for ~2 hours, after which, 5 mL of fresh warm media was added to the petri dish and incubated for 24 hours. The media was removed and the inserts were placed into the slots in the fluidic device with tweezers and immediately connected to a peristaltic pump via a 3D-printed threaded tubing fitting. The peristaltic pump circulated media through the devices while in the incubator at 420 μL/min for 24 hours. After 24 hours, the inserts were removed from fluidic device, fixed with 2.5% glutaraldehyde in PBS for 30 minutes in the incubator, rinsed 3× with PBS, incubated with the staining solution for 30 minutes (1 mL of PBS with 2 drops of Actin Red 555), rinsed 3× with PBS and stored in fresh PBS until confocal imaging was done (within 2 hours of staining).
Macrophage culture on the inserts
Inserts coated with silk fibroin fibers (on PS sheet) were used to culture macrophages. The inserts were first immobilized on the bottom of a 40 mm diameter petri dish using double sided tape, sprayed with 70% ethanol and dried in UV for at least two hours for sterilization. When macrophages (RAW 264.7; ATCC) cultured in regular 100 mm petri dishes were confluent, the cells were scraped off and centrifuged at 500 rpm. The cell pellet was re-suspended in 4 mL fresh media (DMEM with 10% FBS and 1% pen-strep), and transferred to a petri dish containing 8 different inserts (4 mL of a ~2 million cells/mL solution was to the dish). The petri dish was placed in an incubator (37 °C, 5% CO2) for 24 hours without disturbance, after which, the inserts can be removed for subsequent experiments or media can be changed for longer cell culture on the inserts. Flat inserts were also prepared as a control by coating a layer of silk fibroin solution on the PS sheet followed by air drying. The same sterilization and cell seeding procedures were followed for macrophage culture on the flat inserts.
Macrophage stimulation and cytokine quantitation
Lipopolysaccharide (LPS), a commonly used molecule to stimulate macrophages [34], was used to stimulate immune responses of macrophages. A stock LPS solution was prepared by dissolving LPS powder (Sigma-Aldrich, MO, USA) in PBS at 100 μg/mL. Upon using, the stock solution was diluted by 100× in cell culture DMEM media to a final concentration of 1 μg/mL. In a sterile cell hood, four inserts cultured with the macrophages were placed into the pre-designed slots in the 3D-printed fluidic device. For macrophage stimulation under flowing conditions, the assembled device was first connected to Tygon tubing (0.02” i.d., 0.06” o.d.) using customized 3D-printed connectors. The loop was then placed in a sterile test tube preloaded with 2 mL of the LPS containing DMEM media. This setup was placed in a cell culture incubator (37 °C, 5% CO2) with the middle section of the Tygon tubing fed into the peristaltic pump on the benchtop. The media was then circulated through the cells immobilized on inserts at 1 mL/min. After 1, 2, 4, 6, 8, 10, 12, and 24 hours stimulation, an aliquot of 50 μL of the media was sampled and stored immediately at −20 °C, after which 50 μL of fresh media was added to keep the total volume the constant. For static macrophage simulation, a fluidic device with cell-laden inserts was soaked in 2 mL of LPS containing media in a test tube, after which, the same sampling process was conducted. The TNF-α in the samples was measured with ELISA (PeproTech, NJ, USA) following the manufacturer’s instructions. Briefly, a 96-well plate was prepared by immobilizing the capture antibody on the bottom of each well for overnight. After loading standards and samples and subsequent rinsing, the detection antibody (biotinylated) was added. Streptavidin-HRP (horse radish peroxidase) conjugate was then added to link to the detection antibody. Lastly, 100 μL TMB liquid substrate (3,3′,5,5′-Tetramethylbenzidine) was added to each well, which can turn its color to blue via the HRP conjugate (with subsequent absorbance readings in a plate reader, MolecularDevices, CA, USA). Four conditions were measured: macrophages on fibrous scaffold + flowing LPS stimulation; macrophages on fibrous scaffold + static LPS stimulation; macrophages on flat surface + flowing LPS stimulation; and macrophages on flat surface + static LPS stimulation. The measured TNF-α value for each device was divided by the specific cell count in the device and a final result of TNF-α release per cell was reported.
After a 24-hour stimulation, the inserts from each cell module were easily removed with a pair of tweezers, and immersed in 500 μL of DI water to lyse the cells for 2 hours. The lysate was then used to quantitate the amount of DNA using the Hoechst fluorescence assay as a measure of cell count [35]. The 10 mg/mL stock Hoechst solution (Sigma-Aldrich, MO, USA) was diluted in TNE buffer (50 mM Tris-HCl, 100 mM NaCl, 0.1 mM EDTA, pH=7.4) to a final concentration of 20 μg/mL. An aliquot of 200 μL of the cell lysate was pipetted into a well of a 96 well pate, followed by addition of 50 μL of the prepared Hoescht assay. Fluorescence was detected at 460 nm (with 350 nm excitation) by a plate reader (MolecularDevices, CA, USA). Standards were prepared by lysing 0, 2, 5, 10, and 20 thousand cells in DI water for a calibration curve.
PDMS-based absorbance detection module
A U-shaped channel structure was 3D printed with Acrylonitrile butadiene styrene (ABS). The design of this structure can be found in Fig S5 in the ESM. This structure was immersed in a mixture of PMDS pre-polymer and the curing reagent (10:1). After the PDMS cured, the whole piece was soaked in acetone in a closed container for about 5 hrs, after which, the ABS part was dissolved and a channel was formed in the PDMS (process depicted in Fig. S7 in the ESM).
The 3D-printed fluidic device that can house inserts was connected to a commercially available mixing tee using Tygon tubing and customized adaptors. The side arm of the mixing tee was connected to a syringe loaded with Griess reagent. Eluent from the fluidic device and the Griess reagent was combined in the mixing tee, which was then pumped through the PDMS detector. Two optical fibers were immobilized in the fabricated holes on both sides of the channel. One optical fiber was connected to a tungsten lamp while the other connected to a modular spectrometer (OceanOptics, FL, USA). The detector uses a photodiode array to detect a whole spectrum between 400 nm and 1000 nm. For NO2− detection, an absorbance wavelength of 530 nm was used. Due to possible fluctuation of the light source, a reference wavelength of 650 nm (no absorbance at this wavelength) was utilized. The absorbance difference between the two wavelengths (A530-A650) was used for subsequent quantitation. NO2− standards of 0, 2, 5, 10, and 20 μM were first pumped through the setup to generate a calibration curve. Cells cultured on both fibrous and flat inserts were studied. Phenol red free media that contains 1 μg/mL LPS was pumped through the fluidic device containing cell-laden inserts at 15 μL/min, which was then mixed with the Griess reagent at the T mixer and then introduced into absorbance detection module for real time absorbance detection.
Results and Discussion
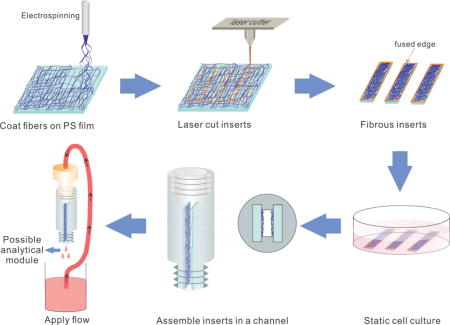
In this work, we developed a new approach to make a scalable and multiplexable 3D cell culture microfluidic device. As shown in Figure 1, fibers are first electrospun onto polystyrene sheets, after which, inserts are laser cut into desired dimensions (for this study 15 × 3 mm inserts with ~100 μm thick fibrous scaffolds). Cells statically cultured on the inserts can be simply inserted into a 3D-printed device with locking slots for dynamic culture and stimulation with downstream detection modules. This new approach has several unique advantages. (i): 3D-cell culture: the fibrous scaffolds coated on the inserts can serve as a mimic of ECM; (ii): scalability: The number of inserts (cells) can be increased to suit the limit of detection (LOD) of a certain downstream detection scheme, or be decreased when it is costly and time consuming to culture a certain cell line. After flow based stimulation, the inserts can also be easily removed for subsequent studies such as re-seeding, imaging, and DNA extraction; (iii): multiplexing: although it is not included in this manuscript, different cell types cultured on different inserts can be integrated in one fluidic device for cell-to-cell studies; (iv): adjustable channel size: The space between adjacent inserts re-defines the effective channel size. 3D-printing enables various distances between slots where the inserts will slide into and thus various effective channel sizes. This is especially useful for studies where shear stress needs to be tuned.
Figure 1.
Schematic showing the process of creating an insert-based microfluidic device for 3D cell culture that can be easily scaled and multiplexed. Fibers are directly electro/blow-spun onto a polystyrene film and subsequently laser-cut into inserts. The fused edges aid in fiber adherence on the polystyrene film. After cells are seeded on the fibrous scaffolds, they can be easily assembled in a 3D-printed fluidic vessel that can be connected to downstream analytical modules.
Fiber Attachment Characterization
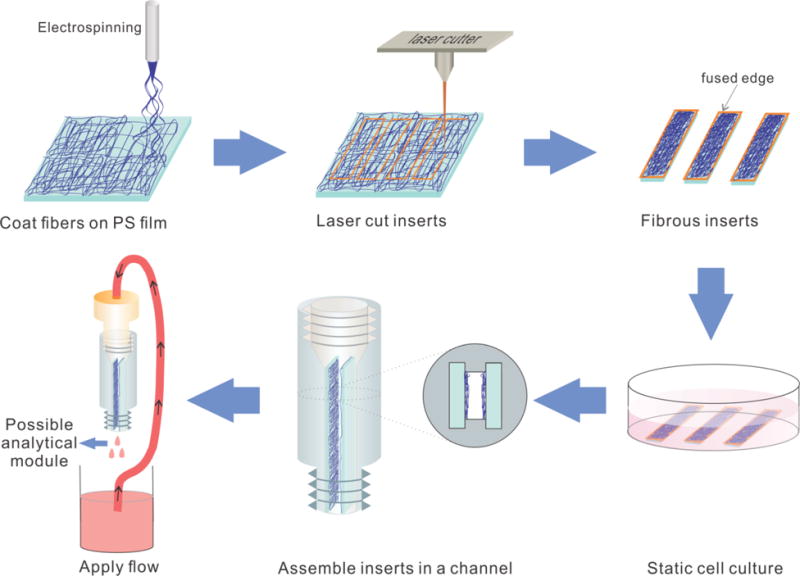
The laser cutting process used in the protocol not only generates inserts of desired size, but also sinters the edges of the fibrous layer, increasing the adhesion between the substrate and fibers. To evaluate how the laser may assist fiber attachment on a substrate, we conducted an N2 blowing experiment and tested the amount of fiber loss by mass. Briefly, an N2 flow (10 psi, directed through a 510 μm diameter pipet tip orifice that was placed ~3 cm from insert) was applied to the fibers on an insert (laser cut or scissor cut) for different time durations. The mass difference from before and after the experiment was used to reflect the amount of lost fibers. Because PS and silk fibroin fibers were mainly used for the cell culture studies here, attachment of those fibers on PS substrate were first measured. Transparency films (made of cellulose acetate) can also be used as substrate for the fibrous inserts and subsequent cell culture. We also tested the fibers coated on the transparency film.
Figure 2A is the SEM image of the edge of a laser cut insert, which clearly shows that the fibers were sintered on the substrate. For a scissor cut insert, the fibers seem like a separate layer on a substrate (SEM image shown in Figure 2B). To make a more quantitative comparison, the fiber loss for both scissor cut and laser cut inserts as a function of air bellowing time was plotted (Figures 2C and D). Overall, the N2 flow can cause fiber loss on both kinds of inserts (as would be expected by high pressure used). However, with laser cutting, the fiber loss is significantly less than a regular scissor cutting during the 60s N2 blowing. For example, for the first 20s, about 90% of the PS fibers were still on the PS substrate for laser cut inserts while only about 40% of the fibers were remaining on scissor cut inserts (p<0.03). After 60s of blowing, the laser sintering could keep more than 50% of the fibers on the inserts. Without the laser sintering, however, only less than 10% of the fibers were still on the substrate (p<0.02). Other fiber material/substrate material combinations were also tested, the results of which are summarized in Fig. S5 in the ESM. Clearly, the use of a laser cutter to form the inserts leads to a more stable 3D scaffold, which is important for the introduction of continuous flow over the scaffold/inserts.
Figure 2.
Characterization of fiber attachment on laser cut and scissor cut inserts. (A) An SEM image of the fused edge of a laser cut insert. It shows that the fiber layer and the substrate are sintered together by the laser. (B) SEM image of polystyrene fibers electrospun on polystyrene, scissor cut (800×, 15° to normal). The edge shows that the fiber layer was not immobilized strongly on the substrate. (C) Detachment of PS fibers off a PS substrate. With 15 psi air blowing for up to 60 seconds, fibers on laser cut inserts show a higher fiber remaining rate. In comparison, more fibers can be lost on a scissor cut insert. (D) Detachment of silk fibroin fibers off a PS substrate. The same trends can be seen as in (C). Other fiber material/substrate material combinations are also studied, the results of which are in Fig. S5 in the ESM
Cell Attachment with Flow
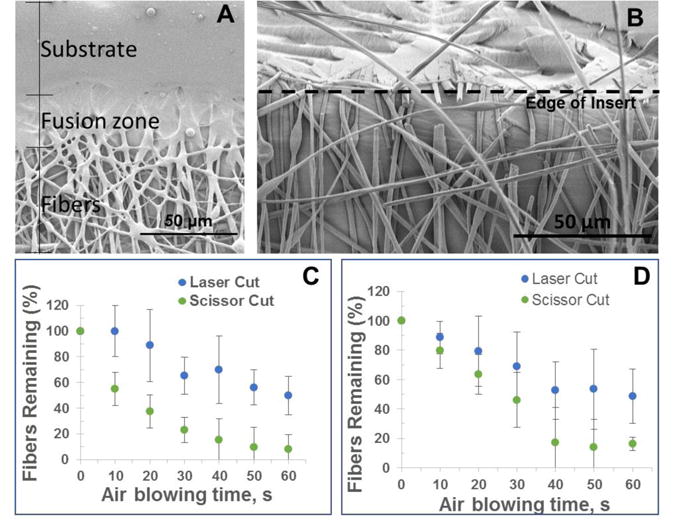
In order to evaluate the biocompatibility of endothelial cells on the fibrous inserts under flowing conditions, endothelial cells were introduced to PS fibers and cell attachment as well as cell penetration through the scaffold was tested. The main purpose of this experiment is not to create a monolayer endothelium mimic. We acknowledge that it is ideal for endothelial cells to form a confluent layer; here we use this adherent cell line to characterize the 3D nature of these inserts/scaffolds. We commonly use this endothelial cell line in our laboratory so it is readily available. The fibrous scaffold used for this study consisted of 15% PS in DMF electrospun for 20 minutes at a voltage of 25 kV and the polymer solution pumped at 0.8 mL/hr. These parameters created a scaffold with 12.8 ± 2 μm pores (calculated by average diameter, n = 50 pores). These relatively large pores allow trypisinized endothelial cells (average diameter 3.5 ± 0.8 μm for the cells used in this study) to penetrate through the scaffold and adhere throughout. Once adhered on fibers, endothelial cells have an average diameter of 6.1 ± 1.0 μm in static conditions (n=50). As shown in Figure 4A, the percent area of the confocal image occupied by endothelial cells was determined at various planes through the z-axis, the top, 20 μm, and 40 μm into the scaffold. The cells coverage was determined to be 10%, 8%, and 6% of the total area (775 μm × 775 μm), respectively. The effects of flow on endothelial cells were tested. The inserts with immobilized cells that were placed into the fluidic device as seen in Figure 3 and a 3D-printed fitting for tubing was connected. The cells on fibers were subject to flow for 24 hours using a peristaltic pump that circulated media at a rate of 420 μL/min. Figure 4B shows another confocal micrograph of the cells on the surface a PS-fiber coated inserts after being subject to 24 hours of flow indicating that the cells still adhere on the fibers after exposure to flow. It is important to note that once the cells elongate, as seen in Figure 4B, they have an average length of 26.3 ± 4.0 μm, which is larger than the average pore size.
Figure 4.
(A) Confocal image endothelial cells immobilized on polystyrene fibers (40 μm mat, 12 μm pores) on a polystyrene film 24 hours after introduction of cell suspension. (B) Confocal image of endothelial cells adhered to polystyrene fibers after 24 hours of flow in two-slot cell module at 420 μL/min. Endothelial cells were stained with Actin Red 555.
Figure 3.
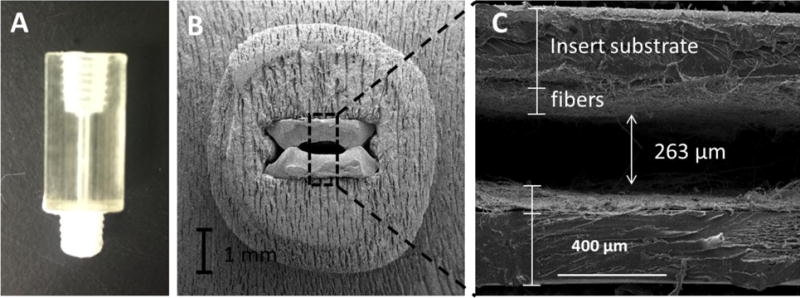
A two-insert configuration that was used for endothelial cell culture and studies. (A) 3D-printed fluidic module with parallel slots along the channel for insert placement. (B) An SEM cross-section of two inserts with fibers integrated into the fluidic device. (C) A magnified view of the two fibrous inserts. In this application, a distance of 260 μm between the inserts was used although a customized distance can be achieved within the resolution of a 3D-printer.
Macrophage studies on the device
Upon stimulation, macrophages can change phenotypes to execute different functions. Unstimulated macrophages are designated as an M0 state [36]. M0s can be activated to an M1 phenotype (pro-inflammatory state), which can produce various pro-inflammatory cytokines (i.e.,TNF-α) to effectively remove necrotic cells or debris. M2 is another phenotype of activated macrophages, which produces increased pro-healing cytokines such as vascular endothelial growth factor (VEGF) [37]. Interaction of activated macrophages with other cell types has been found to be related to many diseases such as atherosclerosis and cancer [38, 39]. Therefore, a microfluidic-based investigation of the activation of macrophages that yields clinically relevant results would be of great interest to the medical field. We have recently reported that macrophages cultured on polycaprolactone (PCL) fibers directly coated on the inner wall of a fluidic device can release more cytokines than those cultured on a flat surface [31]. However, the activation speed of macrophages (i.e., M0 to M1), which determines how fast the immunological responses will take place, has not been studied in a microfluidic device (to our knowledge). It has been reported that macrophages in vivo can transition to the M1 state within 1 hour after being stimulated by LPS [40], while those cultured in vitro with culture flasks can take much longer to do so. To investigate if fibrous scaffolds and flow can cause macrophage activation at a more in vivo-like rate, we used the fluidic device with 4 inserts as shown in Figure 5A. Cells were first cultured on the inserts in a petri dish with fresh DMEM media. Before integrating the cell-laden inserts into the device, a sample insert can be examined to ensure normal cell morphology and confluency. As shown in Figure 5B-C, macrophages cultured on the fibrous ECM analogue (made of silk fibroin) can fuse to form giant cells as they do in vivo [41].
Figure 5.
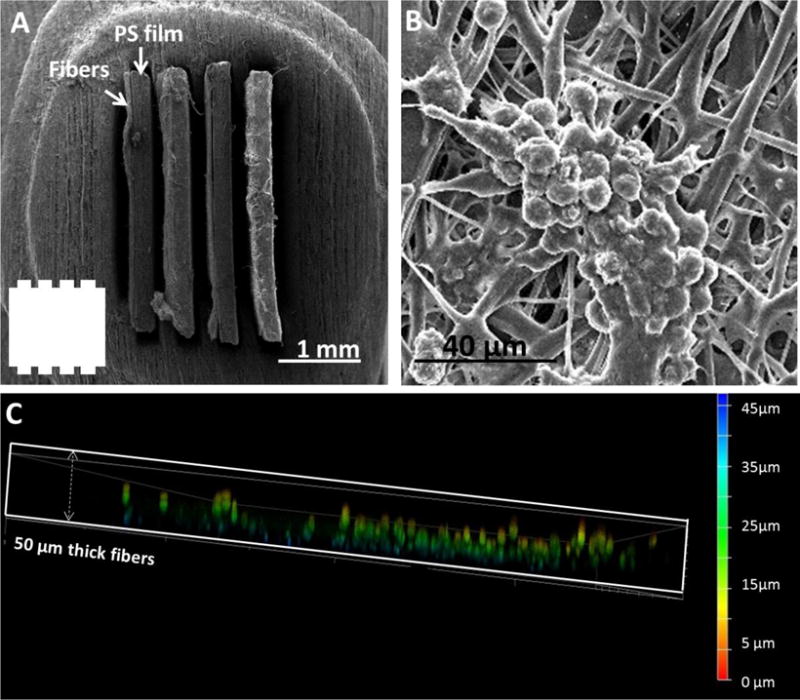
A four-insert configuration that was used for macrophage studies. (A) An SEM cross-view of fibrous inserts (fibers on PS films) assembled in a channel. The number of inserts and space in between can be customized. The inset shows the shape of the channels without inserts. (B) Macrophages on fibers tend to fuse to form cell clusters as is observed in vivo. (C), A side confocal view of the fibrous scaffold of an insert cultured with macrophages. The color bar indicates the depth in the scaffold. It can be seen that the macrophages can penetrate into the scaffold up to 40 μm to form a true 3D culture.
To study the macrophage activation under flowing condition, macrophages were cultured on both fibrous and flat inserts and assembled in separate fluidic devices. Each assembled device was configured in a loop using a 3D-printed adapter and Tygon tubing, through which, 2 mL of media with 1 μg/mL LPS was circulated at 1 mL/min to stimulate the cells (Figure 1). After 1, 2, 4, 6, 8, 10, 12, and 24 hours of stimulation, an aliquot of 50 μL of the stimulating solution was sampled, followed by adding 50 μL of fresh media (with LPS) back to the circulation. The cytokine TNF-α (M1 marker) in the samples were measured using ELISA kits. As a control, cells cultured on flat inserts were also prepared and stimulated. Because different devices may have different amount of cells cultured, it is necessary to count the cells in each device so that the final results can be normalized to cytokine release per cell. The removable inserts of the device design allows an easy way to conduct this measurement. After the stimulation, the inserts in a device were removed with tweezers and the cells were lysed. The Hoechst assay was then used to quantitate DNA amount in the lysate as a measurement of cell count [35]. To study the macrophage activation with under static condition, a fluidic device containing 3D or 2D cell-laden inserts was soaked (no flow) in 2 mL media that contains 1 μg/mL LPS, respectively, and the same sampling and quantitation procedures were followed.
Chensue and colleagues once reported that after LPS stimulation, TNF-α production in vivo was maximal after 1 hr of LPS stimulation, which then decayed because of scavenging by receptor cells [40]. With our device, TNF-α release from macrophages cultured on the 3D scaffold (blue solid line in Figure 6) increased immediately after 1 hour of LPS stimulation, which then quickly plateaus. The cells cultured on a flat insert, however, released TNF-α in a slower fashion (red solid line), with the plateau not showing until 6-8 hours of stimulation. Because there were no TNF-α scavenging cells in our system, the TNF-α accumulated and eventually levels out at a steady state. However, by comparing the activation speed, it is apparent that the fused macrophages on the 3D scaffold can more closely mimic the in vivo M0/M1transition speed. For the static stimulation (no flow) controls, cells cultured on the 3D scaffolds (blue dashed line) were also activated quicker than those cultured on 2D surfaces (red dashed line). However, the overall release of TNF-α in a static condition was lower than in the flowing condition. There has been evidence showing that certain molecules produced by activated macrophages can inhibit TNF-α production as a feedback [42]. Therefore, one possible reason for the difference between the flowing and static stimulation is that the flowing media acts as a continuous clearing mechanism to remove (dilute) macrophage derived molecules accumulated around the cells. These results also show that cells cultured on fibrous scaffolds can release more TNF-α at the plateau stage. Garg and colleagues have studied macrophage activation by taking measurements after 1 and 3 days of LPS stimulation. They found that a more fibrous scaffold can increase the expression of both M1 and M2 markers in macrophages [43].
Figure 6.
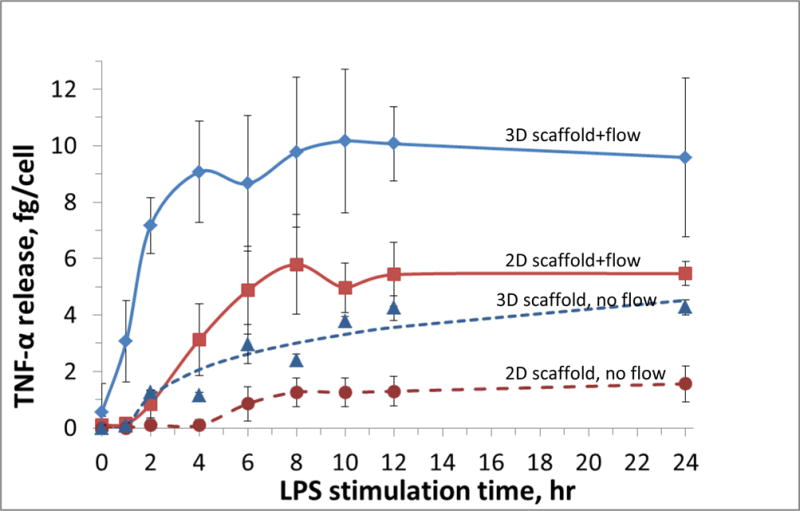
TNF-α release from LPS-stimulated macrophages under different conditions (n = 4 for each data point). While flowing media with LPS for stimulation (1 μg/mL LPS in media; 1 mL/min flow rate), macrophages cultured on fibers inserts in the fluidic device (blue solid line) exhibited a faster TNF-α release to LPS stimulation, as compared to use of a 2D flat inserts (solid red line). Literature results indicate that after LPS administration, TNF-α release in vivo reaches the maximum within 1 hour. These results suggest that macrophages cultured on a fibrous scaffold can mimic in vivo immune response speed more realistically than those on flat surfaces. Static LPS stimulation of macrophages (on fibrous and flat surfaces, dashed blue and red lines, respectively) was also quantitated. A difference in activation speed was also observed with these conditions. However, the overall TNF-α release was less than macrophages stimulated under flow.
This macrophage study proves that the new device design represents a versatile and powerful way for cell studies in vitro. Cells cultured on different substrates (i.e., 3D vs. 2D) can be examined before being combined on a microfluidic device. Although our study used four inserts of one cell type in a device, the amount of inserts and the types of cells can be easily multiplexed. More importantly, after cellular stimulation, the inserts can be removed by tweezers for morphology observation and lysis studies. We also proved that the following stimulation can affect the biology of the macrophages as compared to static culture.
Integration of Detection Module
We and others have previously shown that 3D printed microfluidics devices can be made with thread for connection with tubing [44, 45]. We used this advantage to help integrate downstream detection modules with the insert-based cell culture device. With this new approach, once assembled in the fluidic device, the inserts and scaffold redefine the width and height of a fluidic channel (Figure 3). The images in Figure 3 B-C show that an insert can be immobilized in the slots tightly, Using ImageJ, the area of the space not occupied by the inserts was determined as well as the area of the center channel in between the inserts. The inserts occupy 73% of the total open space in the device (once inserted). The average total available space for fluid to fill the device is 0.52 mm2, with 38% of the available space being the channel in between the inserts (see ESM Fig. S6).
With a module approach, absorbance detection can be easily integrated with the cell culture device. This was added by creation of a transparent PDMS device using a 3D-printed sacrificial mold, which can be connected to the macrophage module for downstream NO2− quantitation using visible absorbance detection. LPS simulated macrophages can produce nitric oxide (NO) [46], which can be oxidized to NO2− within a few seconds [47]. Therefore, by monitoring the amount of NO2−, the activation of macrophages can be measured. The Griess reagent has been widely used for NO2− quantitation, which can react with NO2− to form a pink product with maximum absorption at 530 nm [48]. Fig. S7 (see ESM) shows the fabrication process of the PDMS device. Briefly, a U-shaped part was 3D-printed with Acrylonitrile butadiene styrene (ABS), which was then immersed in a mixture of PDMS pre-polymer and the curing reagent. After the PDMS is cured, the whole device was soaked in acetone so that the ABS part can be dissolved and a void channel can be fabricated. Compared to clean room lithography, this method is simpler and enables fabrication of fully enclosed channels without a sealing step. We made a single U-shaped channel (1 mm diameter; 1 cm long) in the device; but more complicated and intricate structures should be possible. Upon using, two optical fibers can be inserted to the pre-molded void spaces outside both ends of the channel using plastic sleeves (Figure 7A). An optical fiber connected to a tungsten lamp for incident light was placed at one end of the channel, while another fiber at the other end of the channel coupling to a modular spectrometer for real time absorbance detection. To make a calibration curve, a T mixer was used to mix NO2− standards and the Griess reagent before they were delivered into the channel. Figure 7B shows initial results for online detection of NO2− of 0, 2, 5, 10, 20 μM, with a calculated LOD of 0.3 μM. Next, a device that contained macrophage laden inserts (fibrous or flat) was connected to the PDMS-based absorbance detector module. The eluent from the cell module was also mixed with Griess reagent online while being pumped through the detector. The amount of NO2− after 0, 10, 20, 30, 40, and 50 min of LPS stimulation was quantitated. As shown in Figure 7C, for macrophages cultured on 3D fibrous scaffold, after 20 min stimulation, a significant amount of NO2− was detected. For cells cultured on 2D flat surface, however, during the 50-min experiment, there was not significant production of NO2−. These results further confirmed that macrophages on fibrous scaffolds can be activated faster by LPS than those on a flat surface. However, with this analytical module, we could detect the macrophage activation at a higher temporal resolution (in this work 2 pts/min, only specific time points are averaged and shown in Figure 7C), which is not very practical by off line sampling (i.e., pipetting sample, etc).
Figure 7.
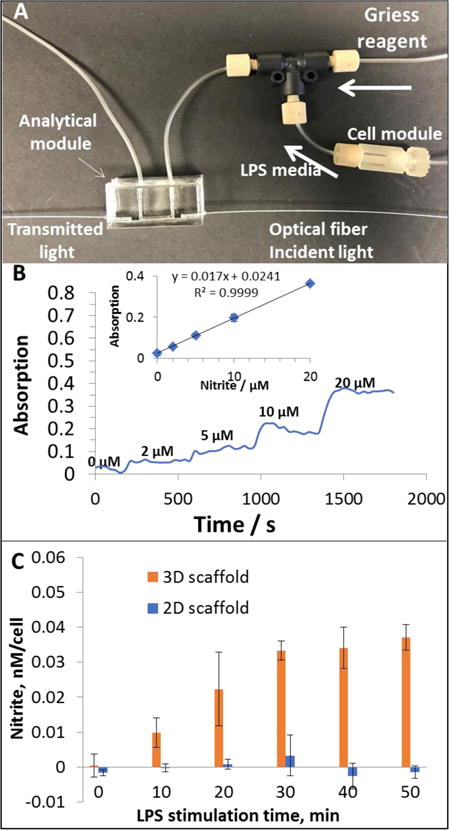
Analytical module for nitrite quantitation downstream the cell culture module. (A), eluent from the cell module was mixed with Griess reagent in the T mixer, which was then flowing through the U-shaped channel in the PDMS device. The two holes on both sides of the channel can immobilize optical fibers connected to a tungsten lamp and a spectrometer, respectively. (B), nitrite standards were pumped through the cell module device (without cells) to generate a calibration curve. Elevated absorption reading can be observed as nitrite concentration increases. The absorption values on each plateau were averaged and plotted vs. the concentrations (inset). (C), Macrophages cultured on fibrous or flat inserts were stimulated by LPS containing media (phenol red free). Nitrite was quantitated downstream for 50 min. For macrophages cultured on fibrous scaffold, significant amount of nitrite was detected after 20 min, which then maintained on a higher level. Macrophages cultured on flat surface, however, did not show significant nitrite production during the 50 min. A control (blank) where LPS media was flowed through a cell free insert showed no detectable signal at any of the time points.
Conclusion
In this work, we present a new approach to assemble a scalable and multiplexable microfluidic device for 3D cell culture and studies. The assembled device contains two main parts: removable fibrous inserts and a 3D-printed fluidic device. The inserts were cut into rectangular strips by laser-cutting, which also helps immobilize (sinter) the fibers on the PS substrate. The fluidic device has pre-designed locking slots along the channel, in which the inserts can be integrated and immobilized. The number of inserts and the distance between adjacent inserts can be customized by changing designs of the locking slots in the 3D-printed fluidic device. For example, in this work, we applied a two-insert model (distance between inserts = 360μm) and a four-insert model (distance between inserts = 180 μm). Endothelial cells were then cultured on the fibrous inserts and integrated in the fluidic device. After circulating media for 24 hours at 420 μL/min, the endothelial cells were still adhered to the fibers. Finally, a device containing 4 inserts was used for macrophage activation studies. It was found that macrophages cultured on fibers made of silk fibroin can be polarized to M1 state by LPS at a more in vivo mimic speed than those cultured on flat surfaces. 3D-printing enables the fabrication of connection mechanism such as threads so that a customized analytical device can be connected to the cell culture device for online detection.
Supplementary Material
Acknowledgments
Support from the National Institute of General Medical Sciences (Award Number R15GM084470-04) is acknowledged.
Footnotes
Compliance with Ethical Standards
Conflict of interest: The authors declare that they have no conflict of interest.
References
- 1.Mehling M, Tay S. Microfluidic cell culture. Curr Opin Biotechnol. 2014;25:95–102. doi: 10.1016/j.copbio.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Selimovic A, Erkal JL, Spence DM, Martin RS. Microfluidic device with tunable post arrays and integrated electrodes for studying cellular release. Analyst. 2014;139(22):5686–94. doi: 10.1039/c4an01062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson AS, Selimovic A, Martin RS. Microchip-based electrochemical detection for monitoring cellular systems. Anal Bioanal Chem. 2013;405(10):3013–20. doi: 10.1007/s00216-012-6682-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menon NV, Chuah YJ, Cao B, Lim M, Kang YJ. A microfluidic co-culture system to monitor tumor-stromal interactions on a chip. Biomicrofluidics. 2014;8(6) doi: 10.1063/1.4903762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selimovic S, Dokmeci MR, Khademhosseini A. Organs-on-a-chip for drug discovery. Curr Opin Pharmacol. 2013;13(5):829–33. doi: 10.1016/j.coph.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Petersen NJ, Mogensen KB, Kutter JP. Performance of an in-plane detection cell with integrated waveguides for UV/Vis absorbance measurements on microfluidic separation devices. Electrophoresis. 2002;23(20):3528–36. doi: 10.1002/1522-2683(200210)23:20<3528::AID-ELPS3528>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Sassa F, Morimoto K, Satoh W, Suzuki H. Electrochemical techniques for microfluidic applications. Electrophoresis. 2008;29(9):1787–800. doi: 10.1002/elps.200700581. [DOI] [PubMed] [Google Scholar]
- 8.Chung BG, Flanagan LA, Rhee SW, Schwartz PH, Lee AP, Monuki ES, et al. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip. 2005;5(4):401–6. doi: 10.1039/b417651k. [DOI] [PubMed] [Google Scholar]
- 9.Bennett MR, Pang WL, Ostroff NA, Baumgartner BL, Nayak S, Tsimring LS, et al. Metabolic gene regulation in a dynamically changing environment. Nature. 2008;454(7208):1119–22. doi: 10.1038/nature07211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao JB, Wu L, Wu JZ, Zheng YH, Zhao H, Jin QH, et al. Integrated microfluidic chip for endothelial cells culture and analysis exposed to a pulsatile and oscillatory shear stress. Lab Chip. 2009;9(21):3118–25. doi: 10.1039/b909312e. [DOI] [PubMed] [Google Scholar]
- 11.van der Meer AD, Poot AA, Feijen J, Vermes I. Analyzing shear stress-induced alignment of actin filaments in endothelial cells with a microfluidic assay. Biomicrofluidics. 2010;4(1) doi: 10.1063/1.3366720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sung KE, Su G, Pehlke C, Trier SM, Eliceiri KW, Keely PJ, et al. Control of 3-dimensional collagen matrix polymerization for reproducible human mammary fibroblast cell culture in microfluidic devices. Biomaterials. 2009;30(27):4833–41. doi: 10.1016/j.biomaterials.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sell S, Barnes C, Smith M, McClure M, Madurantakam P, Grant J, et al. Extracellular matrix regenerated: tissue engineering via electrospun biomimetic nanofibers. Polymer International. 2007;56(11):1349–60. [Google Scholar]
- 14.Yang F, Murugan R, Ramakrishna S, Wang X, Ma YX, Wang S. Fabrication of nano-structured porous PLLA scaffold intended for nerve tissue engineering. Biomaterials. 2004;25(10):1891–900. doi: 10.1016/j.biomaterials.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 15.Naba A, Pearce OMT, Del Rosario A, Ma DD, Ding HM, Rajeeve V, et al. Characterization of the Extracellular Matrix of Normal and Diseased Tissues Using Proteomics. J Proteome Res. 2017;16(8):3083–91. doi: 10.1021/acs.jproteome.7b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlaepfer DD, Hunter T. Signal transduction from the extracellular matrix - A role for the focal adhesion protein-tyrosine kinase FAK. Cell Struct Funct. 1996;21(5):445–50. doi: 10.1247/csf.21.445. [DOI] [PubMed] [Google Scholar]
- 17.Sheetz MP, Felsenfeld DP, Galbraith CG. Cell migration: Regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8(2):51–4. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 18.Mo XM, Xu CY, Kotaki M, Ramakrishna S. Electrospun P(LLA-CL) nanofiber: a biomimetic extracellular matrix for smooth muscle cell and endothelial cell proliferation. Biomaterials. 2004;25(10):1883–90. doi: 10.1016/j.biomaterials.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 19.Lebaron RG, Athanasiou KA. Extracellular matrix cell adhesion peptides: Functional applications in orthopedic materials. Tissue Eng. 2000;6(2):85–103. doi: 10.1089/107632700320720. [DOI] [PubMed] [Google Scholar]
- 20.Zhang WJ, Zhang YS, Bakht SM, Aleman J, Shin SR, Yue K, et al. Elastomeric free-form blood vessels for interconnecting organs on chip systems. Lab Chip. 2016;16(9):1579–86. doi: 10.1039/c6lc00001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YF, Chan HN, Michael SA, Shen YS, Chen Y, Tian Q, et al. A microfluidic circulatory system integrated with capillary-assisted pressure sensors. Lab Chip. 2017;17(4):653–62. doi: 10.1039/c6lc01427e. [DOI] [PubMed] [Google Scholar]
- 22.Skardal A, Shupe T, Atala A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov Today. 2016;21(9):1399–411. doi: 10.1016/j.drudis.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu YL, Chen CP, Summers S, Medawala W, Spence DM. C-peptide and zinc delivery to erythrocytes requires the presence of albumin: implications in diabetes explored with a 3D-printed fluidic device. Integr Biol. 2015;7(5):534–43. doi: 10.1039/c4ib00243a. [DOI] [PubMed] [Google Scholar]
- 24.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21(12):745–54. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theberge AB, Yu JQ, Young EWK, Ricke WA, Bushman W, Beebe DJ. Microfluidic Multiculture Assay to Analyze Biomolecular Signaling in Angiogenesis. Anal Chem. 2015;87(6):3239–46. doi: 10.1021/ac503700f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golden AP, Tien J. Fabrication of microfluidic hydrogels using molded gelatin as a sacrificial element. Lab Chip. 2007;7(6):720–5. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 27.Pham QP, Sharma U, Mikos AG. Electrospinning of polymeric nanofibers for tissue engineering applications: A review. Tissue Eng. 2006;12(5):1197–211. doi: 10.1089/ten.2006.12.1197. [DOI] [PubMed] [Google Scholar]
- 28.Greiner A, Wendorff JH. Electrospinning: A fascinating method for the preparation of ultrathin fibres. Angew Chem Int Edit. 2007;46(30):5670–703. doi: 10.1002/anie.200604646. [DOI] [PubMed] [Google Scholar]
- 29.Sill TJ, von Recum HA. Electrospinning: applications in drug delivery and tissue engineering. Biomaterials. 2008;29(13):1989–2006. doi: 10.1016/j.biomaterials.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Li WJ, Laurencin CT, Caterson EJ, Tuan RS, Ko FK. Electrospun nanofibrous structure: A novel scaffold for tissue engineering. J Biomed Mater Res. 2002;60(4):613–21. doi: 10.1002/jbm.10167. [DOI] [PubMed] [Google Scholar]
- 31.Chen CP, Mehl BT, Sell SA, Martin RS. Use of electrospinning and dynamic air focusing to create three-dimensional cell culture scaffolds in microfluidic devices. Analyst. 2016;141(18):5311–20. doi: 10.1039/c6an01282e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.http://www.stratasys.com/materials/material-safety-data-sheets/polyjet/transparent-materials.
- 33.Yang YM, Chen XM, Ding F, Zhang PY, Liu J, Go XS. Biocompatibility evaluation of silk fibroin with peripheral nerve tissues and cells in vitro. Biomaterials. 2007;28(9):1643–52. doi: 10.1016/j.biomaterials.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Doe WF, Henson PM. Macrophage stimulation by bacterial lipopolysaccharides. I. Cytolytic effect on tumor target cells. J Exp Med. 1978;148(2):544–56. doi: 10.1084/jem.148.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rago R, Mitchen J, Wilding G. DNA Fluorometric Assay in 96-Well Tissue-Culture Plates Using Hoechst-33258 after Cell-Lysis by Freezing in Distilled Water. Anal Biochem. 1990;191(1):31–4. doi: 10.1016/0003-2697(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 36.Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4(3):281–6. doi: 10.2174/1568010054022024. [DOI] [PubMed] [Google Scholar]
- 37.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 40.Chensue SW, Terebuh PD, Remick DG, Scales WE, Kunkel SL. In vivo biologic and immunohistochemical analysis of interleukin-1 alpha, beta and tumor necrosis factor during experimental endotoxemia. Kinetics, Kupffer cell expression, and glucocorticoid effects. Am J Pathol. 1991;138(2):395–402. [PMC free article] [PubMed] [Google Scholar]
- 41.Vignery A. Macrophage fusion: are somatic and cancer cells possible partners? Trends Cell Biol. 2005;15(4):188–93. doi: 10.1016/j.tcb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 42.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281(5379):1001–5. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 43.Garg K, Pullen NA, Oskeritzian CA, Ryan JJ, Bowlin GL. Macrophage functional polarization (M1/M2) in response to varying fiber and pore dimensions of electrospun scaffolds. Biomaterials. 2013;34(18):4439–51. doi: 10.1016/j.biomaterials.2013.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Au AK, Huynh W, Horowitz LF, Folch A. 3D-Printed Microfluidics. Angew Chem Int Edit. 2016;55(12):3862–81. doi: 10.1002/anie.201504382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CP, Mehl BT, Munshi AS, Townsend AD, Spence DM, Martin RS. 3D-printed microfluidic devices: fabrication, advantages and limitations-a mini review. Anal Meth. 2016;8(31):6005–12. doi: 10.1039/C6AY01671E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–50. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 47.Lancaster JR. A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide. 1997;1(1):18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 48.Ivanov VM. The 125th anniversary of the Griess reagent. J Anal Chem. 2004;59(10):1002–5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.