

Abstract
Unlike cytosolic processing and presentation of viral antigens by virus-infected cells, antigens first expressed in an infected non-professional antigen-presenting cell, such as CD4+ T cells in the case of HIV, and then taken up by dendritic cells are cross-presented. This generally requires entry through the endocytic pathway, where endosomal proteases have first access for processing. Thus, understanding virus escape during cross-presentation requires an understanding of resistance to endosomal proteases such as cathepsin S. We have modified HIV-1MN gp120 (gp120MN) by mutating a key cathepsin S cleavage site T322T323 in the V3 loop of the immunodominant epitope IGPGRAFYTT to IGPGRAFYVV to prevent digestion. We found this mutation to facilitate cross-presentation, and provide evidence from both MHC-binding and X-ray crystallographic structural studies that this results from preservation of the epitope rather than an increased epitope affinity for the class I MHC molecule. In contrast, when the protein is expressed by a vaccinia virus in the cytosol, the wild type protein is immunogenic without this mutation. These results demonstrate proof-of-concept that a virus like HIV, infecting predominantly non-professional presenting cells, can escape T cell recognition by incorporating a cathepsin S cleavage site that leads to destruction of an immunodominant epitope when the antigen undergoes endosomal cross-presentation.
Keywords: MHC, antigen presentation, HIV, X-ray crystallography
Introduction
Worldwide, in 2016 there were 36.7 million people living with HIV and 1 million deaths due to HIV-related illness (ref. WHO global health observatory data http://www.who.int/gho/en/, http://unaids.org). Despite progress, there remains an urgent need for an effective HIV vaccine (1). To date, however, current HIV vaccine strategies have offered only modest protection (2). Research efforts to improve on these have focused on designing novel HIV immunogens and understanding the role of antigen processing and presentation of various HIV proteins, including Tat and gp120 (3).
It is well recognized that HIV exploits multiple mechanisms to evade immune recognition, including a high mutation rate, glycosylation of the envelope protein gp120 and the virus’ ability to manipulate host antigen processing and presentation mechanisms (4). While much research has explored HIV’s manipulation of the classical MHC class I (MHC-I) pathway, recent work has begun to focus on the role of the much less-explored endosomal route which involves both the MHC-I cross presentation and MHC-II dependent pathways (5). Because the predominant cell infected by HIV is the CD4+ T cell, which is not a professional antigen-presenting cell (APC), much of the presentation of HIV antigens from infected cells requires cross-presentation by uninfected dendritic cells (DCs) to prime specific CD8+ CTL responses. Thus, escape from cross-presentation becomes a potentially critical issue for HIV.
Typically, following immunization with a whole protein in adjuvant, protein is phagocytized, processed, and presented through the endosomal pathway (6, 7). Once in the phagolysosome, a range of endosomal proteases digest it into peptide fragments that can be loaded onto MHC class II (MHC-II) molecules and presented to CD4+ T cells (8). As a result, this immunization strategy typically selects for CD4+ T cell presentation and B cell responses. However, during endosomal processing, peptides can be processed and presented on MHC- I molecules to CD8+ T cells, a process known as cross-presentation (9). Endosomal acid proteases known as cathepsins contribute to the production of such antigenic peptides (8–10). Many cathepsins have redundant specificity; however, the cathepsin S protease is crucial for removing the invariant chain from the MHC- II molecule and thus regulating CD4+ T cell immunity (11–13), and a recent study found that cathepsin S is also important for cross-presentation to CD8+ T cells (9).
Although the CD8+ response in HIV model systems has been examined with respect to MHC-I-dependent presentation to CD8+ T cells, and the importance of antigen processing in HIV CTL epitope dominance has been well documented for the endogenous class I MHC processing pathway (14), the role of endosomal antigen processing in cross-presentation of viral proteins leading to the MHC-I presentation has been incompletely explored (7). In this regard, it is striking that the cathepsin cleavage sites are highly conserved among many HIV gp120 isolates even though they are in regions that otherwise have high mutation rates, such as the V1/V2 and V3 loops (5). This raises the question whether these sites have been conserved through selective pressure, for example to permit escape from immune responses especially during endosomal cross-presentation (5). To investigate this question and the role of endosomal processing in induction of CD8+ T cells specific for HIV envelope, we focus here on the immune response in BALB/c mice to HIVMN gp120 glycoprotein (gp120MN) and variants engineered to affect processing in known cathepsin S sensitive sites. gp120MN was shown early to be a target for neutralizing antibodies (15) and was used at that time as an experimental vaccine in monovalent or bivalent form (15, 16). Further, it was a key component in the first successful phase III human HIV vaccine trial, RV144 (2), albeit with modest 31% vaccine efficacy. It is tempting to ask whether modification of cathepsin cleavage sites could make the modified gp120MN a more effective vaccine immunogen and improve on the efficacy of the protein used in the RV144 vaccine trial.
We used a repeated immunization strategy employing either wild type (WT) gp120MN protein or a cathepsin S cleavage site mutant gp120MN protein (VV gp120MN) and the liposomal Cationic Adjuvant Formulation 09 (CAF09) to compare both CD4+ and CD8+ T cell responses to peptides spanning the gp120MN sequence, since CAF09 is known to induce strong CD4+ as well as CD8+ T cell responses (17). We mapped the CD8+ response to an immunodominant epitope (18–20) IGPGRAFYTT encompassing the cathepsin S site at T322T323 that we had mutated to V322V323 to prevent cathepsin cleavage. Whole protein immunization with the cathepsin S mutant protein (VV gp120MN) elicits a strong CD8+ T cell response to the WT IGPGRAFYTT (gp120314–323) epitope, a response lacking in mice immunized with the wild type (WT gp120MN) protein. We hypothesized that this enhanced CD8+ T cell response in mice immunized with the VV gp120MN protein results from preservation of the immunodominant epitope, allowing it to be presented to CD8+ T cells. Peptide-MHC binding assays surprisingly excluded the alternative explanation that the mutation improved MHC-binding of the epitope. To understand the binding, we crystallized and determined the three-dimensional structure of H2-Dd complexed with three MN-derived peptides. Thus, manipulation of the cathepsin S cleavage site in the gp120MN can alter the cross-presentation of the immunodominant epitope, IGPGRAFYTT, and the induction of antigen-specific CD8+ T cells in BALB/c mice. In contrast, a vaccinia virus that expresses various HIV proteins, including the wild type gp160MN protein, can elicit robust CD8+ T cell responses to the IGPGRAFYTT epitope (18–20), as vaccinia expresses the gp160 protein in the cytosol and does not require passage through endosomes, which may account for its evasion of cathepsin S cleavage. We conclude that augmentation of CD8+ responses induced by whole gp120 protein in adjuvant benefits from judicial introduction of a cathepsin S-resistant site within or adjacent to the location of an immunodominant epitope, preventing escape by this mechanism. This conceptually simple strategy may lead to improvement in vaccine design and efficacy.
Materials and Methods
Recombinant HIV-1MN envelope mutants and cathepsin S digestion studies
Plasmids encoding recombinant gp120MN (UCSC358) and recombinant gp120MN mutated to replace threonine at positions 322 and 323 with valine (VV gp120MN) (UCSC435) were expressed in 293HEK cells and purified by immunoaffinity chromatography as described previously (21). Purified human cathepsin S (CatS) was purchased from Biomol (Philadelphia, PA). 6 μg samples of protein were treated with a 1:100 dilution of CatS and incubated for the times indicated. The reaction conditions for the protease digestion and the method for stopping the reactions were similar to those described previously (5). The irreversible inhibitor of cathepsins B, L and S, Z-FA-FMK (Enzo Life Sciences), was used in control experiments to demonstrate the specificity of the protease. Because there are 4 CatS cleavage sites in recombinant glycoprotein 120 (rgp120MN), the T322V and T323V mutations did not completely prevent protease cleavage. However, N-terminal sequence analysis demonstrated that the fragments detected resulted from cleavages between positions 208 and 209, 261 and 262, and 435 and 436 as described by (5).
Cell culture and peptide induction of surface MHC-I expression
A TAP2-deficient cell line, RMAS-Dd, expressing the MHC- I H2-Dd (gift of Dr. David Raulet, U. California, Berkeley) was cultivated in serum free, X-VIVO medium (Lonza). Cell cultures were incubated with indicated peptides overnight either with or without the addition of human β2-microglobulin (hβ2m)(Sigma-Aldrich). Following culture, cells were stained with the mAb 34-5-8S (AbCam), which binds H2-Dd MHC- I molecules in a peptide-dependent, but not peptide-specific fashion (22). Stained cells were analyzed using a BD LSR II flow cytometer with FlowJo© software. The results are expressed as the fluorescence index (FI), the ratio of mean fluorescence intensity (MFI) in the presence vs absence of peptide, minus 1, as previously described (23). This method was an adaptation from previously described methods (23, 24).
HIV gp120MN wild type and recombinant proteins and peptides
Proteins for immunization were produced by a previously described method (25). We prepared one wild type recombinant protein, WT gp120MN, and one mutant gp120MN envelope glycoprotein, referred to as VV gp120MN. The VV recombinant g120 protein differs from the WT gp120 protein by substitution at the cathepsin S cleavage site T322T323 in the V3 domain, where the native sequence has been mutated to V322V323. The following synthetic peptides, obtained from Genescript, were >95% pure, dissolved in water and diluted in culture medium for in vitro studies: IGPGRAFYTT, IGPGRAFYVV, IGPGRAFYT, IGPGRAFYV, IGPGRAFYTI, IGPGRAFYVI, IGPGRAFYTV, and IGPGRAFYVT. To avoid confusion, peptide pools are denoted p.p. followed by the number, and single peptides are denoted s.p. followed by the residue numbers or name.
Whole protein immunization
Studies were performed on 6–12 wk old BALB/c mice purchased from Charles River Laboratories and maintained in the National Cancer Institute Animal Care Unit under pathogen-free conditions. Animal studies were approved by the National Cancer Institute Animal Care and Use Committee and conducted in accordance with all federal and National Institutes of Health policies and regulations. Mice were immunized three times at two-week intervals i.p. with experimental vaccines containing 100 μg of either WT gp120MN or the mutant VV gp120MN formulated with the cationic adjuvant formulation 09 (CAF09) consisting of dimethyldioctadecylammonium bromide (DDA, NCK, Copenhagen, Denmark), synthetic monomycoloyl glycerol (MMG, NCK, Copenhagen, Denmark) analogue MMG-1 and the TLR-3 agonist polyinosinic-polycytidylic acid (pI:C, Sigma-Aldrich, Copenhagen, Denmark), formulated by the film method in a volume consisting of 0.1 mL CAF09 and 0.1 mL of antigen in 10 mM TRIS-buffer (pH 7.4) as described (17, 26). In CAF09, the lipid DDA forms the backbone of the liposomes along with MMG, a synthetic analogue of a mycobacterial cell wall component that is highly immunostimulatory and also stabilizes the liposomes. Incorporation of poly(I:C) enables type I interferon induction and hence licensing of DCs to cross prime a CD8+ T cell response (26).
Vaccinia immunization
Studies were performed on 6–8 wk old BALB/c mice purchased from Charles River Laboratories. Mice were immunized once i.p. with vaccinia virus (vMN) (a kind gift of Patricia Earl and Bernard Moss, NIAID) expressing the gp160MN protein, at 20 × 106 PFU/mouse. At either day 9 or 13 post infection (p.i.), splenocytes were isolated and stimulated in vitro with minimal peptides or stained with dextramers loaded with desired peptides.
Flow cytometric analysis of dextramer staining
The following H2-Dd dextramers were purchased from Immudex (Copenhagen, Denmark, and Fairfax, VA): IGPGRAFYTT (PE), IGPGRAFYT (APC), IGPGRAFYVV (PE), and IGPGRAFYV (APC). For dextramer staining, spleen cells (1–5 × 106) were isolated and directly stained for CD3, CD4, CD8, CD16/32 (Biolegend), Yellow Viability or AquaBlue (Life Technologies) plus the indicated dextramers. For intracellular staining, single cell suspensions (106 cells/μL) from spleen were isolated and stimulated with indicated peptides or ionomycin and phorbol 12-myristate 13-acetate (PMA), as previously described (27–29). Subsequently, cells were removed and stained for CD3, CD4, CD8, CD16/32, IL2, IFNγ (Biolegend) and TNFα (BD Pharmingen). Intracellular cytokine production was quantified using the previously described method (27–29). Stained cells were analyzed using a BD LSR II flow cytometer with FlowJo software.
Antibody blocking H2-Dd MHC- I molecules
Splenocytes from VV gp120MN-immunized mice were distributed to a 96 well plate (106 cells/200 μL) and incubated for 15 min with one of the following anti-mouse mAbs: H2-Dd (34-2-12), H2-Ld/H2-Db (28-14-8), H2-Kd (SF1-1.1) (Biolegend), or an isotype control, mouse IgG2a (AbCam). After incubation, the indicated peptide was added at 1.5 μg/ml, the cells were cultured with peptide for 6 hours and then were stained for CD3, CD4, CD8, IL2, IFNγ (Biolegend), AquaBlue or Yellow Viability Dye (Life Technologies) and TNF (BD Pharmigen). Intracellular cytokine production was quantified using the previously described method (27–29). Stained cells were analyzed using a BD LSR II flow cytometer with FlowJo software.
Protein expression and purification, crystallization and data collection and refinement
The luminal domain of H2-Dd, expressed in E. coli BL21(DE3) from plasmid pET21, was refolded with murine β2m and peptide s.p.V9 (IGPGRAFYV), peptide s.p.T9 (IGPGRAFYT), or peptide s.p.VI10 (IGPGRAFYVI), as described previously (30, 31). Following dialysis and concentration by ultrafiltration, the proteins were purified by size exclusion chromatography on Superdex 75 followed by ion exchange chromatography on mono Q (GE Healthcare Life Sciences). The purified complexes were concentrated to ~6 mg/ml and crystallization screens were set up in hanging drops at 4 °C. Several small (0.05 × 0.01 mm) crystals of the s.p.V9 and s.p.T9-containing complex were observed after two weeks in solutions of 12% PEG 20,000 in 0.1M (2-(n-morpholino)-ethanesulfonic acid) (MES) pH 6.5, and those with peptide s.p.VI10 were obtained in 15% PEG 20,000 and 0.1 M cacodylate at pH 6.0. Crystals were flash frozen in 10% ethylene glycol in crystallization buffer and diffraction data were collected remotely on beamline SER-CAT 22-ID at the Argonne Photon Source. Data were indexed with XDS (32), and analyzed with Xtriage in PHENIX (33). For the complexes with s.p.T9 and s.p.V9, space groups were P212121 and P1 respectively and for s.p.VI10 analysis indicated pseudo-merohedral twinning with a twin fraction of 0.38, in space group P21. Molecular replacement solutions for complexes were readily found with Phaser (34) using 3ECB (H2-Dd complexed with P18-I10 (RGPGRAFVTI) and murine β2m) as a search model, with the peptide removed. Following rigid body and individual B-factor refinement in PHENIX, SA-omit maps of the peptides were calculated which readily permitted manual building of the peptides. Further refinement of the complex with peptide s.p.V9 resulted in a final model with R-work/R-free of 0.198/0.249 at 2.35 Å, for the complex with s.p.T9, R-work/R-free of 0.195/0.239 at 1.96 Å, and for the complex with peptide s.p.VI10 a final model with R-work/R-free of 0.184/0.224 at 3.05 Å. Data collection and refinement statistics are listed in Table 1. Final models are illustrated with the calculated SA-omit maps in Fig. S1. Structure factors and coordinates have been deposited in the protein data bank (PDB) (URL: https://www.rcsb.org/pdb/home/home.do) under accession numbers 5T7G, 5KD7, and 5KD4 for s.p.T9, s.p.V9, and s.p.VI10 respectively. Molecular graphics were produced with PyMOL (The PyMOL Molecular Graphics System, Version 1.7, Schrödinger, LLC., unpublished). Analysis of crystal structures was performed with Protein interfaces, surfaces and assemblies’ (PISA) program at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html), (35).
Table 1.
X-ray data collection and refinement statistics
| H2-Dd-spVI10 | H2-Dd-spV9 | H2-Dd-spT9 | |
|---|---|---|---|
|
| |||
| PEPTIDE | IGPGRAFYVI | IGPGRAFYV | IGPGRAFYT |
|
| |||
| PDB idcode | 5KD4 | 5KD7 | 5T7G |
| Data collection | |||
| Space group | P21 | P1** | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 47.13, 90.30, 120.07 | 51.42, 75.36, 120.03 | 50.24, 120.50, 143.74 |
| α, β, γ (°) | 90.00, 113.19, 90.00 | 93.52, 87.14, 96.50 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 47–3.05 (3.16–3.05) * | 43–2.35 (2.43–2.35)* | 39–1.96 (2.03–1.96)* |
| Rsym or Rmerge (%) | 18.9 (95.0)* | 5.5 (77.1)* | 6.0 (97.2)* |
| I/σI | 13.0 (2.6)* | 14.7 (1.8)* | 12.9 (1.3)* |
| Completeness (%) | 99.1 (98.6)* | 97.9 (96.5)* | 99.5 (98.6)* |
| Redundancy | 7.6 (7.6)* | 3.9 (4.0)* | 4.0 (3.9)* |
| Rpim(%) | 7.4 (36.8)* | 3.2 (44.7)* | 3.3 (56.6)* |
| CC1/2(%) | 99.3 (75.8)* | 99.8 (75.7)* | 99.9 (54.6)* |
| Pseudo-merohedral-twin | 0.389 (h,-k,-2h-l) | none | none |
| Refinement | |||
| Resolution (Å) | 47–3.05(3.16–3.05)* | 43–2.35(2.43–2.35)* | 39–1.96 (2.03–1.96)* |
| No. reflections (unique) | 17625 | 72767 | 61428 |
| Rwork/Rfree (%) | 18.4(33.4)*/22.4(35.9)* | 19.8(28.8)*/24.9(32.2)* | 19.5(30.7)*/23.9(36.0)* |
| No. atoms | 6278 | 12407 | 6615 |
| H2-Dd/β2m | 6122 | 12117 | 6043 |
| Peptide | 156 | 276 | 140 |
| B-factors (average) | |||
| H2-Dd/β2m | 51.5 | 52.8 | 38.8 |
| Peptide | 43.3 | 38.3 | 33.6 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.007 | 0.005 | 0.009 |
| Bond angles (°) | 1.17 | 0.79 | 1.20 |
| Ramachandran favored/outlier(%) | 94.1/0.3 | 95.0/0.6 | 96.0/0.1 |
Highest resolution shell is shown in parenthesis.
In space group P1 there are two p/H2-Dd/β2m complexes per asymmetric unit.
Data analysis and statistics
All comparisons between mutant gp120MN and WT gp120MN immunization groups were conducted using the Mann-Whitney non-parametric test or unpaired Student t test except where ANOVA was used as noted. Data are expressed as mean +/-SEM. P < 0.05 was considered significant. * = p<0.05; ** = p<0.01; *** = p<0.001. For binding curves in Figs. 4 and 5, a stratified Wilcoxon rank sum test was used, with a Hochberg correction for multiple comparisons were such occurred.
Figure 4.
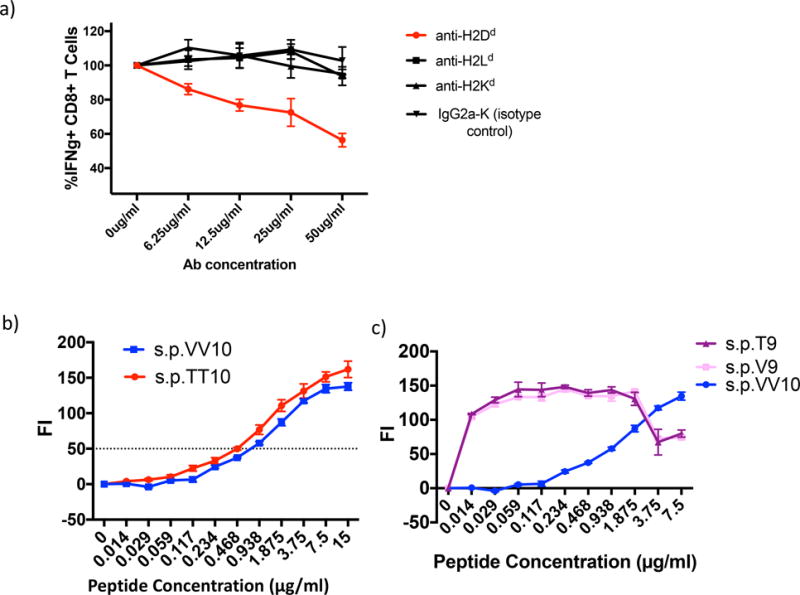
The IGPGRAFYTT epitope is presented on the H2-Dd molecule. Moreover, mutating the last two amino acid residues in the V3 minimal epitope does not significantly alter peptide binding affinity with the H2-Dd MHC-I molecule. a) Splenocytes were isolated from BALB/c mice 14 days after the final immunization with either VV gp120 or WT gp120 in CAF09 and stimulated in vitro for 6 hours with the 15-mer peptide s.p.79 (which contains the IGPGRAFYTT epitope) as well as varying concentrations of anti-H-2Dd, H-2Ld, H-2Kd, and IgG2a-K (isotype control). After 6 hour stimulation, CD8+ T cells were analyzed for the following intracellular IFNγ.
Data were calculated as the % difference over the untreated Ab control ((treated CD8+ IFNγ amount/untreated IFNγ amount)*100). Anti-H2-Dd produced significant inhibition compared to the other antibodies and control (p = 0.0001 by ANOVA on log-transformed data). b) Peptide induced surface expression of H2-Dd with peptides s.p.TT10 and s.p.VV10. TAP-deficient, RMAS-Dd were incubated with the indicated concentrations of each peptide and stained with the antibody 34-5-8 (an anti-H2-Dd antibody) as described in Materials and methods. Results are shown as the fluorescence index, the mean fluorescence intensity (MFI) over background (∆ mean fluorescence intensity) × 100. The horizontal line indicts the concentration at which the level of H2-Dd staining is increased by 50% (FI0.5). c) Binding titration of 9-mer peptides s.p.T9 and s.p.V9 compared with that of s.p. VV10. Binding assay was performed identically to that in panel b. The curve for s.p.VV10 is significantly different by ANOVA from the other two at p < 0.0001, and the other two curves do not differ from each other.
Figure 5.
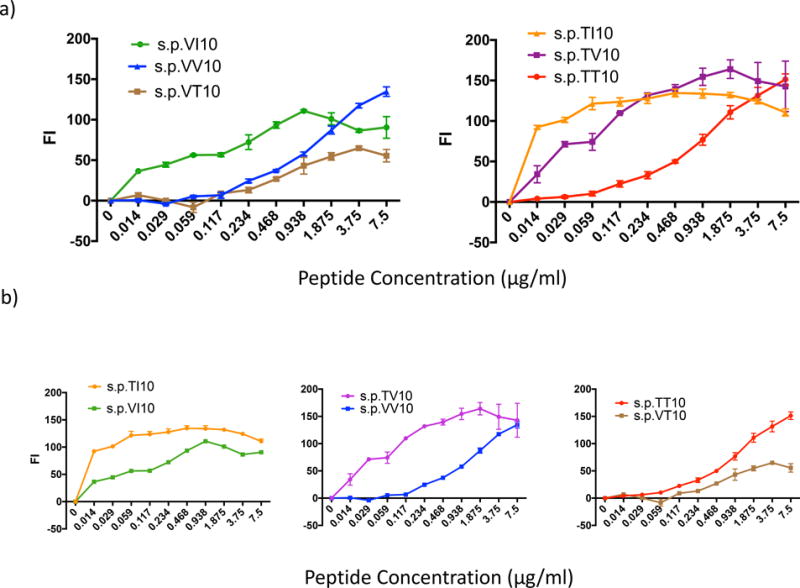
A more hydrophobic amino acid at the 323 anchor residue enhances peptide binding to the H2-Dd MHC-I molecule, while altering the 9th amino acid residue or penultimate position from the hydrophilic Thr (T322) to a more hydrophobic Val (V322) can inhibit peptide MHC I binding. Peptide induced surface expression of H-2Dd with peptides PTT10 (IGPGRAFYTT), PTI10 (IGPGRAFYTI), PTV10 (IGPGRAFYTV), PVI10 (IGPGRAFYVI), PVV10 (IGPGRAFYVV), PVT10 (IGPGRAFYVT). TAP-deficient, RMAS-Dd were incubated with indicated concentrations of each peptide and stained with the antibody 34-5-8 (an anti-H2-Dd antibody) as described in Materials and methods. Results are shown as the fluorescence index (FI), the mean fluorescence intensity (MFI) over background (∆ mean fluorescence intensity) × 100 as in Fig 4b. a) Peptide affinity for the H2-Dd MHC-I molecule with the penultimate position is held constant as Val322 or Thr322 and the anchor residue is altered to either Ile323, Val323 or Thr323. Statistics: left panel, s.p.VV10 vs VT10 p = 0.0023; s.p. VV10 vs VI10 and s.p.VT10 vs VI10, p < 0.0001 by stratified Wilcoxon rank sum with Hochberg correction for multiple comparisons; right panel, s.p.TT10 differs from TV10 and TI10 at p<0.0001, but s.p.TV10 and TI10 do no differ from each other greatly. b) Peptide:MHCI binding affinity with the anchor residue (10th amino acid) held constant at Ile323, Val323 or Thr323 while the penultimate position (9th amino acid) is either Thr322 or Val322. Statistics: left panel, p < 0.0001; middle panel, p<0.0001; right panel, p<0.0001, all by the stratified Wilcoxon test.
Results
Immunization with recombinant VV gp120MN generates a strong IFNγ+ CD8+ T cell response to an immunodominant epitope in gp120MN
Cathepsin S plays a critical role in MHC-II antigen endosomal processing and presentation (36, 37) and has been suggested to play a role in cross-presentation on MHC-I molecules to CD8+ T cells (9). To evaluate the role of cathepsin S in modulating the HIV-1-specific cellular responses, we took advantage of a previously described recombinant HIVMN gp120 protein, which has a mutation in the cathepsin S cleavage site, from T322T323 to V322V323 in the V3 domain (VV gp120)(38) (Fig 1a). The recombinant VV gp120 protein is completely resistant to cathepsin S digestion at that site (Fig 1b), although three other cathepsin S digestion sites within gp120 such as 208–209, 261–262, and 435–436 remain digestion sensitive. We compared the ability of WT gp120 with VV gp120 to induce antigen specific cellular immune responses. Mice were immunized with either of these recombinant proteins in a cationic liposomal adjuvant, CAF09 (26) three times with two weeks between immunizations (see schematic in Fig 1c). Approximately two weeks after the final immunization, splenocytes were isolated and stimulated in vitro with 15-mer HIV gp120MN peptides (each peptide overlapping by 10 residues) spanning the entire WT gp120MN protein (for a total of 212 peptides, divided into 10 peptide pools (p.p.1-p.p.10) of 12–30 peptides each, distributed as shown in Fig 2a).
Figure 1.
a) Overview of the HIV gp120MN V3 domain. The cathepsin S lysosomal protease cleaves (as indicated by the black arrow) between the amino acids T322T323 (red) in the WT gp120 protein. In the mutant protein that inhibits cathepsin S cleavage, referred to as the VV gp120 protein, the T322T323 has been mutated to V322V323 (blue). The sequence IGPGRAFYTT (purple) is a minimal epitope that has previously been reported to elicit CD8+ T cell responses in BALB/c mice immunized with modified vaccinia virus expressing HIV gp160MN. b) Effect of mutations in the V3 domain of rgp120MN on cathepsin S digestion. Purified rgp120MN and rgp120MN mutated to replace threonine (T) at positions 322 and 323 with valine (V) were digested with cathepsin S (CatS) for the times indicated. The reactions were stopped by the addition of SDS-PAGE sample buffer and stored overnight at −80°C. The samples were loaded onto gels and the protein digests were visualized by Coomassie blue staining. Controls included incubation of the proteins in CatS digest buffer without added enzyme, and incubation in Cat S digest buffer containing the irreversible cathepsin inhibitor Z-FA-FMK. Because there are 4 CatS cleavage sites in rgp120MN the T322V and T323 mutations did not completely prevent protease cleavage. However, N-terminal sequence analysis demonstrated that the fragments detected resulted from cleavages between positions 208 and 209, 261 and 262, and 435 and 436 as described by Yu et al (5). c) The repeated immunization strategy. At day 0 (D0), BALB/c mice were immunized I.P. with 100μg of either wild type (WT) gp120MN or the cathepsin S mutant, VV gp120MN protein in a cationic liposome, CAF09. At 14 day intervals (or at D14 and D28), mice were re-immunized. On D38, splenocytes were isolated and stimulated in vitro with desired peptides for 12 hours (overnight). After 12 hours, splenocytes were stained for intracellular cytokines: TNF, IFNγ and IL-2.
Figure 2.
CD8+ T cells from BALB/c mice immunized with the cathepsin S mutant, VV gp120, generate strong IFNγ responses to peptides containing the IGPGRAFYTT sequence and recognize an IGPGRAFYTT dextramer when compared to mice immunized with the WT gp120 protein. a) BALB/c mice were immunized with either WT gp120 or VV gp120 in CAF09, as described in the Materials and Methods. Splenocytes were isolated 14 days after the final immunization and stimulated in vitro for 6 hrs with (7.5 μg/ml) HIV gp120MN overlapping 15-mer peptides spanning the length of the gp120MN protein, and then stained for IFNγ. CD4+ T cell responses to peptide pools are shown in Fig S1 b) BALB/c mice were immunized with either WT gp120 or VV gp120 in CAF09 and 14 days later splenocytes were isolated, stimulated in vitro for 6 hrs with (7.5 μg/ml) single peptides 78 (s.p.78), s.p.79, s.p. 80/81 and s.p.TT10 (IGPGRAFYTT) and stained for intracellular cytokines: TNFα, IFNγ and IL-2. c) Mice were immunized with either WT gp120 or the cathepsin S mutant, VV gp120 in CAF09, as described in legends to Figure 1c, and Ag-specific CD8+ T cells were detected with Dd-s.p.T10 dextramers. d) Frequency of H-2Dd-s.p.T10 peptide dextramer-binding CD8+ T cells isolated from immunized BALB/c mice splenocytes that were directly stained for surface markers, CD3, CD4, CD8 and an H2-Dd s.p.TT10 (IGPGRAFYTT) dextramer. * indicates p < 0.05.
Priming with exogenous protein provided a greater IFNγ+ CD8+ T cell response in BALB/c mice immunized with the VV gp120 protein than in animals given the WT gp120 protein after in vitro stimulation with peptides spanning the wild type HIV gp120MN protein (p = 0.0286). These responses were directed primarily to p.p.3 (s.p. 61–90) (Fig 2a). Interestingly, this pool contained peptides s.p.78 (RKRIHIGPGRAFYTT) and s.p.79 (HIGPGRAFYTTKNII), both of which contain a previously described immunodominant epitope, IGPGRAFYTT (s.p.TT10) for mice immunized with recombinant vaccinia virus (vMN) expressing the entire gp160MN protein (19, 20). Further epitope mapping revealed that only mice immunized with the VV gp120MN protein, but not WT gp120MN, developed strong IFNγ+CD8+ T responses to s.p.78 and s.p.79 (Fig 2b). To determine whether the IFNγ+CD8+ T cell response was directed against the s.p.TT10 epitope, splenocytes were stained with the s.p.TT10/H2-Dd (Dd-s.p.TT10) dextramer. This dextramer was loaded with the minimal epitope, s.p.TT10, which is the wildtype sequence and hence differs from the sequence in the VV gp120MN protein (IGPGRAFYVV (s.p.VV-10)), by the substitution of TT for VV. Mice immunized with VV gp120MN had a significantly greater number of s.p.TT10/H2-Dd- binding CD8+ T cells as well as significantly greater IFNγ+ CD8+ T cell responses to epitope peptides s.p.78, s.p.79, s.p.VV10, s.p. T9, and s.p.V9 (Fig 2c, d and Fig 3a). It is well known that immunization with whole protein can elicit a CD8+ T cell response through cross-presentation (7, 39–41), and previous studies have demonstrated that cathepsin S can affect cross-presentation (9, 42). To explain this enhanced IFNγ+ CD8+ T cell response in VV gp120MN-immunized mice, we proposed the following hypotheses: (1) the mutation from T322T323 to V322V323 in the V3 domain of gp120 had prevented the epitope IGPGRAFYVV (s.p.VV10) from being cleaved by cathepsin S in the endosome (and thus also prevented further endosomal degradation), thus enhancing the frequency with which the epitope could be cross-presented; (2) the mutation at the final residue from a hydrophilic residue T323 to a more hydrophobic V323, enhanced the binding of the mutant Val anchor residue within the F pocket in the MHC-I molecule, thus increasing the immunogenicity of the s.p.VV10 epitope; or, (3) the mutation from T322T323 to V322V323 did both.
Figure 3.
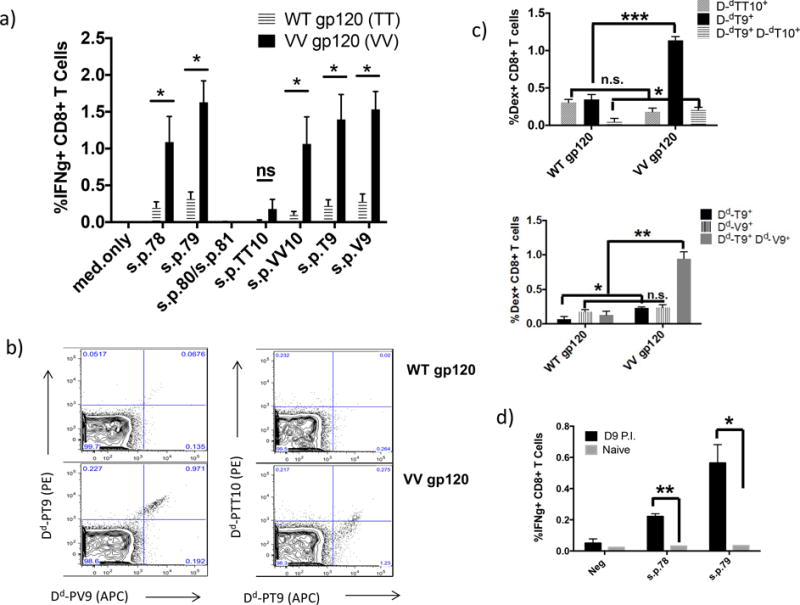
CD8+ T cells from BALB/c mice immunized with Cat S mutant, VV gp120 protein induce strong IFNγ+ CD8+ T cell response to peptides IGPGRAFY-T, TT, V and VV. Furthermore, these CD8+ T cells are H2-Dd peptide-positive for s.p.TT10, s.p.T9 and s.p.V9. a) BALB/c mice were immunized with either WT gp120 or VV gp120 in CAF09, as described in the Material and Methods. 14 days following the last immunization, splenocytes were isolated and stimulated with (7.5μg/mL) minimal peptides s.p.TT10 (IGPGFAYTT), s.p.T9 (IGPGRAFYT), s.p.VV10 (IGPGRAFYVV), and s.p.V9 (IGPGRAFYV). b) representative data of CD8+ T cells positive for dextramers, Dd-s.p.TT10, Dd-s.p.T9, and Dd-s.p.V9. c) Percent of CD8+ T cells positive for dextramers, d) BALB/c mice were immunized with 108 PFU of modified vaccinia virus expressing the HIV gp160MN protein, 7 days post immunization, splenocytes were isolated and stimulated in vitro for 6 hours with (7.5μg/mL) peptides s.p.78, s.p.79, s.p.80 and s.p.81. Following stimulation, cells were stained for intracellular cytokine IFNγ. * indicates p < 0.05; ** indicates p < 0.01; and *** indicates p < 0.001.
Immunization with the VV gp120 protein elicits greater IFNγ+ CD8+ T cell responses to minimal epitopes than immunization with wild type gp120 protein
We observed that CD8+ T cells from VV gp120MN immunized mice had a greater cross-reactive response to the minimal epitopes s.p.TT10 and s.p.VV10, with s.p.VV10 providing the greatest response. However, we were interested in determining whether the same CD8+ T cells were cross-reactive to the s.p.VV10 and s.p.TT10 as well as the alternative minimal epitopes s.p.V9 and s.p.T9 (Fig 3a). We utilized several dextramers with different fluorescent labels, and compared the cross-reactivity of CD8+ T cells from WT gp120MN and VV gp120MN-immunized mice. Interestingly, CD8+ T cells from mice immunized with VV gp120 bound the s.p.V9 and s.p.T9 dextramers with similar frequency and almost all the T cells were double positive, indicating that the same T cells reacted with both epitopes; while the s.p.T9 and s.p.TT10 dextramers were recognized unequally with a greater response to s.p.T9, only some of which cross reacted with s.p.TT10. By contrast to those immunized with VV gp120, cells from mice primed with the WT gp120 protein did not bind any of the three dextramers, s.p.V9, s.p.T9 and s.p.TT10 (Fig 3b and c). The dextramer positive cells for DdT9, DdV9 and double positive for DdT9 and DdTT10 or for DdT9 and DdV9 were significantly higher for VV-gp120-immunized mice than for WT-gp120-immunized mice (Fig. 3c).
Vaccinia virus infection bypasses endosomal destruction of the TT epitope
Previous work using recombinant vaccinia virus expressing the gp160MN protein has elicited CD8+IFNγ+ T cells that recognize the immunodominant s.p.TT10 (IGPGRAFYTT) epitope (19, 20). Vaccinia virus can infect antigen-presenting cells (APCs), where the resulting recombinant gp160 protein is produced and processed in the cytosol through the typical MHC-I proteasomal pathway. In contrast, we hypothesized that immunization with WT gp120MN protein processed through the endosomal pathway would result in the degradation of the s.p.TT10 (IGPGRAFYTT) epitope, via cathepsin S cleavage during cross-presentation. However, the VV gp120MN protein with the mutation of the cathepsin S cleavage site would result in preservation of the epitope during cross-presentation and induction of CD8+ T cells that cross-reactively recognized 15-mer peptides containing the IGPGRAFYTT epitope. Thus, in contrast to mice immunized with recombinant protein, those immunized with the recombinant vaccinia expressing the WT gp160MN even once without boosting or in vitro expansion raised a response to both s.p.78 and s.p.79 (Fig 3d), consistent with our earlier results (19, 20). Thus, it appears that immunization with vaccinia virus, which is followed by the processing and presentation of the gp160MN protein through the classical, MHC-I pathway, preserves the s.p.TT10 epitope from cathepsin S cleavage and subsequent degradation, thereby enabling s.p.TT10 presentation to T cells. This is in stark contrast to the cross-presentation pathway where the s.p.TT10 epitope seems to be cleaved by cathepsin S and immediately further degraded so that even the s.p.T9 9-mer is not presented.
The s.p.TT10 epitope, contained within the P79 15-mer peptide, is presented by H2-Dd
Immunization with a whole protein may elicit CD8+ T cell responses through cross-presentation. Based on our results, it appears that immunization with the cathepsin S resistant mutant, VV gp120MN, can elicit CD8+ T cell responses specific for the s.p.TT10 epitope through cross-presentation. However, this mutant VV gp120MN protein has the mutation T323 to V323 at the H2-Dd C-terminal anchor residue. It is well known that a more hydrophobic amino acid can bind more efficiently to the F-pocket in the H2-Dd MHC-I molecule (43, 44). Thus, we expected that the cathepsin S-site mutation from T323 to V323 also enhanced the binding affinity of the mutant epitope, s.p.VV10 (IGPGRAFYVV) for the MHC-I molecule. We first confirmed that H2-Dd presented the s.p.79 15-mer peptides that contain the PTT10 minimal epitope from the gp120MN protein. Previous studies of immune responses to the gp160MN protein in BALB/c mice demonstrated the immunodominant minimal epitope s.p.TT10 was H2-Dd restricted (38, 45). Since BALB/c mice express three classical MHC- I molecules, H2-Dd, H2-Ld and H2-Kd that could potentially present the s.p.TT10 peptide, we evaluated the presenting element with an in vitro antibody blocking study. Only the blocking antibody to H2-Dd, but not those to the other class I molecules, resulted in a significant reduction (p = 0.0215) in the IFNγ+ CD8+ T cell response against s.p.79 (HIGPGRAFYTTKNII) in mice immunized with the VV gp120 protein (Fig 4a). Additionally, s.p.TT10 binding to H2-Dd was further confirmed in an RMA-S/H2-Dd epitope stabilization assay (Fig 4b). Thus, the binding was to H2-Dd as predicted.
In vitro binding of minimal epitopes to H2-Dd MHC-I molecule
Following confirmation that the s.p.79 epitope, containing the IGPGRAFYTT minimal epitope, is presented by H2-Dd, we evaluated the relative binding affinities of s.p.VV10, s.p.TT10 and related peptides to H2-Dd. Previous studies demonstrated that substitution of amino acid residues with greater hydrophobicity at the C-terminal anchor residue position can enhance peptide binding to the H2-Dd MHC-I molecule (24, 43, 46). Therefore, we expected that s.p.VV10, with the more hydrophobic amino acid, Val, at the C-terminus would have greater binding affinity to H2-Dd than s.p.TT10 with the more hydrophilic Thr at the anchor residue site. Surprisingly, however, there was no significant difference in the apparent binding affinities of peptides s.p.TT10 and s.p.VV10 to H2-Dd, both of which give a 50% increase in H2-Dd surface expression at about 1.0 μg/ml with nearly superimposable curves (Fig 4b). Moreover, the slight difference (< 1 log2) was in the direction that s.p.VV10 was a slightly weaker binder than s.p.TT10, not supporting an improvement in binding. To explore the basis of the similarity in affinity between s.p.TT10 and s.p.VV10, we compared several peptides that have a Val or Thr penultimate residue (position nine, immediately adjacent to the anchor residue) and varied anchor residues: Thr, Val, and Ile as a positive control. The following single peptides (s.p.) were tested: IGPGRAFYTI (s.p.TI10), IGPGRAFYTV (s.p.TV10), IGPGRAFYVI (s.p.VI10) and IGPGRAFYVT (s.p.VT10) (Table 1). First, we compared the binding affinity of the peptides with the same penultimate position (either T or V) and varied the anchor amino acid residues T, V, and I (as a positive control for its previously demonstrated ability to enhance peptide/MHC binding) (Fig 5). As expected, when we compared peptides with T at the penultimate position, we observed the following hierarchy of binding: s.p.TI10> s.p.TV10> s.p.TT10, demonstrating that the more hydrophobic anchor residues, I and V, enhance the interaction of the peptide with H2-Dd more than the less hydrophobic T (Fig 5a right panel). Similarly, when we compared peptides with different terminal anchor residues all with V at the penultimate position, we observed the following binding: s.p.VI10 > s.p.VV10 = s.p.VT10, indicating the more hydrophobic I bound best, while V and T bound with similar affinity (Fig 5a left panel). Unexpectedly, we noticed peptides with V at the penultimate position had significantly worse binding to H2-Dd molecule than peptides with T at the penultimate position. Keeping the anchor residue constant, and comparing the penultimate position between T and V, T always bound with greater affinity to H2-Dd: s.p.TI10 > s.p.VI10, s.p.TV10> s.p.VV10, and s.p.TT10 > s.p.VT10 (Fig 5b).
This binding behavior of peptides with substitutions at the penultimate position from T322 to V322 in the VV gp120 protein helps explain the inability of the VV mutation to improve binding affinity. While the Val at anchor position 10 should increase binding, the Val at position 9 interferes and apparently counters this beneficial effect, thus resulting in similar net binding affinity for s.p.TT10 and s.p.VV10. We also asked how the 9-mers would bind if the proteins were cleaved at the cathepsin S cleavage site in the TT wild type protein, or in the equivalent place (albeit without a cathepsin cleavage site) in the VV protein. In fact, both 9-mers bind equally well and with about 2 logs higher affinity then the corresponding 10-mers (Fig 4c). Therefore, if the TT site were cleaved by cathepsin S and the VV not, then the TT protein would be expected to be more immunogenic, not less, unless that first cleavage led to rapid further degradation of the 9-mer in the endosome. For this reason, the data are not consistent with 9-mer’s binding to explain the immunogenicity, but rather with the hypothesis that the VV mutation protects the site from cleavage and further degradation in the endosome and allows the VV10 peptide to be presented.
X-ray structures provide a basis for differential effects of peptide substitutions IGPGRAFYVI and IGPGRAFYTI peptides in the H2-Dd MHC-I molecule
In order to understand contributions of the different peptides to the level of MHC-I binding, we determined three new peptide/H2-Dd X-ray structures (Table 1, Fig S1) and examined them along with previously reported ones (31, 45). As illustrated in Fig 6, a comparison of the peptide conformation of the three H2-Dd-bound 10-mers, s.p.VI10, s.p.TI10, and P18-I10 (the IIIB strain homolog of s.p.TI10 based in the MN strain) reveals essentially the same backbone configuration (Fig 6a,c) and the same anchors at positions 2 and 3 (GP), 5 (R), and 10 (I) (Fig 6b,d). One apparent difference is the orientation of the sidechain of position 7 Phe of s.p.TI10, which is stacked with the position 8 Tyr, while in the other two structures the position 7 and 8 residues are splayed, with position 7 Phe oriented toward the α1 helix for s.p.VI10 and P18-I10. Careful inspection of the crystal contacts in the s.p.TI10 structure, however, indicates that this is a region that interacts with another molecule in the crystal, and that this most likely represents the result of crystal packing rather than the natural interaction of the position 7 Phe sidechain with solvent. However, a logical basis for the apparent differences in binding affinity of different 9-mer and 10-mer peptides to H2-Dd is suggested by consideration of the size and hydrophobicity of the C terminal residue. Those peptides with I at the carboxyl terminus (10-mers s.p.VI10, s.p.TI10, and s.p.18I10) bury more surface area than peptides with C-terminal V, and thus bind less well. Additionally, we may hypothesize that V at position 9 in the context of s.p.VV10 thus generates a steric effect and exposes a hydrophobic side chain to solvent, making s.p.VV10 less avid for H2-Dd than s.p.TV10.
Figure 6.
Comparison of superposed 10-mers and 9-mers of different peptides in complex with H2-Dd. X-ray structures of (a, b) s.p.VI10, s.p.TI10, and P18-I10 and of (c,d) s.p.T9, s.p.V9, and s.p.A9 are illustrated. (P18-I10 is the HIV-IIIB strain homolog of s.p. TI10 which is based on the MN strain.) s.p.VI10, s.p.T9, and s.p.T9 (PDB codes: 5KD4, 5T7G, and 5KD7) are described in Materials and methods, and Table 1. s.p.TI-10, P18-I10, and s.p.A-9 (3E6H, 3ECB, and 3E6F, respectively) have been reported previously (31, 45). For panels a and b, s.p.VI10, s.p.TI10 and P18-I10 were superposed, but only H2-Dd cartoon of P18-I10 is shown. Panels a and c show the peptides as viewed from the TCR perspective and b and d represent an approximately 90° rotation.
To explore the structural basis of 9-mer peptides binding to H2-Dd, we examined the structures of s.p.T9, s.p.V9 and the previously determined PA-9 (IGPGRAFYA (3E6F, ref (45)) (Fig 6c and d). For the three 9-mer peptides, there is essentially no difference in the backbone conformation (rmsd of Cα atoms < 0.2 Å), or in the orientation of the amino acid side chains. Taken together, the binding and structural studies indicate that there are no major differences in binding of the different 10-mers that have a position 2,3 (GP), position 5 (R), and position 10 (I) anchor motif, nor are there major differences among the 9-mers with position 9 T, V or A. Quantitative differences in binding seem to be related to the degree of hydrophobicity (and buried surface area) of the C terminal anchor and the result of energetic costs of exposing penultimate residues to aqueous solvent. Furthermore, these results help to explain why the mutation from T322T323 to V322V323 did not enhance peptide-binding affinity. Overall, by excluding the hypothesized increased affinity of the mutant VV peptide to the MHC molecule as one explanation for the increased immunogenicity of the VV protein, we are left with the conclusion that the main mechanism by which the VV protein is more immunogenic when administered in a way that requires endosomal cross-presentation is that it eliminates the cathepsin S cleavage site and thus protects the epitope, and the molecule as a whole, from cathepsin cleavage and possible further endosomal degradation from the C-terminus.
Discussion
The most common view of CD8+ T cell responses to viral infections is that viral proteins are synthesized endogenously and thus processed through the endogenous class I MHC processing pathway involving proteasomes and TAP transport into the endoplasmic reticulum, where the peptides are loaded onto class I MHC molecules. That is certainly the case when viruses infect professional antigen-presenting cells such as DCs, or when the infected cell is the target of CTL killing. However, priming of CD8+ T cells in the case of viral infections of cells that are not professional antigen-presenting cells often involves cross-presentation, in which antigen expressed in infected cells is taken up by DCs and presented to T cells. This may be a major mechanism especially relevant in HIV infection because the predominant cells infected are CD4+ T cells, which are not professional antigen-presenting cells. The cross-presentation mechanism usually involves uptake initially into endosomes where whole protein antigens can be processed into fragments (7). This has been mimicked by immunizing with whole proteins in adjuvants that allow endosomal uptake but then transfer to the endogenous pathway for class I MHC presentation, or could be loaded onto recycling class I MHC molecules in the endosome (47). Thus, during cross-presentation, antigens are exposed to endosomal proteases to which they would not be exposed if they had been synthesized in the cytosol and processed by proteasomes initially. We hypothesized that such endosomal processing may provide another opportunity for viruses to escape immune recognition, by promoting destruction of key antigens by endosomal proteases such as cathepsins. Cathepsin S in endosomes has been shown to contribute to the processing of antigens during cross-presentation (9). To address this hypothesis, we took advantage of the fact that an immunodominant determinant IGPGRAFYTT within the V3 loop of HIV-1MN gp120 contains a cathepsin S cleavage site TT. Thus, we asked whether that cleavage site would open up the molecule for endosomal destruction of the epitope, preventing cross-presentation of this dominant epitope. The epitope had been previously mapped as immunodominant when animals were immunized with a recombinant vaccinia virus expressing the HIV envelope (19, 20). As vaccinia synthesizes the protein in the cytosol, we knew that the epitope was active and even immunodominant when presented in the cytosol. Here, we contrasted that with immunization with the whole recombinant protein in an adjuvant, CAF09, that promotes cross-presentation of whole proteins (26). Indeed, we found that CD8+ T cells specific for this epitope were not induced effectively when the whole protein in adjuvant was injected. However, when the epitope was mutated to change the TT to VV, making it resistant to cathepsin S (as shown in Fig 1b), now the epitope was quite immunogenic even with the whole protein as vaccine. This result supported our hypothesis that cathepsin S in endosomes could cause destruction of epitopes during cross-presentation that were not destroyed during classical endogenous processing and presentation, and that this situation could provide the virus with another means of immune escape.
Mutation of the epitope from IGPGRAFYTT to IGPGRAFYVV clearly promoted the immune response not only to the modified VV sequence but also to original wild type (WT) TT sequence. Nevertheless, several explanations remained possible. The C-terminal valine is a stronger anchor residue than threonine for peptides binding to H2-Dd, as previously shown (43, 44). Also, as we hypothesized, the VV mutation could prevent endosomal destruction of the epitope, promoting induction of immunity. These explanations are not mutually exclusive, so both could be acting together. To test these, we first carried out binding studies of these peptides to H2-Dd using the RMA-S binding assay described in Materials and Methods. To our surprise, the VV peptide did not bind with higher affinity than the TT peptide (Fig 4b). To investigate the reason for this surprising result, we examined the binding of several related peptides (Fig 5). What was revealed by these studies was that the penultimate V was deleterious for binding. When the C-terminal residue was held constant as either V, T, or I, the peptide with a V at the penultimate position was always a poorer binder than the one with a T at the penultimate position. On the other hand, when the penultimate position was held constant, the C-terminal V or I resulted in higher affinity binding than the C-terminal T. Thus, the V at the penultimate position canceled out the beneficial effect of the V at the anchor residue in the conversion from TT to VV, and the net effect was no improvement in binding. This result, however, left by default only the remaining interpretation that the VV substitution promoted immunogenicity by preventing destruction during cross-presentation.
This binding study answered one question but raised another: why was the penultimate V deleterious for binding to H2-Dd, especially considering that this residue was not expected to interact directly with the MHC molecule? To address this new question, we prepared crystals of H2-Dd bound to several of the peptides and carried out X-ray crystallographic studies of the structures of these peptide-MHC complexes (Fig 6 and Fig S1). The backbone peptide structures of all the complexes were found to be quite comparable, and the penultimate V was indeed pointing up out of the MHC groove (Fig S1). The only structural explanations we have for the deleterious effect of V at this position is that it is a hydrophobic residue that produces adverse entropic effects when forced to be exposed to aqueous solvent, whereas the threonine at that position has a hydroxyl group that is hydrophilic, and that there are fewer opportunities for H-bonds.
Another question that arose was whether the 9-mer peptides with the C-terminal residue removed would bind as well, and indeed they do, although the differences in affinity of the various 10-mers indicates that the 10-mers are what is binding in those studies and not peptides degraded to 9-mers, which bind with higher but equal affinity. Further, the 9-mers with the C-terminal T or V are recognized by the same T cells, as determined by dextramer staining. Thus, if the 10-mer epitope were cleaved just to remove the terminal T, it should have been immunogenic just like the VV protein. However, the endosome contains many proteases, and we believe that once the TT bond is broken, other proteases will rapidly further destroy the epitope, making the TT protein poorly immunogenic compared to the VV protein which does not get cleaved at that site. Once the site is protected in the endosome and the protein undergoes cross-presentation, the T cells induced by the VV protein clearly cross-react well with the TT version of the epitope, as seen by the recognition of peptides s.p.78 and s.p.79 in Fig 2. Thus, it is the preservation of the epitope from endosomal destruction that determines the difference in immunogenicity.
In conclusion, these studies have revealed a novel mechanism by which a virus like HIV can evade the immune system and avoid inducing an immune response when infecting cells that are not professional antigen-presenting cells, such as CD4+ T cells that are the main target of HIV infection. In such cases, to induce a primary immune response, the viral antigens must be cross-presented by professional antigen-presenting cells like DCs, and exposure of these antigens to endosomal proteases during such cross-presentation allows the virus a stealth mechanism to avoid induction of immunity by selecting for sequences containing cathepsin cleavage sites in critical epitopes. Recognition of this novel mechanism should allow it to be taken into account in designing viral immunogens for more effective AIDS vaccines or vaccines against other viruses that might share this evasion strategy. Furthermore, as rgp120MN was a key component of the RV144 vaccine that is the only phase III human AIDS vaccine trial to show efficacy (albeit a moderate 31% efficacy) (2), such modifications of rgp120 might make it more immunogenic and therefore a more effective vaccine candidate. In addition, an adjuvant like CAF09, that can promote induction of CD8+ T cells to a soluble whole protein antigen through cross-presentation, could also potentially improve the efficacy of the vaccine. Thus, this study provides at least two approaches for potential improvement on the encouraging success so far with the first efficacious human AIDS vaccine.
Supplementary Material
Acknowledgments
We thank Dr. David Venzon, NCI Biostatistics & Data Management Section, for help with statistical analyses of binding curves. We thank the Statens Serum Institute, Copenhagen, Denmark, for the gift of CAF09 adjuvant. Data were collected at SER-CAT (22-ID) beamline at the Advanced Photon Source (APS), Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. The views are those of the authors and not official views of the NIH or the US Department of Health and Human Services.
Footnotes
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This work was supported in part by NCI intramural funding Project NIA-C-04020 to JAB and by NIAID grant: R01 AI089378-01 to PWB, and by the Intramural research program of the NIAID, NIH, and by a grant #12-132230 to RB from the Danish Research Council.
References
- 1.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 3.Martrus G, Altfeld M. Immunological strategies to target HIV persistence. Curr Opin HIV AIDS. 2016;11:402–408. doi: 10.1097/COH.0000000000000289. [DOI] [PubMed] [Google Scholar]
- 4.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 5.Yu B, Fonseca DP, O’Rourke SM, Berman PW. Protease cleavage sites in HIV-1 gp120 recognized by antigen processing enzymes are conserved and located at receptor binding sites. J Virol. 2010;84:1513–1526. doi: 10.1128/JVI.01765-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 7.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh CS, deRoos P, Honey K, Beers C, Rudensky AY. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J Immunol. 2002;168:2618–2625. doi: 10.4049/jimmunol.168.6.2618. [DOI] [PubMed] [Google Scholar]
- 9.Hari A, Ganguly A, Mu L, Davis SP, Stenner MD, Lam R, Munro F, Namet I, Alghamdi E, Furstenhaupt T, Dong W, Detampel P, Shen LJ, Amrein MW, Yates RM, Shi Y. Redirecting soluble antigen for MHC class I cross-presentation during phagocytosis. Eur J Immunol. 2015;45:383–395. doi: 10.1002/eji.201445156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biniossek ML, Nagler DK, Becker-Pauly C, Schilling O. Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. J Proteome Res. 2011;10:5363–5373. doi: 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- 11.Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, De Sanctis GT, Ploegh HL, Chapman HA. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest. 1998;101:2351–2363. doi: 10.1172/JCI1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reise RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, De Sanctis GT, Ploegh HL, Chapman HA. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest. 1998;101:2351–2363. doi: 10.1172/JCI1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beers C, Burich A, Kleijmeer MJ, Griffith JM, Wong P, Rudensky AY. Cathepsin S controls MHC class II-mediated antigen presentation by epithelial cells in vivo. J Immunol. 2005;174:1205–1212. doi: 10.4049/jimmunol.174.3.1205. [DOI] [PubMed] [Google Scholar]
- 14.Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, Chang CH, Harndahl M, Weimershaus M, Gerstoft J, Akkad N, Klenerman P, Fugger L, Jones EY, McMichael AJ, Buus S, Schild H, van Endert P, Iversen AK. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol. 2009;10:636–646. doi: 10.1038/ni.1728. [DOI] [PubMed] [Google Scholar]
- 15.Berman PW, Matthews TJ, Riddle L, Champe M, Hobbs MR, Nakamura GR, Mercer J, Eastman DJ, Lucas C, Langlois AJ, et al. Neutralization of multiple laboratory and clinical isolates of human immunodeficiency virus type 1 (HIV-1) by antisera raised against gp120 from the MN isolate of HIV-1. J Virol. 1992;66:4464–4469. doi: 10.1128/jvi.66.7.4464-4469.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman PW. Development of bivalent rgp120 vaccines to prevent HIV type 1 infection. AIDS Res Hum Retroviruses. 1998;14(Suppl 3):S277–289. [PubMed] [Google Scholar]
- 17.Billeskov R, Wang Y, Solaymani-Mohammadi S, Frey B, Kulkarni S, Andersen P, Agger EM, Sui Y, Berzofsky JA. Low Antigen Dose in Adjuvant-Based Vaccination Selectively Induces CD4 T Cells with Enhanced Functional Avidity and Protective Efficacy. J Immunol. 2017;198:3934–3506. doi: 10.4049/jimmunol.1600965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi H, Cohen J, Hosmalin A, Cease KB, Houghten R, Cornette J, DeLisi C, Moss B, Germain RN, Berzofsky JA. An immunodominant epitope of the HIV gp160 envelope glycoprotein recognized by class I MHC molecule-restricted murine cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1988;85:3105–3109. doi: 10.1073/pnas.85.9.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi H, Merli S, Putney SD, Houghten R, Moss B, Germain RN, Berzofsky JA. A single amino acid interchange yields reciprocal CTL specificities for HIV gp160. Science. 1989;246:118–121. doi: 10.1126/science.2789433. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Nakagawa Y, Pendleton CD, Houghten RA, Yokomuro K, Germain RN, Berzofsky JA. Induction of broadly cross-reactive cytotoxic T cells recognizing an HIV-1 envelope determinant. Science. 1992;255:333–336. doi: 10.1126/science.1372448. [DOI] [PubMed] [Google Scholar]
- 21.Smith DH, Winters-Digiacinto P, Mitiku M, O’Rourke S, Sinangil F, Wrin T, Montefiori DC, Berman PW. Comparative immunogenicity of HIV-1 clade C envelope proteins for prime/boost studies. PLoS One. 2010;5:e12076. doi: 10.1371/journal.pone.0012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otten GR, Bikoff E, Ribaudo RK, Kozlowski S, Margulies DH, Germain RN. Peptide and β2-microglobulin regulation of cell surface MHC class I conformation and expression. J Immunol. 1992;148:3723–3732. [PubMed] [Google Scholar]
- 23.Huang Y, Terabe M, Pendleton CD, Khursigara DS, Bera TK, Pastan I, Berzofsky JA. Identification and enhancement of HLA-A2.1-restricted CTL epitopes in a new human cancer antigen-POTE. PLOS One. 2013;8:e64365. doi: 10.1371/journal.pone.0064365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, Kroemer G. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7:271ps271. doi: 10.1126/scitranslmed.3010473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korsholm KS, Hansen J, Karlsen K, Filskov J, Mikkelsen M, Lindenstrom T, Schmidt ST, Andersen P, Christensen D. Induction of CD8+ T-cell responses against subunit antigens by the novel cationic liposomal CAF09 adjuvant. Vaccine. 2014;32:3927–3935. doi: 10.1016/j.vaccine.2014.05.050. [DOI] [PubMed] [Google Scholar]
- 27.Sui Y, Hogg A, Wang Y, Frey B, Yu H, Xia Z, Venzon D, McKinnon K, Smedley J, Gathuka M, Klinman D, Keele BF, Langermann S, Liu L, Franchini G, Berzofsky JA. Vaccine-induced myeloid cell population dampens protective immunity to SIV. J Clin Invest. 2014;124:2538–2549. doi: 10.1172/JCI73518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamoreaux L, Roederer M, Koup R. Intracellular cytokine optimization and standard operating procedure. Nat Protoc. 2006;1:1507–1516. doi: 10.1038/nprot.2006.268. [DOI] [PubMed] [Google Scholar]
- 29.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 30.Tormo J, Natarajan K, Margulies DH, Mariuzza RA. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature. 1999;402:623–631. doi: 10.1038/45170. [DOI] [PubMed] [Google Scholar]
- 31.Wang R, Natarajan K, Margulies DH. Structural basis of the CD8 alpha beta/MHC class I interaction: focused recognition orients CD8 beta to a T cell proximal position. J Immunol. 2009;183:2554–2564. doi: 10.4049/jimmunol.0901276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Driessen C, Bryant RA, Lennon-Dumenil AM, Villadangos JA, Bryant PW, Shi GP, Chapman HA, Ploegh HL. Cathepsin S controls the trafficking and maturation of MHC class II molecules in dendritic cells. The Journal of cell biology. 1999;147:775–790. doi: 10.1083/jcb.147.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 38.Belyakov IM, Wyatt LS, Ahlers JD, Earl P, Pendleton CD, Kelsall BL, Strober W, Moss B, Berzofsky JA. Induction of a mucosal cytotoxic T-lymphocyte response by intrarectal immunization with a replication-deficient recombinant vaccinia virus expressing human immunodeficiency virus 89.6 envelope protein. Journal of virology. 1998;72:8264–8272. doi: 10.1128/jvi.72.10.8264-8272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, Berglund P, Gibbs J, Bennink JR, Yewdell JW. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. doi: 10.1126/science.1096378. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Hong H, Li D, Ma S, Di Y, Stoten A, Haig N, Di Gleria K, Yu Z, Xu XN, McMichael A, Jiang S. Comparing pooled peptides with intact protein for accessing cross-presentation pathways for protective CD8+ and CD4+ T cells. J Biol Chem. 2009;284:9184–9191. doi: 10.1074/jbc.M809456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matheoud D, Perie L, Hoeffel G, Vimeux L, Parent I, Maranon C, Bourdoncle P, Renia L, Prevost-Blondel A, Lucas B, Feuillet V, Hosmalin A. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood. 2010;115:4412–4420. doi: 10.1182/blood-2009-11-255935. [DOI] [PubMed] [Google Scholar]
- 42.Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity. 2004;21:155–165. doi: 10.1016/j.immuni.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Corr M, Boyd LF, Padlan EA, Margulies DH. H-2Dd exploits a four residue peptide binding motif. J Exp Med. 1993;178:1877–1892. doi: 10.1084/jem.178.6.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Natarajan K, Malchiodi EL, Margulies DH, Mariuzza RA. Three-dimensional structure of H-2Dd complexed with an immunodominant peptide from human immunodeficiency virus envelope glycoprotein 120. J Mol Biol. 1998;283:179–191. doi: 10.1006/jmbi.1998.2091. [DOI] [PubMed] [Google Scholar]
- 45.Honda M, Wang R, Kong WP, Kanekiyo M, Akahata W, Xu L, Matsuo K, Natarajan K, Robinson H, Asher TE, Price DA, Douek DC, Margulies DH, Nabel GJ. Different vaccine vectors delivering the same antigen elicit CD8+ T cell responses with distinct clonotype and epitope specificity. J Immunol. 2009;183:2425–2434. doi: 10.4049/jimmunol.0900581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi H, Houghten R, Putney SD, Margulies DH, Moss B, Germain RN, Berzofsky JA. Structural requirements for class-I MHC molecule-mediated antigen presentation and cytotoxic T-cell recognition of an immunodominant determinant of the HIV envelope protein. J Exp Med. 1989;170:2023–2035. doi: 10.1084/jem.170.6.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gromme M, Uytdehaag FG, Janssen H, Calafat J, van Binnendijk RS, Kenter MJ, Tulp A, Verwoerd D, Neefjes J. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci U S A. 1999;96:10326–10331. doi: 10.1073/pnas.96.18.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.