

Abstract
Despite decades of effort, little progress has been made to improve the treatment of cancer metastases. To leverage the central role of the mechanoenvironment in cancer metastasis, we present a mechanoresponsive cell system (MRCS) to selectively identify and treat cancer metastases by targeting the specific biophysical cues in the tumor niche in vivo. Our MRCS uses mechanosensitive promoter-driven mesenchymal stem cell (MSC)-based vectors, which selectively home to and target cancer metastases in response to specific mechanical cues to deliver therapeutics to effectively kill cancer cells, as demonstrated in a metastatic breast cancer mouse model. Our data suggest a strong correlation between collagen cross-linking and increased tissue stiffness at the metastatic sites, where our MRCS is specifically activated by the specific cancer-associated mechano-cues. MRCS has markedly reduced deleterious effects compared to MSCs constitutively expressing therapeutics. MRCS indicates that biophysical cues, specifically matrix stiffness, are appealing targets for cancer treatment due to their long persistence in the body (measured in years), making them refractory to the development of resistance to treatment. Our MRCS can serve as a platform for future diagnostics and therapies targeting aberrant tissue stiffness in conditions such as cancer and fibrotic diseases, and it should help to elucidate mechanobiology and reveal what cells “feel” in the microenvironment in vivo.
INTRODUCTION
Cancer metastases account for more than 90% of cancer deaths. However, there are currently no effective and selective treatments that directly target metastatic cancer. In particular, about 20 to 30% of women worldwide will develop invasive breast cancer during their lifetime, leading to more than 500,000 deaths a year due to metastasis from the breast to other organs (1,2), with a median survival of only 2 to 3 years (3,4). Surgical resection of widespread metastases is generally not feasible, whereas various classes of chemotherapeutic drugs are ineffective at treating disseminated cancer and often have severe side effects. Current therapy for metastatic breast cancer therefore focuses on prolonging survival and providing palliative care (1, 4–6). In addition, tumors can develop resistance to many existing drugs through various mechanisms that are, in part, due to cancer heterogeneity (1, 7).
Cells constantly interact with their surrounding niche, which includes an array of complex biochemical and biophysical signals from the surrounding extracellular matrix (ECM). Although not appreciated historically, it has recently become evident that the physical and mechanical properties of cellular microenvironments (the so-called “mechano-niche”) regulate essential cell functions (8–12). Important roles for matrix stiffness in driving breast cancer metastasis have been elucidated (13,14). Specifically, increased matrix stiffness, which is primarily driven by increased collagen deposition and cross-linking by lysyl oxidase (LOX) proteins, promotes breast cancer migration, invasion, cell plasticity, and eventual metastasis, primarily through regulation of integrin signaling (15). LOX accumulation spatially correlates with the presence of metastases in both mouse models of metastasis and human patients (13,16). In mouse models of breast cancer metastasis, secretion of LOX by the primary breast tumor stimulates collagen cross-linking in discrete areas of the lung that promote formation of metastases (16–20). Deposition of LOX at the metastatic niche correlates with both collagen linearization and formation of collagen-collagen covalent bonds in the lung parenchyma, both of which markedly increase matrix stiffness (15). Therefore, we reasoned that the distinctive mechanical properties of the metastatic niche might offer a viable target for the development of diagnostics and therapeutics specifically targeting metastases.
We hypothesized that a cell-based system, specifically mesenchymal stem cells (MSCs), can be used for such an approach to generate a mechanoresponsive cell system (MRCS) that responds specifically to mechanoenvironmental cues to target breast cancer metastases (fig. S1). MSCs are multipotent cells that can be derived from multiple adult tissues, including bone marrow and fat (21,22). MSCs are the basis for the first approved stem cell treatment in humans outside of bone marrow transplant (Prochymal, Osiris Therapeutics) and for more than 400 ongoing trials listed on ClinicalTrials.gov with widely demonstrated safety (23, 24). Systemically infused MSCs preferentially home to and integrate with tumors in vivo, including both primary breast tumors and lung metastases (25, 26). Mounting evidence now suggests that MSCs have leukocyte-like, active homing mechanisms for tumor tropism involving a variety of adhesion molecules and tumor-derived cytokines, chemokines, and growth factors (27). This selective and active homing ability makes MSCs an appealing vector for localized delivery of therapeutics in cancer treatment (25, 26).
Tissue mechanical properties regulate MSC fate: Tissue and matrix stiffness is sufficient to drive expression of genes involved in MSC differentiation (28–30). Specifically, soft matrices, similar to the brain (Young’s modulus of less than 1 kPa), direct MSCs into a neurogenic lineage, whereas stiffer matrices (5 to 75 kPa), similar to muscle and bone, direct them into myogenic and osteogenic lineages through integrin- and focal adhesion-dependent mechanisms (28). The range of stiffness to which MSCs respond encompasses those found in normal breast and lung tissues (less than 1 kPa), as well as invasive cancers and metastases (10- to 15-fold higher stiffness) (31). MSC differentiation is inherently a transcriptional program, which allows us to use promoters regulating genes involved in MSC mechanotransduction/differentiation cascades to drive expression of downstream reporters or therapeutics (28).
In particular, Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) have previously been reported as sensors and mediators of mechanical cues via, for instance, the cytoskeleton and Rho guanosine triphosphatase (32, 33). On soft substrates in vitro (<1 kPa), YAP remains inactivated in the cytoplasm, but on stiff substrates in vitro (>10 kPa), YAP localizes to the nucleus and becomes activated as a transcriptional factor (32–34). YAP/TAZ have greater nuclear accumulation in samples from breast cancer patients associated with enhanced desmoplasia (35). YAP/TAZ have also been reported to be key upstream factors that regulate lineage-specific transcription factors (including RUNX2, an osteogenic marker) and drive MSC differentiation, including osteogenesis (36).
In light of the tight correlation between tissue stiffness, breast cancer metastasis, and mechanotransduction-mediated MSC differentiation, we have developed an MRCS to directly target the mechanoenvironmental cues of breast cancer metastases for specific delivery of an antitumor agent, cytosine deaminase (CD), which locally activates the prodrug 5-fluorocytosine (5-FC) to kill cancer (fig. S1). Our study demonstrates that the MRCS, which is engineered to be inducible by biophysical and mechanical cues, specifically responds to matrix stiffness in vitro and can selectively target and kill cancer metastases with minimal side effects in vivo.
RESULTS
MRCS and in vitro validation
We have established an MRCS using a YAP/TAZ stiffness-sensing promoter. When activated, YAP/TAZ can drive the expression of downstream reporters such as enhanced green fluorescent protein (eGFP) (MRCS-eGFP) for in vitro imaging, firefly luciferase (MRCS-Luc) for later in vivo imaging, or antitumor agents (MRCS-CD) as cancer treatment (Fig. 1A and fig. S2). In effect, YAP/TAZ serves as an on/off switch for the MRCS gene expression triggered by the substrate stiffness in our study. For this text, cells that constitutively express a gene, such as CD-MSC or Luc-MSC, will have the gene being expressed first in the nomenclature. For MRCS cells such as MRCS-CD or MRCS-eGFP, MRCS will come first in the name to indicate that it is the engineered stiffness-sensing promoter system that is driving the expression of the downstream gene.
Fig. 1. MRCS in vitro validation.
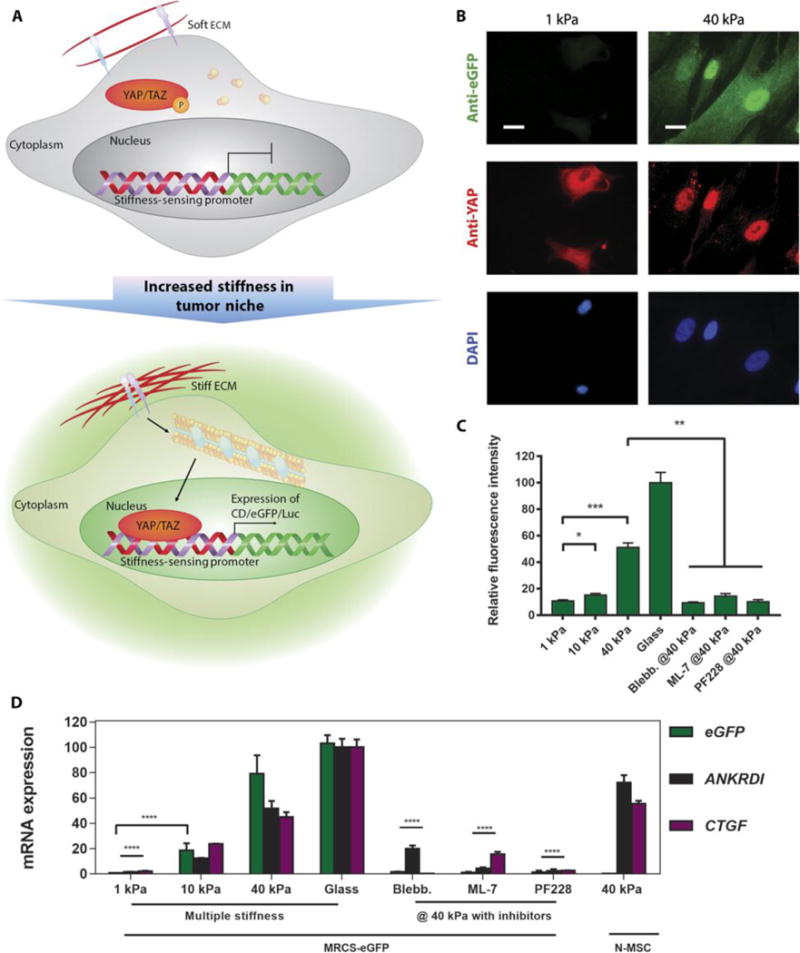
(A) Schematic of a proposed mechanism of how MRCS works. When the stiffness of ECM increases, YAP/TAZ are activated and localize to the nucleus. Then, YAP/TAZ will bind to the synthetic stiffness-sensing promoter in MRCS and drive the expression of downstream reporters (such as eGFP and Luc) and/or therapeutics. Note: This schematic is simplified to clarify the major components in MRCS mechanism. (B) Representative images of MRCS-eGFP plated on soft (~1 kPa) and firm (~40 kPa) polyacrylamide gels. eGFP (stained with anti-eGFP; green) was turned on in response to higher stiffness. YAP (stained with anti-YAP; red) localization is also regulated by stiffness, such that it concentrates in the nuclei on stiffer substrates. 4′,6-diamidino-2-phenylindole (DAPI) (blue; nuclear counterstain) is displayed. Scale bars, 25 μm. (C) Quantification of fluorescence intensity of eGFP (stained with antibody) from MRCS-eGFP seeded on substrates with different stiffness or on firm (~40 kPa) substrates treated with 10 μM ML-7 (MLCK inhibitor) or 20 μM PF228 (FAK inhibitor). Blebb., blebbistatin. Data aremeans ± SEM. (D) RT-qPCR analysis of MRCS-eGFP on hydrogels. Expression of eGFP (green) and YAP/TAZ downstream factors (CTGF, purple; ANKRDI, black) was increased on stiffsubstrate and was down-regulated on soft substrate or with mechanosensing inhibitors, showing that MRCS is stiffness-specific. Quadruplicate samples were used for the analysis. Data are means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To validate the selective activation of our MRCS in response to stiffness, we seeded MRCS-eGFP on tunable polyacrylamide hydrogels with various stiffness (~1, ~10, and ~40 kPa) (28, 32). As expected, on soft hydrogel (~1 kPa), YAP remained in the cytoplasm, and no eGFP signal could be detected (Fig. 1B and fig. S3A), whereas on stiffer hydrogels (>10 kPa), YAP localized to the nuclei and eGFP was expressed, typically within 24 to 48 hours after cell seeding (Fig. 1B and fig. S3, B and C). As a control, MRCS-eGFP plated on glass (the highest stiffness used) showed strong activation of YAP/TAZ and eGFP expression (fig. S3D). MRCS-eGFP treated with blebbistatin, an inhibitor of mechanotransduction, which impedes signaling downstream of matrix stiffness and integrin activation (28, 32), showed no eGFP expression, and YAP remained in the cytoplasm, even on stiff substrates (fig. S3E). Two other mechanotransduction inhibitors, PF228 and ML-7, a focal adhesion kinase (FAK) inhibitor and a myosin light-chain kinase (MLCK) inhibitor, respectively, similarly deactivated YAP and downstream eGFP expression (fig. S3, F and G). A more comprehensive view of the cells in Fig. 1B can be found in fig. S4. Quantification of the stiffness-mediated eGFP expression of MRCS confirms that the intensity of reporter expression correlates positively with the substrate stiffness, such that stiffer hydrogel resulted in stronger eGFP signal, with attenuated expression in the presence of mechanotransduction inhibitors (Fig. 1C). This set of data demonstrates that YAP activation in response to altered stiffness is MLCK/FAK-dependent. We used reverse transcription quantitative polymerase chain reaction (RT-qPCR) to further characterize the expression of eGFP mRNA and two additional genes (CTGF and ANKRDI) that are transcriptionally regulated by YAP/TAZ. Consistent with the imaging data, expression of eGFP, CTGF, and ANKRDI was specifically activated on stiffer hydrogels (Fig. 1D). It is interesting to note the differences in expression of eGFP versus the other YAP-induced markers, which are likely due to different sensitivities of YAP binding to exogenous and endogenous promoters, because we are using a synthetic promoter for eGFP. We similarly prepared and characterized MRCS engineered to produce firefly luciferase (MRCS-Luc) (fig. S5). Collectively, these data indicate that our MRCS is stiffness-specific and can respond to a range of matrix stiffness to drive downstream gene expression.
MRCS-CD killing breast cancer cells in response to matrix stiffness in vitro
To use MRCS to locally treat breast cancer metastasis in the lung, we engineered the cells to express CD instead of a reporter gene (fig. S2). CD is a prodrug convertase that converts the inactive prodrug 5-FC to the active drug 5-fluorouracil (5-FU) (37). This leads to localized tumor killing via the bystander effect in which the apoptotic MRCS locally releases CD (fig. S1) (38). This promising technique is currently being used in clinical trials, for example, with 5-FU delivery by neural stem cells for treatment of glioblastoma (38). To validate the effectiveness of this prodrug system, we first confirmed that MSCs engineered to constitutively express CD (abbreviated as CD-MSC) are able to sufficiently convert 5-FC to kill MDA-MB-231 breast cancer cells in vitro (fig. S6). We next constructed MRCS-CD with the YAP/TAZ promoter to drive the expression of CD in response to matrix stiffness. To validate the stiffness-specific regulation of CD expression and conversion of 5-FC, we seeded MRCS-CD on polyacrylamide hydrogels with different stiffness. On soft hydrogel (~1 kPa), a minimal amount of CD was expressed (fig. S7A), but on stiffer hydrogels and glass (>10 kPa), CD expression was turned on (fig. S7, B to D). This expression pattern also correlated well with the localization of YAP. In the presence of mechanotransduction inhibitors, CD expression was turned off even on stiff hydrogel (~40 kPa) (fg. S7, E to G). Quantification of stiffness-dependent CD expression was also performed and showed increased CD expressed from MRCS on stiffer substrates (Fig. 2A). Additionally, we calculated the proportion of MRCS activated by substrates of varied stiffness from fluorescent signals. On soft substrate (~1 kPa), only about 2% of MRCS was activated, compared to 13% on ~10 kPa, 56% on ~40 kPa substrate, and 100% on glass.
Fig. 2. MRCS-CD activation dependent on substrate stiffness in vitro.
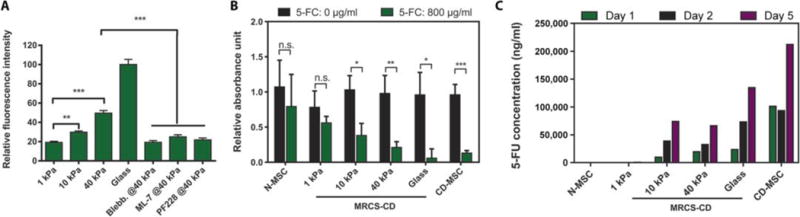
(A) Quantification of fluorescent signals of CD shows MRCS-CD responding to matrix stiffness in vitro. MRCS-CD were stained with antibody after plating on tunable polyacrylamide gels or glass as indicated, or treated with 50 μM blebbistatin, 10 μM ML-7 (MLCK inhibitors), or 20 μM PF228 (FAK inhibitor). The fluorescent signal of CD was analyzed, and the relative fluorescence intensity is shown. Data are means ± SEM. Triplicate samples were used for the analysis. (B) MRCS-CD kill cancer cells in response to matrix stiffness and 5-FC in vitro. MRCS-CD were cocultured with MDA-MB-231 breast cancer cells (231: MRCS = 2:1) with (800 μg/ml; green) or without (black) 5-FC on substrates with different stiffness. Total cell proliferation (XTT assay) is displayed. The data were normalized to breast cancer only (231: MRCS = 1:0) with or without 5-FC on each stiffness. Triplicate samples were used for the analysis. Data are means ± SD. n.s., not significant. *P <0.05, **P <0.01, and ***P <0.001. (C) Conversion of 5-FC to 5-FU by MRCS-CD in response to matrix stiffness in vitro. MRCS-CD were seeded on substrates with different stiffness, with 5-FC (800 μg/ml) in growth medium for 1,2, or 5 days. The concentration of 5-FU in the conditioned medium was detected by LC-MS/MS.
To test whether MRCS-CD could kill cancer specifically on high-stiffness substrates, we cocultured MRCS-CD with MDA-MB-231 breast cancer cells on polyacrylamide hydrogels with or without 5-FC and performed XTT assay to quantify total cell proliferation (Fig. 2B). On soft hydrogel (~1 kPa), there was no significant difference in cancer cell proliferation with or without the addition of 5-FC, consistent with the low expression of CD under these soft matrix conditions. When seeded on hydrogels with increased stiffness (~10 and ~40 kPa) or on glass, cell proliferation was significantly decreased in the presence of 5-FC in proportion to stiffness (P < 0.05), suggesting that CD was expressed and converted the prodrug to its active form to kill the cancer cells. CD-MSC without a stiffness-sensing promoter showed significantly reduced cell proliferation in the presence of 5-FC due to constitutive expression of CD (P < 0.001), whereas native MSCs (N-MSC) showed no difference in total cell proliferation, as expected, because they do not produce CD. These data demonstrate that MRCS-CD can selectively activate CD expression in response to matrix stiffness and convert 5-FC to kill adjacent cancer cells in vitro. To quantify the conversion of 5-FC to 5-FU, we seeded MRCS-CD on substrates with different stiffness with 5-FC in the growth medium for 1, 2, or 5 days, at which point the amount of 5-FU in the growth medium was detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Fig. 2C) (37). The data show that the conversion to 5-FU is stiffness-dependent, with no detectable conversion on soft substrate (~1 kPa) and increased conversion with increased stiffness (~10 and ~40 kPa). This also demonstrates that MRCS-CD can continue to express CD and convert 5-FC to 5-FU over a period of at least several days in vitro.
To further characterize the timing of cancer killing, we cocultured MRCS-CD with MDA-MB-231 using Transwell in the presence of 5-FC (fig. S8). XTT assay was performed to quantify cell proliferation for both cell types. The decreased proliferation of MDA-MB-231 expressing firefly luciferase plus red fluorescent protein (RFP) (Luc-RFP-231) indicates that MRCS-CD began to kill cancer cells (or attenuate cancer growth) within 2 days, with the MRCS-CD themselves dying after. The bystander effect of MRCS-CD also lasted after they were removed from the cancer cells, with a significant decrease in cancer proliferation measured on day 9 (P < 0.01) even when the MRCS-CD were removed as early as day 2 (fig. S9). This suggests that, even if MRCS only transiently interacts with tumor microenvironments and gets cleared after 2 days, cancer growth can be attenuated over a longer period of time.
MRCS homing to and specifically activated at the metastatic niche in vivo
As a model of breast cancer metastasis to the lung, we used an MDA-MB-231 xenotransplantation model in mice. We chose MDA-MB-231 cells because they secrete large amounts of LOX, which increases the cross-linking of collagen fibrils in the lung that is essential for metastasis (16). MDA-MB-231 cells were engineered to express reporters including eGFP (eGFP-231) or firefly luciferase plus RFP (Luc-RFP-231) and seeded via tail vein injection in immunocompromised mice to establish tumor foci in the lung (4 to 6 weeks after cancer infusion) (Fig. 3, A and B, animals on day 0) before MSC infusion. Here, we used two sets of immunocompromised mice: Foxn1nu (nude) and nonobese diabetic/severe combined immunodeficiency gamma (NSG). We primarily focused on the nude mouse system because it is partially immunocompromised and therefore more likely recapitulates the clinical setting than NSG. They also have better health condition than NSG mice after cancer seeding, allowing us to monitor the course of treatment for a longer period (39). On the other hand, NSG mice establish tumors more robustly and rapidly and therefore were also used when we examined MRCS tumor homing and correlation between collagen cross-linking, tumor cells, and MRCS activation in ex vivo immunofluorescence and second harmonic generation (SHG) experiments (39, 40).
Fig. 3. MRCS-CD killing cancer cells in vivo.
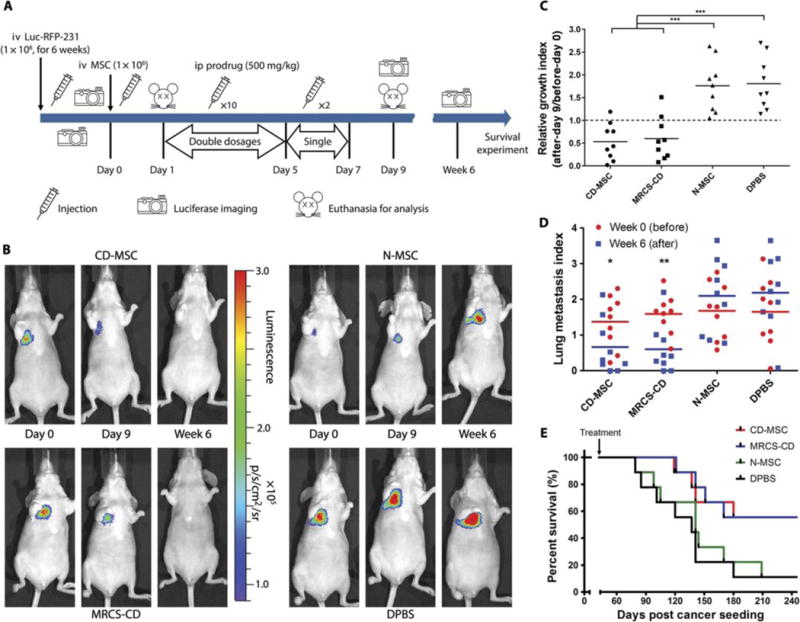
(A) Design and timeline of animal experiment to test MRCS-CD with 5-FC in vivo. iv, intravenous; ip, intraperitoneal. (B) Representative images of nude mice that received MRCS-CD treatments show that MRCS-CD decreased lung metastasis signals in vivo. Luciferase imaging was taken before (day 0, left) and after short-term 5-FC treatment (day 9, middle), as well as long-term 5-FC treatment (6 weeks, right). Quantification of luciferase signals in the lungs in vivo after (C) short-term and (D) long-termtreatments. (E) Mouse survival after MRCS-CD treatment. In (C), relative growth index = luciferase read on day 9 (after)/luciferase read on day 0 (before). In (D), lung metastasis index = log10 [(luciferase read of the tested mouse)/(luciferase read of average for tumor-free mice)]; the lung metastasis index of tumor-free mice = 0. The differences between “week 0” groups are not statistically significant. n = 9 for each group. *P <0.05, **P <0.01, and ***P <0.001. In (E), P = 0.0382, CD-MSC versus DPBS; P = 0.0429, MRCS-CD versus N-MSC; and P = 0.0211, MRCS-CD versus DPBS. Median survival (days): CD-MSC, 260; MRCS-CD, 260; N-MSC, 141; DPBS, 137.
MSCs home to both primary tumor and metastatic sites including breast cancers (26, 41, 42). Here, we first examined whether MSCs engineered to constitutively express firefly luciferase (Luc-MSC) are able to home to the lungs. We systemically infused Luc-MSC into mice hosting human eGFP-231 breast cancer cells in the lung and tumor-free controls. We found that Luc-MSC homed to and persisted in the lungs (fig. S10). Next, we investigated whether MRCS can home to and be specifically activated at the tumor sites using MRCS-Luc, which served as a surrogate for MRCS-CD and allowed us to readily track transplanted MRCS and monitor their activation using induced luciferase in vivo. We demonstrated that systemically infused MRCS-Luc homed to and were induced to express luciferase only in the tumor sites in the lung of eGFP-231 tumor-bearing mice (fig. S11, A and B). The observed luciferase signal, which reflects the collective functional outcome of MRCS homing and activation at tumor sites, persisted in tumor-bearing mice for up to 1 to 2 days (fig. S11B). Given that previous studies, including our own, have consistently demonstrated that systemically transplanted MSCs can persist in tumor sites for up to a week, we suspect that some residual MRCS might exist in tumors after 2 days following transplantation but become undetectable because of the limited sensitivity of in vivo luciferase imaging (42). Finally, we confirmed the in vivo homing and activation of MRCS-CD in Luc-RFP-231 tumor-bearing mice using ex vivo immunohistochemistry (IHC). We demonstrated that MRCS-CD colocalized with and were locally activated to express CD at cancer sites in lung sections of tumor-bearing (but not tumor-free) mice (fig. S11, C and D). Similar results were observed with the infusion of MRCS-eGFP (fig. S12). Collectively, these data suggest that MRCS selectively homes to and is specifically activated at the metastatic niche in vivo. This set of experiments also allowed us to identify time points at which MRCS persisted and was activated in tumors in vivo to guide the schedule of treatment (5-FC).
MRCS-CD killing cancer specifically and with minimal side effects in vivo
To evaluate the efficacy of MRCS-CD for treating breast cancer lung metastasis, mice seeded with Luc-RFP-231 cancer cells for 6 weeks were given MRCS-CD (on day 0), followed by the prodrug 5-FC, and were monitored for therapeutic outcomes (Fig. 3A). MRCS-CD were administered 1 day before the start of prodrug treatment to allow time for colocalization with tumors in the lungs. 5-FC was given in multiple doses for 7 days, which is consistent with the typical MSC persistence period in the tumor (41, 42). The amount of cancer within the lungs was quantified by measuring the cancer luciferase signal using in vivo imaging (Fig. 3B). Compared to initial values before prodrug treatment, luciferase signals were decreased in mice treated with MRCS-CD and MSCs engineered to constitutively express CD (CD-MSC), both shortly after treatment (day 9) and at 6 weeks after treatment (Fig. 3, C and D). N-MSC and Dulbecco’s phosphate-buffered saline (DPBS) control groups failed to decrease lung metastasis signals and showed increase in cancer mass over time as cancer continued to grow, as expected. Cancer signals after prodrug treatment (day 9) were normalized to cancer signals before treatment (day 0) for each mouse, which quantitatively demonstrated that CD-MSC and MRCS-CD significantly decreased the amount of cancer compared to N-MSC and DPBS groups (P < 0.001; Fig. 3C). Long-term (6 weeks after treatment) CD-MSC- and MRCS-CD-treated groups maintained a lower amount of lung metastasis compared to the day 0 baseline values (P <0.05), whereas N-MSC and DPBS groups saw an overall increase in cancer signals over time (Fig. 3D). Survival outcomes were also significantly improved by CD-MSC and MRCS-CD treatments compared to N-MSC and DPBS groups (P < 0.05; Fig. 3E). Note that, without 5-FC injection, MRCS-CD could not attenuate cancer growth in vivo (fig. S13). Note that, starting from day 120 (week 18), the survival rate of the MRCS-treated group started to decline (Fig. 3E), suggesting that, in some animals, lung tumors were decreased rather than totally cleared out by a single MRCS treatment. This demonstrates the potential need for repeated cell infusion together with prodrug administration (37).
Because intravenous delivery of MSCs, used in most clinical trials, results in initial entrapment of large numbers of MSCs in the pulmonary vasculature (43), localized activation of a prodrug, rather than constitutively expressing a drug, at only the metastatic niche is desirable to reduce off-target toxicity in the pulmonary and other organ systems. Although CD-MSC and MRCS-CD had similar treatment outcomes in terms of efficacy, constitutively expressing CD would convert systemically infused 5-FC indiscriminately in tumor-bearing and tumor-free tissue alike. MRCS-CD, however, would only express CD to activate 5-FC conversion at sites of increased stiffness, as found in tumor sites, and therefore have less damaging systemic side effects. To examine the side effects of MRCS-CD and compare it to CD-MSC, we used immunostaining of annexin V and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay (Fig. 4A). Staining for annexin V to measure apoptosis showed the specific activation of MRCS-CD at tumorsites, whereas no comparable annexin V signal could be seen on tumor-free tissue. CD-MSC-treated group stained positive for annexin V nonspecifically, indicating extensive tissue damage. Mice treated with N-MSC or DPBS stained positive for tumor but not for annexin V, indicating that native MSC or DPBS infusion does not cause cytotoxicity. TUNEL analysis for damaged DNA further confirmed higher lung tissue damage in the CD-MSC group than for any other group after treatment, including the MRCS-CD group (Fig. 4B). Specifically, MRCS-CD caused localized cell apoptosis only at the tumor sites with minimal lung tissue damage compared to constitutively CD-expressing control. In tumor-free mice, there was no significant increase in tissue damage after treatment with MRCS-CD, demonstrating specificity of activation only at tumor sites. Collectively, our data suggest that MRCS-CD kill cancer specifically with minimized side effects in vivo compared to MSC constitutively expressing therapeutics.
Fig. 4. MRCS-CD killing cancer cells in vivo with minimal side effects.
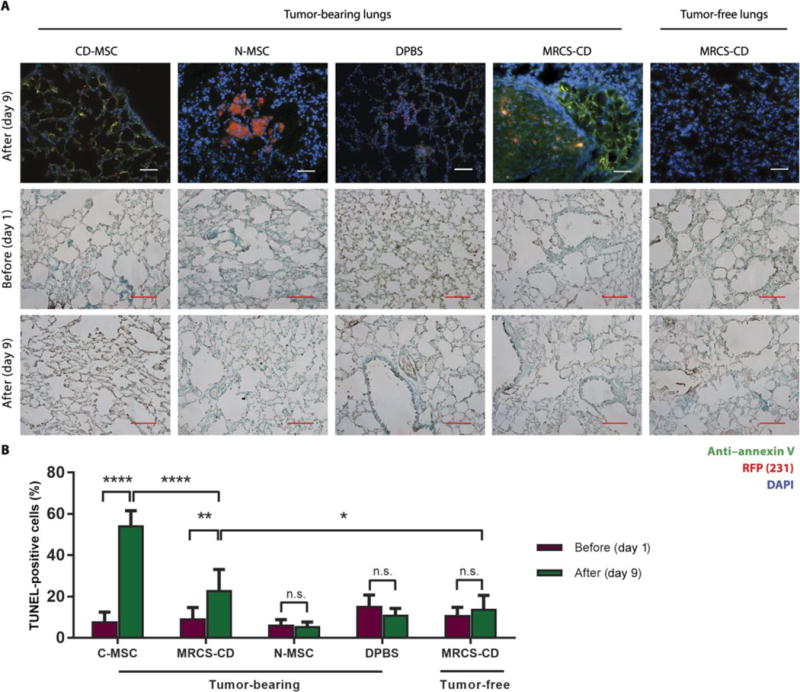
(A) Frozen sections of lungs of Luc-RFP-231 tumor-bearing and tumor-free nude micesacrificed 24 hours after CD-MSC, N-MSC, DPBS, or MRCS-CD infusion were stained with anti-annexin V (green) and DAPI (blue). RFP signal (red) indicates the presence of lung metastasis. Scale bars, 100 μm. Representative images of frozen section samples of tumor-bearing lungs and tumor-free lungs from nude mice treated with CD-MSC, MRCS-CD, N-MSC, or DPBS before and after 5-FC injections by TUNEL assays are shown. Horseradish peroxidase signals (brown) indicate damaged nuclei, and green signals are methyl green counterstain of normal nuclei. Scale bars, 100 μm. (B) Quantification of TUNEL assay data measuring lung tissue damage in vivo. Ten representative images were used per group for quantification. *P < 0.05, **P < 0.01, and ****P < 0.0001. Data are means ± SD.
Potential side effects to other tissues besides the lungs were also evaluated. In particular, we focused on damage to bone marrow, liver, and brain because these are other sites to which MSCs may home (22). Flow cytometry for bone marrow cells showed no significant increase in apoptosis or necrosis after treatment with MRCS-CD in nude mice (fig. S14A). There was also no significant change in the bone marrow cell population after treatment (fig. S14B), showing that the bone marrow was not depleted by the MRCS-CD treatment, unlike in conventional chemotherapy (44). There was also no observable damage in bone marrow (fig. S15), liver (fig. S16), or brain tissue (fig. S17) sections stained by hematoxylin and eosin. The data suggest that MRCS-CD do not induce nonspecific damage to other tissues.
The activation and tumor-killing functions of MRCS in vivo mediated by tumor mechano-cues
Secretion of LOX by primary breast tumor increases the linearization and cross-linking of collagen at the metastatic niche, resulting in increased matrix stiffness (18). Therefore, collagen linearization and cross-linking are robust surrogate markers of matrix stiffness. In addition, exogenous MSCs recruited to the metastatic lung assume an osteogenic differentiation profile not observed in the normal lung (25), although whether this is mediated by matrix stiffness is unclear. To mechanistically elucidate the activation and function of MRCS in the metastatic niche in vivo, we first validated the correlation between LOX expression and collagen expression in metastatic tissues. Up-regulation of LOX expression was observed in tumor-bearing lungs (day 0) compared to that in tumor-free lungs, and it correlated with the location of tumor cells (figs. S18 and S19, A and B). Tumor-bearing lungs (day 0) had higher collagen expression than tumor-free lungs by Masson’s trichrome staining (fig. S19, C and D), which is consistent with a previous study (13). To further explore the correlation between collagen crosslinking and the metastatic niche, we performed SHG imaging to colocalize collagen and lung metastases. SHG microscopy is a powerful modality for imaging collagen fibers (fibrillar network and linearization) in the tissue environment with high specificity (15). With SHG imaging, high collagen expression was observed to colocalize with cancer metastasis (figs. S19, A and B, and S20) and LOX expression (day 0, fig. S19, A and B). We also observed that the collagen networks are significantly (P < 0.0001) more linearized in cancer-specific regions of tumor-bearing lungs than in noncancer regions of tumor-bearing lungs and tumor-free lungs (day 0; Fig. 5, A to D, and fig. S20, A to F), which indicates that the metastatic niches in the lungs have a distinctive mechanomicroenvironment Tumor-bearing lungs (day 0) were also confirmed to have higher stiffness compared to tumor-free lungs by atomic force microscopy (AFM) (Fig. 5, E to H). Results from AFM microindentation of tissue sections showed that, besides having higher overall Young’s modulus (17.68 ± 25.63 kPa), tumor-bearing lungs are more heterogeneous in stiffness than tumor-free lungs that were less stiff (1.61 ± 3.97 kPa) (Fig. 5, G and H). Together, this set of data suggests a strong correlation between metastasis, LOX expression, increased collagen expression/cross-linking/linearization, and increased stiffness at the metastatic sites.
Fig. 5. Specific activation of MRCS in response to mechano-cues in the metastatic niche in vivo.
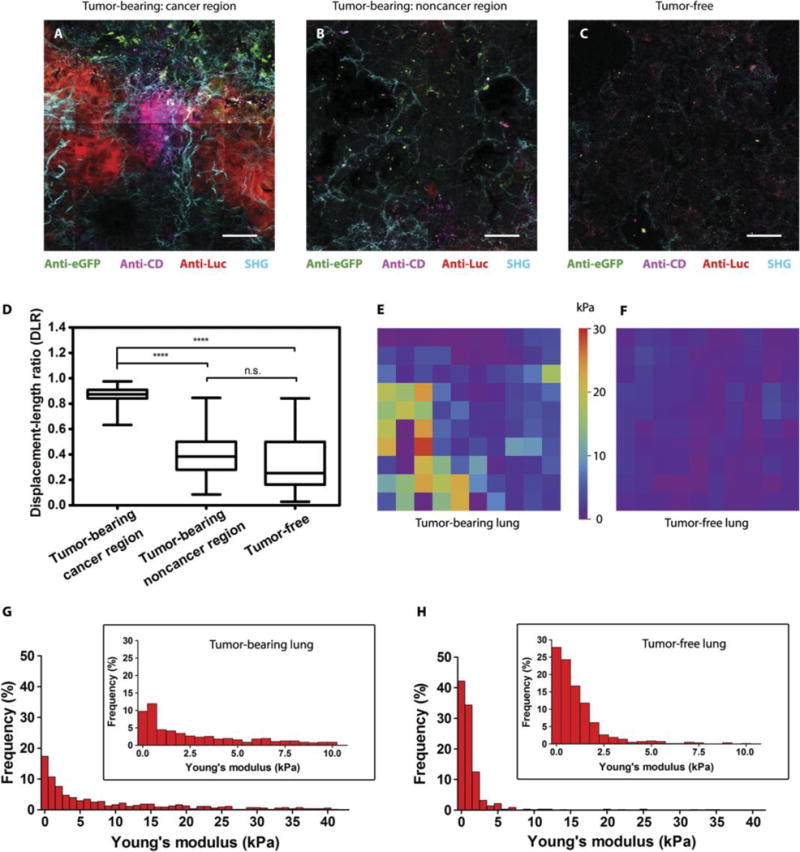
(A to C) Frozen sections of lungs of Luc-RFP-231 tumor-bearing NSG mice [cancer region in (A) and noncancer region in (B)] and tumor-free NSG mice (C) sacrificed 24 hours after infusion of MRCS-CD cotransfected with eGFP were stained with anti-Luc (red) to detect lung metastasis, anti-CD (magenta) for CD expressed by MRCS-CD, and anti-eGFP (green) for MRCS-CD tracking. SHG imaging of collagen networks (cyan) was also overlaid on IHC imaging. Scale bars, 50 μm. Multiple images were tiled into a larger composite image. Each representative image was then extracted from the tiled image. (D) Quantification of collagen linearization using displacement-to-length ratio (DLR) of collagen fibers in SHG images. For a line, DLR = 1, and for a curve, DLR < 1. Representative images are shown in fig. S20. Forty-five fibers per group were used for this analysis. Box and whisker plots are shown as minimum, 25th percentile, median, 75th percentile, and maximum. ****P <0.0001. (E and F) Representative AFM stiffness maps (50 μm × 50 μm) of tumor-bearing (E) and tumor-free (F) lungs. (G and H) Frequency of Young’s modulus values of tumor-bearing (G) and tumor-free (H) lungs from AFM microindentation in the range of 0 to 40 kPa (bin size = 1 kPa), whereas the inset graphs show the frequency within the range of 0 to 10 kPa (bin size = 0.5 kPa). Five hundred measurements per group were analyzed. P <0.001 (Young’s modulus of tumor-bearing lungs versus tumor-free lungs).
To further study how our MRCS interacts with the metastatic niche, we cotransduced the MRCS-CD to constitutively express eGFP as a cell tracker. We then performed SHG imaging with ex vivo IHC staining 24 hours after the systemic infusion of MRCS to tumor-bearing (Fig. 5, A and B) and tumor-free (Fig. 5C) mice (day 1). As observed on the SHG-IHC overlaid images, more MRCS (characterized by the constitutively expressed eGFP) was observed in tumor-bearing lungs. CD of eGFP-labeled MRCS-CD was preferentially activated in the cancer regions that are associated with more linearized collagen cross-linking (Fig. 5A). By contrast, few MRCS-CD were activated to express CD in less linearized noncancer regions (Fig. 5B) or in tumor-free lungs (Fig. 5C). Additionally, in Fig. 5A, although MRCS-CD was recruited to tumor regions, CD expression (magenta) was limited to direct contact with cancer (red) and cross-linking (cyan). The MRCS-CD in the periphery of the tumor region did not express CD, but only the constitutive eGFP (green). Separate images for each color layer are shown in fig. S21. With MRCS-CD, the apoptosis [visualized via poly(adenosine diphosphate-ribose) polymerase (PARP) p85 antibody staining] is correlated with the presence of cancer and increased cross-linked collagen (fig. S22). With CD-MSC, there is apoptosis regardless of the tissue environment, and PARP signals are present not only in highly cancerous and cross-linked regions (fig. S23A) but also in less cross-linked regions (fig. S23B) and in healthy controls (fig. S23C).
To demonstrate the translatability and broad applicability of our MRCS platform, we also evaluated MRCS in a spontaneous model of breast cancer metastasis to the lungs. The establishment of the spontaneous model is shown in fig. S24, demonstrating that lung metastasis occurs within about 6 weeks after implantation of cancer cells to the mouse fat pads. We then demonstrated that MRCS-CD can specifically target cancer regions (RFP) with higher tissue collagen cross-linking (SHG) and induce specific tissue damage (apoptosis via PARP p85 staining) in the metastatic niches in vivo (Fig. 6A). In contrast, we observed minimal off-target damage in noncancer regions of tumorbearing lungs (Fig. 6B and fig. S25) or in healthy lungs (Fig. 6C and fig. S25) where there is low cross-linking compared to the cancer regions of the lungs.
Fig. 6. Cross-linking-specific tissue damage by MRCS in response to mechano-cues in the metastatic niche in vivo in spontaneous lung metastasis model.
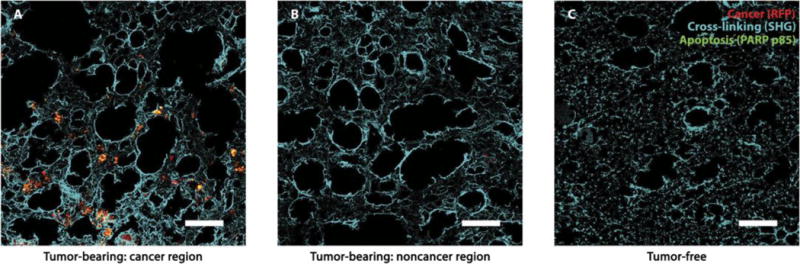
Frozen sections of lungs of tumor-bearing NSG mice with Luc-RFP-231 spontaneous lung metastasis from primary tumors [cancer region in (A) and noncancer region in (B)] and tumor-free NSG mice (C) sacrificed after MRCS-CD infusion and 5-FC treatment as indicated (day 9) were stained with anti-PARP p85 (green) for tissue apoptosis. RFP signal (red) indicates the presence of lung metastasis, and colocalization of red and green appears yellow. SHG imaging of collagen networks (cyan) was presented and overlaid with IHC imaging. Scale bars, 100 μm.
DISCUSSION
Despite decades of effort, little progress has been made to improve the diagnosis and treatment of cancer metastases. In particular, because of the heterogeneity of cancer and its ability to develop resistance to current treatments that target biochemical markers, new targeting strategies are urgently needed. Inspired by the tight correlation between increases in tissue stiffness and breast cancer metastatic niches found in recent studies (13,15,17,18,45) and the fact that MSCs differentiate to specific lineages depending on the stiffness of the microenvironment (28), we have developed a class of cancer therapeutics that directly target the mechanoenvironmental cues of cancer metastases. The MRCS is an attempt to directly interrogate the mechano-niche in vivo and apply it for localized delivery of agents including imaging reporters and therapeutics.
Mechano-niches play vital roles in development, homeostasis, and disease progression, including many types of cancer, and therefore serve as an emerging target for next-generation therapeutics (14, 46). In particular, matrix stiffness is an appealing target for cancer therapeutics due to its long persistence in the body (measured in years), making it refractory to development of resistance to treatments (47, 48). Furthermore, cancer and cancer biomarkers are highly heterogeneous within the population, making it difficult to developtreatments that can accurately target the disease. Mechanical markers such as matrix stiffness, however, manifest similarly in most cases and present a more universal target for therapeutics (14, 49). Given the enormous challenge in the search of specific cancer biomarkers, matrix stiffness may present an opportunity, especially when used in combination with chemical biomarkers, to improve the sensitivity and specificity in cancer targeting. In addition, the natural ability of MRCS to actively home to and integrate into tumors and metastases enables the efficient delivery of therapeutics to the target-site. Together, our MRCS could have major clinical implications in increasing the effectiveness of therapies for patients with metastatic cancer while also ameliorating the deleterious side effects associated with chemotherapy (50) or other less specific cell-based delivery systems that are engineered to constitutively express therapeutics. In addition, our system could potentially be used to prevent metastasis by targeting the LOX-mediated, stiff premetastatic niche (13,15,18), for example, by engineering the MRCS to secrete matrix remodeling enzymes, such as metalloproteinases, to reduce the stiffness of the niche.
MSCs have been proven safe for transplantation into humans in the clinic (23, 24), which paves the way for clinical translation of the proposed MRCS. Transplanted MSCs themselves have previously been proposed to regulate cancer progression, both positively and negatively (21, 51, 52). We do not consider it as a major issue because MRCS only stays in tumors transiently (<7 days) and would be eliminated by suicide genes (CD). Our data show that MRCS can continue to functionally convert 5-FC to 5-FU for at least 5 days while in contact with stiff substrate, although the process of cancer killing could begin as early as 2 days after treatment, with the MRCS-CD themselves dying shortly after. This suggests that MRCS can be a transient yet efficient drug delivery system for treating cancer. Future studies will need to further investigate the activity of both CD-MSC and MRCS-CD in vivo, in particular with regard to conversion of 5-FC, to determine the time points of conversion and killing of the drug source (MSCs), as well as dosages required to ensure efficacy while still minimizing side effects.
Although several organs, including muscle (12 kPa) and bone (25 to 60 kPa) (28,53), approach or exceed the tissue stiffness of invasive breast cancer and may promote activation of our MRCS, we anticipate that this will not be a major issue because of the inherent homing ability of MSCs to cancer and metastases and their rapid clearance from non-inflamed or noninjured tissues (21,22). Our data showed no significant damage to bone, bone marrow, liver, or brain tissues as a result of systemic treatment with MRCS (although we emphasize that MSC type and administration route may affect their homing profile). Although in transit to the metastatic niche MSCs will encounter blood vessel endothelial cells, basement membrane, and ECM components, each with their own characteristic stiffness, we do not expect this to irreversibly influence MRCS activity (28). In particular, many of these mechanical interactions involve shear stress, which does not regulate MSC differentiation. Previous studies have also established that expression of mechanoresponsive genes is rapidly reversible (28, 54). The specificity of MRCS colocalization and activation on tumors was also demonstrated in our study, with targeted release of CD only in regions with cancer cells and high collagen cross-linking, resulting in localized tissue damage in these tumor regions, but not in other nontumor regions. This, combined with data showing that MRCS will not release CD to convert 5-FC while on soft substrates, demonstrates that MRCS should not result in off-target damage in healthy tissues.
In contrast, treatment with CD-MSC resulted in increased tissue damage in the lungs, which was not only restricted to tumor regions but was also observed in healthy tissues. This could be at least partially attributed to the initial entrapment of MSCs in the pulmonary vasculature (43), where CD-MSC could still express CD to cause nonspecific damage, whereas MRCS-CD would be inactive. Although CD-MSC and MRCS-CD displayed similar in vivo treatment efficacy and similar survival curves, this phenomenon may be attributed to the cell therapies reaching a threshold level of cancer killing, beyond which survival curves and whole-body imaging may not be sensitive enough to distinguish the two treatment groups. Further optimization of treatment parameters such as initial tumor burden, timing, and dosage of treatment and using more sensitive imaging modalities may elucidate these differences in future studies. Future studies will also need to further investigate the activation of MRCS in vivo to fully characterize the conditions in the tumor microenvironment, which trigger the conversion of 5-FC. However, it should be emphasized that MRCS-CD attenuated tumor growth. Together, the data suggest that MRCS is effective as a cancer therapeutic and has advantages over CD-MSC with respect to reducing side effects.
Our approach to targeting the mechano-niche in vivo by MRCS may also be relevant for the treatment of other types of fibrotic diseases through, for example, delivery of metalloproteinases. Moreover, by using cells engineered to respond to variations in matrix stiffness to drive expression of diagnostic reporters [such as the HSV-1-tk gene coupled with positron emission tomography imaging (55)], the MRCS could also be used to detect micrometastases at a higher resolution than current imaging techniques. Our system potentially has major advantages over current methods of identifying micrometastases in that it can amplify the signal from smaller numbers of cells by detecting the properties of the local microenvironment and that it can be used in vivo without a need for biopsy or other invasive techniques. Finally, our MRCS could also serve as an approach to elucidate mechanobiology, specifically the interplay of biomechanical cues (56) with cells in their native environment in vivo in the context of cancer and other conditions such as inflammation and injury. Note that MRCS is not restricted to lung metastasis of breast cancer, although it was the focus of this study due to its high morbidity and its robustness as a model to test our hypothesis. However, future studies using other models of metastasis, especially in sites other than the lung, will need to be investigated to evaluate the broad applicability of our approach. Although previous studies have established that matrix stiffness is tightly linked to invasiveness and metastasis, current methods of measuring stiffness rely on elastography or ex vivo measurements with AFM or compression devices (14,15). Unfortunately, these techniques lack the resolution to directly measure the stiffness of the ECM with which the cells interact; instead, they measure the average stiffness of larger regions encompassing both ECM and cellular components of the tissues of interest. A cell-based stiffness sensor should reveal what cells actually “feel” in the microenvironment and dynamically interrogate the mechanoenvironment of primary tumors, metastases, and changes in matrix stiffness during disease progression and response to therapies at a cellular resolution in vivo.
MATERIALS AND METHODS
Study design
This study was designed to investigate the hypothesis that a cell-based system can be engineered to respond specifically to mechanoenvironmental cues (MRCS) to target cancer metastases. In vitro experiments (including tunable hydrogels, qPCR, cocultures, and mass spectrometry)and in vivo and ex vivo experiments using nude and NSG mice (including luciferase imaging, antibody staining, SHG, and AFM) were performed to demonstrate that MRCS can sense and kill cancer cells in response to biophysical cues. For all in vitro studies except for mass spectrometry, three independent experiments with at least three samples per condition were performed. n = 9 for the in vivo cancer killing and survival assays. n ≥ 3 for ex vivo characterization of the biophysical cues in the metastatic niche and the validation of MRCS. For the survival experiment, the end point for mice was defined as “found dead” or euthanasia criteria stated in University of California, Irvine (UCI) Institutional Animal Care and Use Committee protocol 2012–3062 described in Supplementary Materials and Methods. Groups for animal experiments were randomized, except for the cancer killing study where manual group adjustments were performed to keep the differences in initial cancer burden between all “week 0” group animals not statistically significant. In vitro experiments were not blinded. In vivo and ex vivo experiments were blinded.
Statistical analysis
Data were analyzed by Student’s t test when comparing two groups and by analysis of variance (ANOVA) when comparing more than two groups. Log-rank (Mantel-Cox) test was performed for animal survival data analysis by two-way ANOVA, and Mann-Whitney test was used for AFM data. Outliers in AFM data were removed by Grubbs’ test (P < 0.05). Data were expressed as means ± SD or means ± SEM. Two-sided testing with normal-based 95% confidence interval was performed for each analysis, and differences were considered significant at P <0.05.
Supplementary Material
Fig. S1. Concept of MRCS for targeting breast cancer metastases in the lung.
Fig. S2. Construction of MRCS.
Fig. S3. MRCS-eGFP activation in response to substrate stiffness in vitro.
Fig. S4. MRCS-eGFP in vitro validation with immunostaining.
Fig. S5. Further MRCS-Luc in vitro validation.
Fig. S6. CD-MSC able to kill cancer cells in the presence of 5-FC in vitro.
Fig. S7. MRCS-CD responding to matrix stiffness in vitro.
Fig. S8. Bystander effect from MRCS-CD starting at 24 hours in vitro on stiff substrate.
Fig. S9. Bystander effect from MRCS-CD lasting after MSC removal in vitro on stiff substrate.
Fig. S10. Luc-MSC homing to the metastatic niche in vivo.
Fig. S11. MRCS homing and specific activation in response to the metastatic niche in vivo.
Fig. S12. Specific activation of MRCS-eGFP in response to the metastatic niche in vivo.
Fig. S13. MRCS-CD unable to attenuate cancer growth in the absence of 5-FC in vivo.
Fig. S14. No detectable side effects in bone marrow cell populations after systemic treatment with MRCS-CD.
Fig. S15. MRCS-CD causing no detectable side effects in vivo in bone marrow.
Fig. S16. MRCS-CD causing no detectable side effects in vivo in livers.
Fig. S17. MRCS-CD causing no detectable side effects in vivo in brains.
Fig. S18. Up-regulation and colocalization of LOX expression with tumor in tumor-bearing lungs.
Fig. S19. LOX expression up-regulated with increased collagen expression in the metastatic niche.
Fig. S20. SHG imaging showing up-regulated and more linearized collagen in tumor-bearing lungs.
Fig. S21. Split-channel views of MRCS activation in the metastatic niche in vivo.
Fig. S22. Cross-linking-specific tissue damage by MRCS in response to mechano-cues in the metastatic niche in vivo.
Fig. S23. Constitutively CD-expressing MSCs causing nonspecific tissue damage in vivo.
Fig. S24. Spontaneous lung metastasis model establishment.
Fig. S25. Split-channel views of cross-linking-specific tissue damage by MRCS in the metastatic niche in vivo in spontaneous lung metastasis model.
Table S1. Primary antibodies.
Table S2. Secondary antibodies.
Table S3. Primers used in qPCR.
Acknowledgments
We thank D.-K. Kang, M. M. Ali, and J. Zimak for discussion and comments. We are grateful to K. Zhang and X. E. Guo for help on in vitro experiments and discussion. We thank F. Grun, the director of the UCI Mass Spectrometry Facility (Department of Chemistry, UCI), for his advice and help with the mass spectrometry training and analysis. We thank K. Aboody, T. Synold, and M. Metz from the City of Hope for sharing their LC-MS/MS method for prodrug conversion. We thank C. Schmidt-Dannert, C. Contag, S. Piccolo, and D. Trono for the plasmids. We thank California NanoSystems Institute at the University of California, Los Angeles for AFM.
Funding: This work was supported by the NIH (1DP2CA195763-01) and the Department of Defense (W81XWH-13-1-0326 to W.Z.), NIH (2P41GM103540 to M.A.D.), and American Cancer Society (IRG-98-279-10) and NIH/National Cancer Institute (1K22CA190511-01A1 to D.A.L.) S.X.Z. was supported by a Cardiovascular Applied Research and Entrepreneurship fellowship [NIH/National Heart, Lung, and Blood Institute (T32)]. W.L. was supported by a California Institute for Regenerative Medicine (CIRM) fellowship (TG2-01152). C.C.C. was supported by NSF Graduate Research Fellowship. A.I.S. received a fellowship from the Fondation ARC pour la recherche sur le cancer (SAE20150602901). G.P. was supported by a CIRM training grant (TB1-01182).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/400/eaan2966/DC1
Materials and Methods
References (57–68)
Author contributions: L.L. designed and performed experiments, analyzed and interpreted data, drafted the manuscript, and coordinated the project. S.X.Z. performed the in vivo experiments, tissue processing, histology, mass spectrometry exams, and AFM microindentation test, analyzed data, and drafted the manuscript. W.L. designed and performed the in vivo experiments and edited the manuscript. H.P.F. performed tissue processing, IHC, and SHG imaging and participated in the in vivo experiments. C.W.W. engineered the cells, conducted the immunocytochemistry, qPCR, XTT and Transwell assays, in vitro luciferase assays, mass spectrometry exams, bone marrow flush, and flow cytometry and analyzed the data. C.C.C. carried out the qPCR and XTT and in vitro luciferase assays, analyzed the data, and participated in the mass spectrometry exams. A.I.S. designed and performed the mass spectrometry exams, designed the histology assays, conducted the flow cytometry assay, and analyzed the data. J.V.C. and M.A.D. performed SHG imaging and edited the manuscript. L.P.N. participated in the in vivo experiments and the mass spectrometry exams, performed the histology assays, and acquired the data. M.L. engineered the cells, helped to prepare the figures, and performed cloning and manuscript editing. G.P. contributed to in vivo experiments and participated in IHC and manuscript editing. T.L.D. edited the manuscript. D.A.L. edited the manuscript. E.J.P. conducted the flow cytometry assay, edited the manuscript, and interpreted the data. W.Z. was responsible for conception and design of the research, interpreted the results, revised the manuscript, and supervised the overall project.
Competing interests: W.Z., L.L., S.X.Z., W.L., E.J.P., and M.L. are inventors on patent application (PCT/US2016/028675) held/submitted by the Regents of the University of California that covers making and uses of mechanosensing cells. The other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature. 1980;283:139–146. doi: 10.1038/283139a0. [DOI] [PubMed] [Google Scholar]
- 3.Ali SM, Harvey HA, Lipton A. Metastatic breast cancer: Overview of treatment. Clin Orthop Relat Res. 2003;2003:S132–S137. doi: 10.1097/01.blo.0000092981.12414.7b. [DOI] [PubMed] [Google Scholar]
- 4.Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Cancer Res. 2009;102:67–101. doi: 10.1016/S0065-230X(09)02003-X. [DOI] [PubMed] [Google Scholar]
- 5.Jones SE. Metastatic breast cancer: The treatment challenge. Clin Breast Cancer. 2008;8:224–233. doi: 10.3816/CBC.2008.n.025. [DOI] [PubMed] [Google Scholar]
- 6.Irvin W, Jr, Muss HB, Mayer DK. Symptom management in metastatic breast cancer. Oncologist. 2011;16:1203–1214. doi: 10.1634/theoncologist.2011-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butcher DT, Alliston T, Weaver VM. A tense situation: Forcing tumour progression. Nat Rev Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–812. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Downing TL, Soto J, Morez C, Houssin T, Fritz A, Yuan F, Chu J, Patel S, Schaffer DV, Li S. Biophysical regulation of epigenetic state and cell reprogramming. Nat Mater. 2013;12:1154–1162. doi: 10.1038/nmat3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox TR, Bird D, Baker AM, Barker HE, Ho MWY, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73:1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong CCL, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IOL, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, Chi JTA, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 18.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bondareva A, Downey CM, Ayres F, Liu W, Boyd SK, Hallgrimsson B, Jirik FR. The lysyl oxidase inhibitor, β-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLOS ONE. 2009;4:e5620. doi: 10.1371/journal.pone.0005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong CCL, Zhang H, Gilkes DM, Chen J, Wei H, Chaturvedi P, Hubbi ME, Semenza GL. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med. 2012;90:803–815. doi: 10.1007/s00109-011-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ankrum J, Karp JM. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol Med. 2010;16:203–209. doi: 10.1016/j.molmed.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell. 2009;4:206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Trounson A, Thakar RG, Lomax G, Gibbons D. Clinical trials for stem cell therapies. BMC Med. 2011;9:52. doi: 10.1186/1741-7015-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Eckert MA, Riazifar H, Kang DK, Agalliu D, Zhao W. From blood to the brain: Can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013;2013:435093. doi: 10.1155/2013/435093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Cao F, De A, Cao Y, Contag C, Gambhir SS, Wu JC, Chen X. Trafficking mesenchymal stem cell engraftment and differentiation in tumor-bearing mice by bioluminescence imaging. Stem Cells. 2009;27:1548–1558. doi: 10.1002/stem.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reagan MR, Kaplan DL. Concise review: Mesenchymal stem cell tumor-homing: Detection methods in disease model systems. Stem Cells. 2011;29:920–927. doi: 10.1002/stem.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597–606. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 28.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 29.Park JS, Chu JS, Tsou AD, Diop R, Tang Z, Wang A, Li S. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials. 2011;32:3921–3930. doi: 10.1016/j.biomaterials.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tse JR, Engler AJ. Stiffness gradients mimicking in vivo tissue variation regulate mesenchymal stem cell fate. PLOS ONE. 2011;6:e15978. doi: 10.1371/journal.pone.0015978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samani A, Zubovits J, Plewes D. Elastic moduli of normal and pathological human breast tissues: An inversion-technique-based investigation of 169 samples. Phys Med Biol. 2007;52:1565–1576. doi: 10.1088/0031-9155/52/6/002. [DOI] [PubMed] [Google Scholar]
- 32.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 33.Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- 34.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, Mohanan S, Morris PG, Du B, Zhou XK, Vahdat LT, Verma A, Elemento O, Hudis CA, Williams RM, Gourdon D, Dannenberg AJ, Fischbach C. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7:301ra130. doi: 10.1126/scitranslmed.3010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, Mueller E, Benjamin T, Spiegelman BM, Sharp PA, Hopkins N, Yaffe MB. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074–1078. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- 37.Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats RA, Garcia E, Aramburo S, Valenzuela VV, Frank RT, Barish ME, Brown CE, Kim SU, Badie B, Portnow J. Neural stem cell-mediated enzyme/prodrug therapy for glioma: Preclinical studies. Sci Transl Med. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aboody KS, Najbauer J, Schmidt NO, Yang W, Wu JK, Zhuge Y, Przylecki W, Carroll R, Black PM, Perides G. Targeting of melanoma brain metastases using engineered neural stem/progenitor cells. Neuro Oncol. 2006;8:119–126. doi: 10.1215/15228517-2005-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fidler IJ. Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev. 1986;5:29–49. doi: 10.1007/BF00049529. [DOI] [PubMed] [Google Scholar]
- 40.Xie X, Briinnert N, Jensen G, Albrectsen J, Gotthardsen B, Rygaard J. Comparative studies between nude and scid mice on the growth and metastatic behavior of xenografted human tumors. Clin Exp Metastasis. 1992;10:201–210. doi: 10.1007/BF00132752. [DOI] [PubMed] [Google Scholar]
- 41.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, Battula VL, Weil M, Andreeff M, Marini FC. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu L, Zhang SX, Aeran R, Liao W, Lu M, Polovin G, Pone EJ, Zhao W. Exogenous marker-engineered mesenchymal stem cells detect cancer and metastases in a simple blood assay. Stem Cell Res Ther. 2015;6:181. doi: 10.1186/s13287-015-0151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, Shimamura T, Rauh D, Mermel C, Fischer S, Stückrath I, Heynck S, Beroukhim R, Lin W, Winckler W, Shah K, LaFramboise T, Moriarty WF, Hanna M, Tolosi L, Rahnenführer J, Verhaak R, Chiang D, Getz G, Hellmich M, Wolf J, Girard L, Peyton M, Weir BA, Chen TH, Greulich H, Barretina J, Shapiro GI, Garraway LA, Gazdar AF, Minna JD, Meyerson M, Wong KK, Thomas RK. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaikh A, Bhartiya D, Kapoor S, Nimkar H. Delineating the effects of 5-fluorouracil and follicle-stimulating hormone on mouse bone marrow stem/progenitor cells. Stem Cell Res Ther. 2016;7:59. doi: 10.1186/s13287-016-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erler JT, Giaccia AJ. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006;66:10238–10241. doi: 10.1158/0008-5472.CAN-06-3197. [DOI] [PubMed] [Google Scholar]
- 46.Gasiorowski JZ, Murphy CJ, Nealey PF. Biophysical cues and cell behavior: The big impact of little things. Annu Rev Biomed Eng. 2013;15:155–176. doi: 10.1146/annurev-bioeng-071811-150021. [DOI] [PubMed] [Google Scholar]
- 47.Sherratt MJ. Tissue elasticity and the ageing elastic fibre. Age. 2009;31:305–325. doi: 10.1007/s11357-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McPherson JM, Sawamura SJ, Conti A. Preparation of [3H]collagen for studies of the biologic fate of xenogenic collagen implants in vivo. J Invest Dermatol. 1986;86:673–677. doi: 10.1111/1523-1747.ep12275992. [DOI] [PubMed] [Google Scholar]
- 49.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at aglance. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol. 2010;24:359–369. doi: 10.1210/me.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Droujinine IA, Eckert MA, Zhao W. To grab the stroma by the horns: From biology to cancer therapy with mesenchymal stem cells. Oncotarget. 2013;4:651–664. doi: 10.18632/oncotarget.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, Csizmadia E, Mariani O, Zhu C, Campagne A, Toner M, Bhatia SN, Irimia D, Vincent-Salomon A, Karnoub AE. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci USA. 2012;109:17460–17465. doi: 10.1073/pnas.1206653109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai RM, Madl CM, Xu M, Zhao X, Chaudhuri O, Verbeke C, Kim WS, Alim K, Mammoto A, Ingber DE, Duda GN, Mooney DJ. Matrix elasticity of void-forming hydrogels controls transplantedstem-cell-mediated bone formation. Nat Mater. 2015;14:1269–1277. doi: 10.1038/nmat4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang C, Tibbitt MW, Basta L, Anseth KS. Mechanical memory and dosing influence stem cell fate. Nat Mater. 2014;13:645–652. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brader P, Kelly K, Gang S, Shah JP, Wong RJ, Hricak H, Blasberg RG, Fong Y, Gil Z. Imaging of lymph node micrometastases using an oncolytic herpes virus and [18F]FEAU PET. PLOS ONE. 2009;4:e4789. doi: 10.1371/journal.pone.0004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh AK, Upadhyay RC, Malakar D, Kumar S, Singh SV. Effect of thermal stress on HSP70 expression in dermal fibroblast of zebu (Tharparkar) and crossbred (Karan-Fries) cattle. J Therm Biol. 2014;43:46–53. doi: 10.1016/j.jtherbio.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 57.Vick JE, Johnson ET, Choudhary S, Bloch SE, Lopez-Gallego F, Srivastava P, Tikh IB, Wawrzyn GT, Schmidt-Dannert C. Optimized compatible set of BioBrick™ vectors for metabolic pathway engineering. Appl Microbiol Biotechnol. 2011;92:1275–1286. doi: 10.1007/s00253-011-3633-4. [DOI] [PubMed] [Google Scholar]
- 58.Patel MR, Chang YF, Chen IY, Bachmann MH, Yan X, Contag CH, Gambhir SS. Longitudinal, noninvasive imaging of T-cell effector function and proliferation in living subjects. Cancer Res. 2010;70:10141–10149. doi: 10.1158/0008-5472.CAN-10-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tse JR, Engler AJ. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1016s47. Chapter 10, Unit 10.16. [DOI] [PubMed] [Google Scholar]
- 60.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 61.Kucerova L, Matuskova M, Pastorakova A, Tyciakova S, Jakubikova J, Bohovic R, Altanerova V, Altaner C. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J Gene Med. 2008;10:1071–1082. doi: 10.1002/jgm.1239. [DOI] [PubMed] [Google Scholar]
- 62.Bianchini P, Diaspro A. Three-dimensional (3D) backward and forward second harmonic generation (SHG) microscopy of biological tissues. J Biophotonics. 2008;1:443–450. doi: 10.1002/jbio.200810060. [DOI] [PubMed] [Google Scholar]
- 63.Campagnola PJ, Millard AC, Terasaki M, Hoppe PE, Malone CJ, Mohler WA. Three-dimensional high-resolution second-harmonic generation imaging of endogenous structural proteins in biological tissues. Biophys J. 2002;82:493–508. doi: 10.1016/S0006-3495(02)75414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Altendorf H, Decencière E, Jeulin D, De sa Peixoto P, Deniset-Besseau A, Angelini E, Mosser G, Schanne-Klein M-C. Imaging and 3D morphological analysis of collagen fibrils. J Microsc. 2012;247:161–175. doi: 10.1111/j.1365-2818.2012.03629.x. [DOI] [PubMed] [Google Scholar]
- 65.Bredfeldt JS, Liu Y, Pehlke CA, Conklin MW, Szulczewski JM, Inman DR, Keely PJ, Nowak RD, Mackie TR, Eliceiri KW. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J Biomed Opt. 2014;19:16007. doi: 10.1117/1.JBO.19.1.016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bredfeldt JS, Liu Y, Conklin MW, Keely PJ, Mackie TR, Eliceiri KW. Automated quantification of aligned collagen for human breast carcinoma prognosis. J Pathol Inform. 2014;5:28. doi: 10.4103/2153-3539.139707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu F, Tschumperlin DJ. Micro-mechanical characterization of lung tissue using atomic force microscopy. J Vis Exp. 2011;2011:e2911. doi: 10.3791/2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, Oertle P, Hyotyla JT, Aebi U, Bentires-Alj M, Lim RYH, Schoenenberger CA. The nanomechanical signature of breast cancer. Nat Nanotechnol. 2012;7:757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Concept of MRCS for targeting breast cancer metastases in the lung.
Fig. S2. Construction of MRCS.
Fig. S3. MRCS-eGFP activation in response to substrate stiffness in vitro.
Fig. S4. MRCS-eGFP in vitro validation with immunostaining.
Fig. S5. Further MRCS-Luc in vitro validation.
Fig. S6. CD-MSC able to kill cancer cells in the presence of 5-FC in vitro.
Fig. S7. MRCS-CD responding to matrix stiffness in vitro.
Fig. S8. Bystander effect from MRCS-CD starting at 24 hours in vitro on stiff substrate.
Fig. S9. Bystander effect from MRCS-CD lasting after MSC removal in vitro on stiff substrate.
Fig. S10. Luc-MSC homing to the metastatic niche in vivo.
Fig. S11. MRCS homing and specific activation in response to the metastatic niche in vivo.
Fig. S12. Specific activation of MRCS-eGFP in response to the metastatic niche in vivo.
Fig. S13. MRCS-CD unable to attenuate cancer growth in the absence of 5-FC in vivo.
Fig. S14. No detectable side effects in bone marrow cell populations after systemic treatment with MRCS-CD.
Fig. S15. MRCS-CD causing no detectable side effects in vivo in bone marrow.
Fig. S16. MRCS-CD causing no detectable side effects in vivo in livers.
Fig. S17. MRCS-CD causing no detectable side effects in vivo in brains.
Fig. S18. Up-regulation and colocalization of LOX expression with tumor in tumor-bearing lungs.
Fig. S19. LOX expression up-regulated with increased collagen expression in the metastatic niche.
Fig. S20. SHG imaging showing up-regulated and more linearized collagen in tumor-bearing lungs.
Fig. S21. Split-channel views of MRCS activation in the metastatic niche in vivo.
Fig. S22. Cross-linking-specific tissue damage by MRCS in response to mechano-cues in the metastatic niche in vivo.
Fig. S23. Constitutively CD-expressing MSCs causing nonspecific tissue damage in vivo.
Fig. S24. Spontaneous lung metastasis model establishment.
Fig. S25. Split-channel views of cross-linking-specific tissue damage by MRCS in the metastatic niche in vivo in spontaneous lung metastasis model.
Table S1. Primary antibodies.
Table S2. Secondary antibodies.
Table S3. Primers used in qPCR.