

Abstract
Kimura’s disease is a rare disease of unknown aetiology, commonly presenting with slow-growing head and neck subcutaneous nodules, lymphadenopathy, eosinophilia and elevated immunoglobulin E. This report describes a very rare case of a 41-year-old female, of White-British ethnicity, with a new diagnosis of Kimura’s disease of the parotid gland and associated cutaneous features. The patient was investigated for 3 years before a diagnosis of Kimura’s disease was reached. A superficial parotidectomy was undertaken and no recurrence was observed in the 20 months following surgery. Kimura’s disease is easily misdiagnosed, owing to lack of clinical awareness. This case report highlights the troubling symptomatology as well as complexities of diagnosis and management of Kimura’s disease. A high level of clinical suspicion is required, for patients of any ethnicity and sex presenting with features consistent with the disease, in order for prompt diagnosis, investigation and management to be achieved.
INTRODUCTION
Kimura’s disease (KD) is a rare chronic inflammatory disease, of unknown aetiopathogenesis, commonly presenting with slow-growing head and neck subcutaneous nodules, lymphadenopathy, peripheral blood eosinophilia and raised serum immunoglobulin (Ig) E levels [1]. It was first reported in China in 1937; since then, ~400 cases have been described in the literature [2]. It has a predilection for males of Asian descent, although sporadic cases have been reported in non-Asians [1].
This report describes a rare case of a 41-year-old female, of White-British ethnicity, with a new diagnosis of Kimura’s disease of the parotid gland. Owing to the complexity of the case, the patient was investigated for 3 years by various specialties, including maxillofacial, dermatology, rheumatology, otorhinolaryngology and haematology, before a diagnosis was reached.
CASE REPORT
A 41-year-old White-British female presented with intermittent left parotid gland swelling, associated with tenderness and pruritus. She also reported recent onset recurrent urticarial/eczematous lesions involving the trunk and limbs. No other systemic features were present. Her past medical history included hay fever, childhood atopic eczema and hypothyroidism.
Examination revealed a slightly enlarged, tender left parotid gland. Intra-oral examination was normal. Urticated erythematous plaques and papules were present on the trunk and limbs.
The patient had already undergone a parotid gland biopsy, in another hospital. The results of this were inconclusive, demonstrating benign lymphoid aggregates within the parotid associated with follicular hyperplasia of the intra/peri-parotid lymph nodes.
Laboratory investigations including inflammatory markers, renal function, autoimmune screen, serum ACE, protein electrophoresis and viral serology were unremarkable. Allergen patch testing was negative.
Ultrasound scan of the parotid gland was consistent with sialadenitis and a sialogram demonstrated normal whole unstimulated flow rate. Sjogren’s syndrome was excluded and the patient was managed empirically, with a course of Co-amoxiclav and Metronidazole, for chronic sialadenitis. She was commenced on cetirizine for her cutaneous features, which were presumed to be chronic urticaria.
Her symptoms were still persistent a year after presentation. A parotid gland sialoendoscopy with corticosteroid washout was hence undertaken, which was of limited benefit.
A positron emission tomography (PET) scan was subsequently arranged, which demonstrated increased uptake of the left parotid gland and several enlarged lymph nodes in the neck bilaterally (Figs 1 and 2).
Figure 1:

Axial PET scans (A and B) showing increased uptake of the left parotid gland and enlarged left-sided lymph nodes in the neck.
Figure 2:
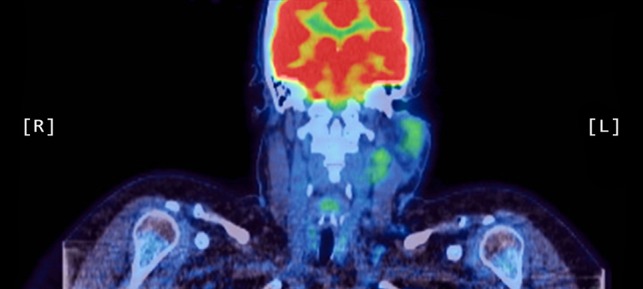
Coronal PET scan showing increased uptake of the left parotid gland and enlarged left-sided lymph nodes in the neck.
The patient developed further subcutaneous pruritic masses in the head and neck, with palpable cervical lymphadenopathy. A left cervical and parotid tail biopsy of lymph nodes and tissue was arranged, to exclude lymphoma. A biopsy of cutaneous lesions was also undertaken. The results of these were non-specific, demonstrating no obvious pathological disease.
A computed tomography (CT) scan demonstrated left parotid multifocal soft tissue abnormality and enlarged left-sided cervical lymph nodes, with prominent superficial left supraclavicular fossa lymph nodes (Figs 3 and 4).
Figure 3:
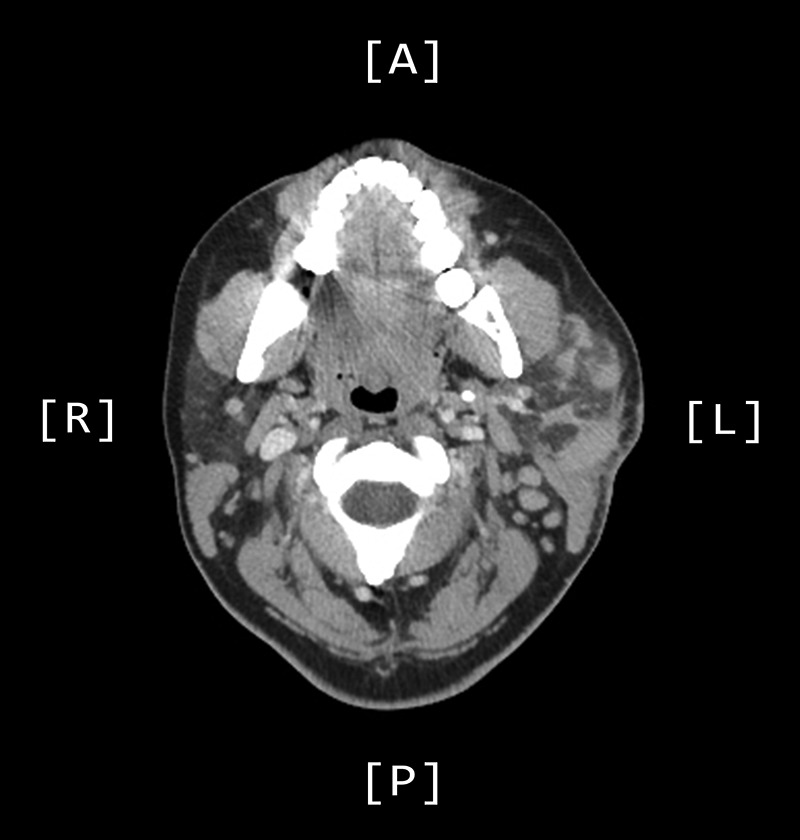
Axial CT sinuses (with contrast) showing left parotid multifocal soft tissue abnormality and enlarged left-sided cervical lymph nodes.
Figure 4:

Coronal CT sinuses (with contrast) showing left parotid multifocal soft tissue abnormality (A) with enlarged left-sided cervical and superficial supraclavicular fossa lymph nodes (B).
At this point IgE was found to be raised at 541 KU/L (normal range: 0–81 KU/L) with normal complement C3/C4. New onset eosinophilia (0.9 × 109/L, normal range: 0.0–0.4 × 109/L) was also noted.
The patient’s symptoms gradually worsened. A left superficial parotidectomy was hence undertaken, almost 3 years following presentation, which left the patient with modest pan-facial palsy (House Brackmann III).
On histological examination the lymph node tissue demonstrated preserved normal architecture. Capsular fibrosis and follicular hyperplasia was noted. The secondary follicles showed polarized germinal centres with tingible body macrophages and well defined mantle zones. The paracortex was expanded by a mixed chronic inflammatory cell infiltrate. Scattered eosinophils were also observed. The parotid parenchyma was mostly replaced by a diffuse lymphoid infiltrate. The residual parotid gland showed lymphoid aggregates in a periductal location. The lymphoid component demonstrated prominent follicular hyperplasia and well developed mantle zones. Parafollicular expansion by an eosinophil-rich mixed inflammatory cell infiltrate was present. Prominent hyalinization of the sinusoidal vessels with thick collagen bundles within the surrounding stroma was also observed. Eosinophil clumps were seen in areas, infiltrating the follicle locally (Figs 5 and 6).
Figure 5:
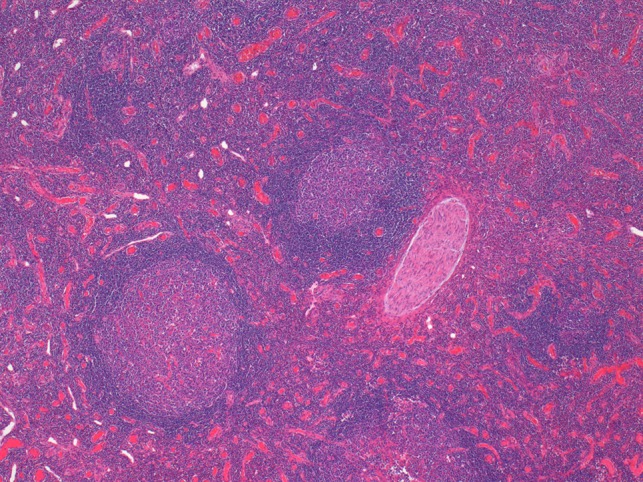
Histological examination of the parotid gland demonstrating diffuse lymphoid infiltrate with prominent follicular hyperplasia and well developed mantle zones (haematoxylin and eosin stain, ×2.5).
Figure 6:
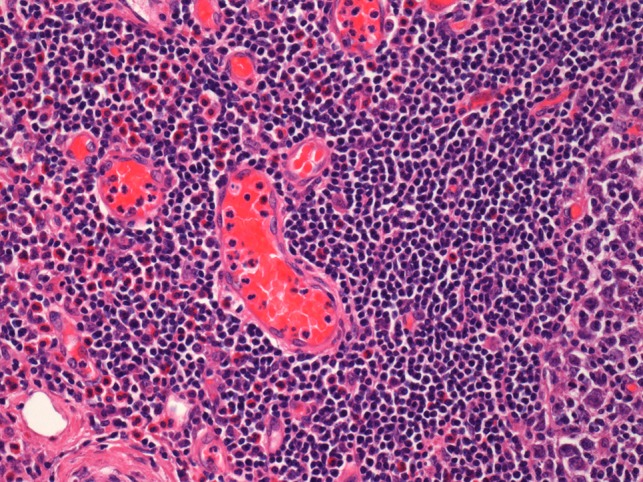
Histological examination of the parotid gland demonstrating eosinophil-rich mixed inflammatory cell infiltrate. Prominent hyalinization of the sinusoidal vessels with thick collagen bundles within the surrounding stroma can be observed (haematoxylin and eosin stain, ×20).
A diagnosis of Kimura’s disease was made, based on clinical and histopathological findings. A pharmacological agent, such as immunosuppressive therapy, biologics, monoclonal antibodies was not commenced, in accordance with the patient’s wishes. The patient has been followed up and no recurrence was observed in the twenty months since surgery.
DISCUSSION
Kimura’s disease presents with progressively enlarging deep subcutaneous nodules, mostly in the head and neck. Subcutaneous nodules have also been reported in the epicranium, orbit, eyelid, inner canthus, auricle, axilla, oral cavity, nasal sinuses, nerves and groin. Malignant transformation of the nodules has not been reported. Salivary gland involvement and local or distal lymphadenopathy is frequently observed in KD. Biochemically, the disease is characterized by peripheral blood eosinophilia and elevated serum IgE levels [1, 3]. KD has been associated with atopic conditions, unspecified dermatitis, lichen simplex, urticaria, prurigo nodularis [4]. Co-existing variable renal pathology is present in up to 60% of patients [5]. Although the aetiology of Kimura’s disease remains yet to be elucidated, several theories have been proposed including autoimmune, allergic and infective components [1, 3, 5, 6].
Radiological findings and fine-needle aspiration cytology are considered to be of limited value in the diagnosis of KD. Accurate diagnosis can only be achieved through excision biopsy and histological examination of the lesions [7].
Consensus regarding the most appropriate management for Kimura’s disease has yet to be reached. Surgical excision is usually performed first line. Recurrence has been reported in up to 25% of patients, occurring 1.3–2.5 years following surgery [8]. Conservative management options include radiotherapy [9], steroids, interferon-a, ciclosporin, thalidomide, leflunomide, intravenous immunoglobulins [4], cetirizine, imatinib [3] and omalizumab [10]. All of the aforementioned have demonstrated partial or complete success; however, recurrence rates appear to be high, especially upon withdrawal of treatment [2].
The differentials for KD are broad and include sarcoidosis, epithelioid haemangioma, atypical infections and neoplastic conditions. Kimura’s disease is easily misdiagnosed, owing to its rarity and lack of clinical awareness of the condition. Although rare, medical practitioners should be aware of the condition because of its troubling symptomatology and the entwined cosmetic and psychosocial disturbance. This case highlights the complexities of diagnosis and management of KD as well as the importance of a multidisciplinary approach in patient management.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Chen H, Thompson LD, Aguilera NS, Abbondanzo SL. Kimura disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2004;28:505–13. [DOI] [PubMed] [Google Scholar]
- 2. Deng WY, Ye SB, Luo RZ, Yan SM, Gao YF, Yang YZ, et al. . Notch-1 and Ki-67 receptor as predictors for the recurrence and prognosis of Kimura’s disease. Int J Clin Exp Pathol 2014;7:2402–10. [PMC free article] [PubMed] [Google Scholar]
- 3. Sun QF, Xu DZ, Pan SH, Ding JG, Xue ZQ, Miao CS, et al. . Kimura disease: review of the literature. Intern Med J 2008;38:668–72. [DOI] [PubMed] [Google Scholar]
- 4. Kottler D, Barete S, Quereux G, Ingen-Housz-Oro S, Fraitag S, Ortonne N, et al. . Retrospective multicentric study of 25 Kimura disease patients: emphasis on therapeutics and shared features with cutaneous IgG4-related disease. Dermatology 2015;231:367–77. [DOI] [PubMed] [Google Scholar]
- 5. Fouda MA, Gheith O, Refaie A, El-Saeed M, Bakr A, Wafa E, et al. . Kimura disease: a case report and review of the literature with a new management protocol. Int J Nephrol 2011;2010:673908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katagiri K, Itami S, Hatano Y, Yamaguchi T, Takayasu S. In vivo expression of IL-4, IL-5, IL-13 and IFN-gamma mRNAs in peripheral blood mononuclear cells and effect of cyclosporin A in a patient with Kimura’s disease. Br J Dermatol 1997;137:972–7. [PubMed] [Google Scholar]
- 7. Iwai H, Nakae K, Ikeda K, Ogura M, Miyamoto M, Omae M, et al. . Kimura disease: diagnosis and prognostic factors. Otolaryngol Head Neck Surg 2007;137:306–11. [DOI] [PubMed] [Google Scholar]
- 8. Bobinskas AM, Chandu A, Nastri AL. Kimura’s disease: an uncommon cause of head and neck masses with potentially serious sequelae. J Surg Case Rep 2015;2015:rjv131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ye P, Wei T, Yu GY, Wu LL, Peng X. Comparison of local recurrence rate of three treatment modalities for Kimura disease. J Craniofac Surg 2016;27:170–4. [DOI] [PubMed] [Google Scholar]
- 10. Nonaka M, Sakitani E, Yoshihara T. Anti-IgE therapy to Kimura’s disease: a pilot study. Auris Nasus Larynx 2014;41:384–8. [DOI] [PubMed] [Google Scholar]