

Short abstract
Over the past few years, nucleosides have maintained a prominent role as one of the cornerstones of antiviral and anticancer therapeutics, and many approaches to nucleoside drug design have been pursued. One such approach involves flexibility in the sugar moiety of nucleosides, for example, in the highly successful anti-HIV and HBV drug tenofovir. In contrast, introduction of flexibility to the nucleobase scaffold has only more recently gained significance with the invention of our fleximers. The history, development, and some biological relevance for this innovative class of nucleosides are detailed herein.
Keywords: Coronaviridae, dengue virus, filoviridae, flaviviridae, nucleoside analogues
Introduction
Nucleosides have played a prominent role in medicinal chemistry as one of the cornerstones of antiviral and anticancer therapeutics.1–3 Because their structures are based on the naturally occurring nucleosides (Figure 1), they have the ability to be recognized by biologically significant enzymes. As a result, even minor changes to the nucleoside scaffold can have profound effects—or can result in no activity at all.
Figure 1.
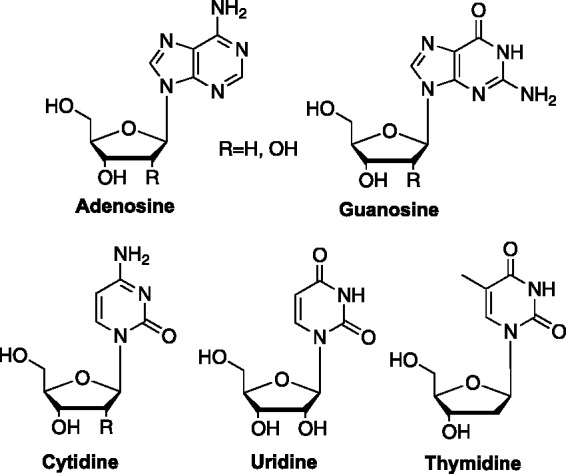
The five most common naturally occurring nucleosides.
Many approaches to nucleoside drug design have evolved over the years, ranging from removal, rearrangement, or addition of functional groups or atoms in the base or sugar scaffolds, to changes in shape and/or size of the sugars and bases.1,3–5 In the 1980s, another modification was introduced by Antonin Holý and Erik De Clercq—imparting flexibility to the sugar scaffold.6–8 In an effort to explore just how much of the sugar was necessary for recognition and activity, they systematically removed the 2′ and/or 3′ carbons (and corresponding hydroxyl groups) of the ribose sugar, which led to the acyclic and subsequently, the acyclic phosphonate classes of nucleosides.6–8 Several of these are still used in the clinic today, including acyclovir,9 ganciclovir,10 and penciclovir,11 as well as the acyclic phosphonates cidofovir,12 adefovir13 and tenofovir (Figure 2).9,10,12,14–17
Figure 2.

Acyclic nucleosides and nucleoside phosphonates.
Due to its inherent flexibility, tenofovir has demonstrated the ability to overcome point mutations related to the development of drug resistance and retain its activity.6,18 Although the idea of flexibility in the sugar moiety became commonplace, until our introduction of flexibility to the heterocyclic purine base19 little had been done with regard to investigating the flexibility for the nucleobases. This is not surprising since it is common knowledge that the heterocyclic bases are critical for base pairing and base stacking, the latter of which is reliant upon the polar aromatic surface of the base.
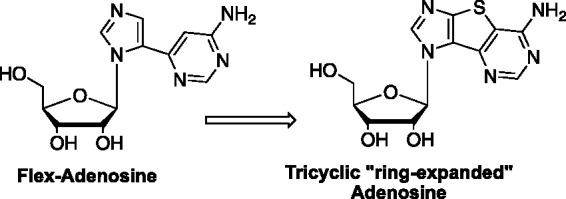
In an effort to explore the concept of introducing flexibility to the heterocyclic bases, we developed a class of nucleosides known as “fleximers,” where the bicyclic purine ring is “split” into its two components, for example, a pyrimidine and imidazole ring, that remain connected by a single bond as exemplified in Figure 3.19–21 This allows for rotation of the two pieces while still retaining the aromatic system and functional groups needed for recognition and biological activity.19,20
Figure 3.
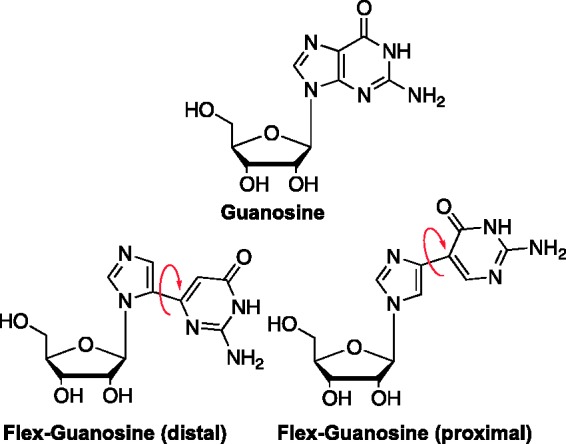
The fleximer concept.
Our initial hypotheses were that this increase in flexibility and repositioning would provide the fleximers with some advantages not possible with rigid nucleobases. For example, the inherent flexibility allows the nucleoside the ability to rotate and reposition in a binding site, particularly when faced with an electronic or steric interference. Since point mutations typically involve these sorts of amino acid changes, we also hypothesized this would allow the fleximer to adjust and engage a secondary amino acid residue not previously involved in the mechanism of action. This would in turn allow it to retain its activity, something a rigid analogue would not be able to do. In addition, this flexibility might also lead to an increase in binding affinity since the fleximer could sample more of the binding site to find more favorable interactions. In addition, the possibility for a “mutually induced fit” model between the binding site and the ligand might lead to the two working together as “dance partners” to find the best/lowest energy conformation for both.
Finally, we also hypothesized that because the fleximer could change conformations, it might be able to be recognized by enzymes that it would not necessarily have interacted with because it might be able to mimic a different nucleoside. We viewed the fleximers as “molecular chameleons”—just as chameleons have to adapt to their surroundings in order to survive, the fleximers would also have to adapt if confronted with a steric or electronic clash in the binding site they were targeting.
Initial results
The first distal fleximers developed were obtained via a ring opening reaction on a series of tricyclic nucleosides.19,20 By treating the thiophene-expanded tricyclic nucleoside with Raney nickel, the sulfur in the spacer ring was removed, leaving the two heterocycles free to rotate about the remaining C5–C6 carbon–carbon bond of the middle ring (Figure 4).19,20 These were designated as “distal” fleximers, based on similar naming for Nelson Leonard’s benzyl-expanded purine nucleosides,22 which were the inspiration for our tricyclic nucleosides. The synthesis proved to be quite challenging, with more than 20 steps on average to achieve the distals, a problem that we have yet to fully overcome despite a number of attempts to optimize the route.23–26
Figure 4.
Retrosynthetic approach to the distal fleximers.
In contrast, the second series, denoted as “proximal” fleximers, were much more facile to achieve since the requisite substitution patterns on the two heterocyclic pieces needed for metal catalyzed cross coupling methods were available.23,27,28 This allowed for use of Stille, Suzuki and other organometallic coupling reactions as depicted in Figure 5.23,29,30
Figure 5.
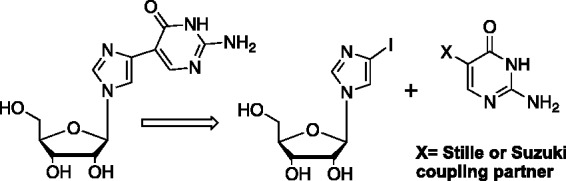
Retrosynthetic approach to the proximal fleximers.
Once the first set of compounds were in hand, we screened them against S-adenosylhomocysteine hydrolase (SAHase), an enzyme target we were focused on at the time. Inhibition of SAHase can lead to incomplete biological methylations that utilize S-adenosylmethionine (SAM) as the methyl donor.31 For example, the cap structures of the mRNA in viruses and parasites in particular, necessarily possess N7- and 2′-O-methylated nucleotides.32,33 These methylations are critical for stability and recognition and are accomplished by methyltransferases (MTases) that utilize SAM as the methyl donor. When this occurs, SAM is converted to S-adenosylhomocysteine (SAH). As shown in Figure 6, when SAHase is inhibited, it leads to a buildup of SAH, which in turn, inhibits the methyltransferases. This subsequently leads to incomplete translation and transcription.31 As a result, inhibiting SAHase (and indirectly MTases) is an important target for antiviral and antiparasitic therapeutics.28,34
Figure 6.
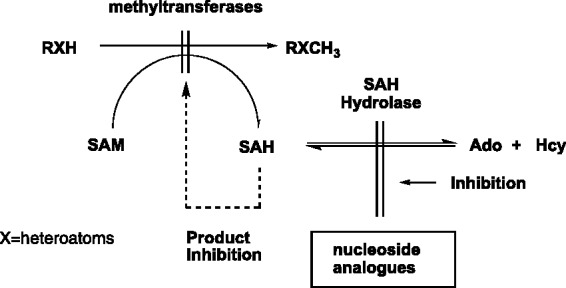
Inhibition of SAHase and indirectly, MTases.
SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine.
Interestingly, our preliminary computational results showed that the distal guanosine fleximer (Flex-G, Figure 3) was predicted to have a more favorable binding energy in SAHase than the adenosine analogue, Flex-A (Figure 4).35 Once the compound was made and actually tested, the Flex-G proved to be an inhibitor of SAHase.35 To our knowledge, this is the first and only report of a guanosine nucleoside inhibiting this adenosine-metabolizing enzyme. In addition, computational results in the binding site of SAHase revealed a curved syn-like conformation for the fleximer (Figure 7), which seemed at first glance to be unlikely since nucleosides with bulky substituents often prefer to reside in the more favorable anti conformation.36 Upon closer examination, however, it became clear why this was occurring—due to the flexibility of the base, the fleximer was able to form an intramolecular hydrogen bond between the carbonyl of the pyrimidine and the 5′-OH of the sugar ring (Figure 7).35 In addition, this in turn swung the amino group of the Flex-G up into the area of the binding site where the amino group of adenosine typically resides—thus essentially creating an adenosine mimic, which would explain the ability of Flex-G to act as an SAHase inhibitor.35
Figure 7.

Flex-G in SAHase.
In an effort to further explore this interesting phenomenon, we undertook an NMR study in collaboration with Plavec et al.37 The results showed that in solution, the fleximer indeed preferred the expected anti conformation; however in the enzyme, the syn conformation was dominant (Figure 8).37 This lends support to our hypothesis that the enzyme and flex-nucleoside are acting as “dance partners,” and that the enzyme is having a direct influence on the conformation of the fleximer.
Figure 8.

Syn vs. anti conformation for Flex-G in SAHase binding site vs. in solution.
As part of a project initiated in a collaborator’s laboratory, we found ourselves involved in an investigation of GDP-L-fucose pyrophosphorylase (GFPP). GFPP is an enzyme involved in the formation of GDP-fucose38 which is utilized by a number of fucose-metabolizing enzymes and is produced through two separate and distinctly different pathways.38 The de novo pathway from GDP-L-mannose is well characterized; however, the salvage pathway via GTP and L-fucose-1-phosphate was poorly understood (Figure 9).38,39
Figure 9.

Two pathways to GDP-fucose.
Moreover, little was known about GFPP other than it had very little sequence identity in common with other nucleotide metabolizing enzymes and that it preferred guanosine analogues as substrates. In an effort to further characterize the substrate requirements for this intriguing enzyme, we performed an extensive SAR study using a wide variety of different modified carbohydrates, furanoses and heterocyclics in various combinations.39 To our surprise, the Flex-GTP was preferred twice as much as the natural substrate GTP as indicated by the discrimination factor shown in Table 1.39
Table 1.
Comparison of GTP vs. Flex-GTP in GFPP.
| Substrate | Ka (M−1 × 106) | KM (μM) | kcat (s−1) | kcat/KM (μM−1s−1 × 103) | D |
|---|---|---|---|---|---|
| GTP | 7.3 | 50.2 | 2.5 | 49.8 | 1.0 |
| Flex-GTP | 4.6 | 39.8 | 3.5 | 87.9 | 0.57 |
D: discrimination factor; (kcat/KM) substrate/(kcat/KM) analogue.
In an effort to identify which residues were responsible for this phenomenon, a series of mutated strains were constructed. The results showed that in the mutant K478A, if the lysine at 478 was mutated to an alanine, Flex-GTP was not recognized while in the K482A mutant, GTP was no longer recognized (Figure 10).40 These amino acids are only four residues apart, so Flex-GTP’s ability to readjust and reposition itself allowed it to engage a secondary amino acid residue not previously involved in the mechanism of action in order to stay active. In addition, this also led to a significant increase in affinity, thereby confirming two more of our initial hypotheses.39,40
Figure 10.

Differential Activities of GTP and Flex-GTP.
Returning to our original focus on SAHase, we decided to explore a series of carbocyclic fleximers as potential antiparasitic agents. Parasites all rely on a heavily methylated “cap four” structure at the end of their mRNA for stability in replication, thus incomplete methylation leads to disruption of transcription and translation.41,42 These methylations are accomplished by various MTases, thus inhibition of SAHase can, as previously mentioned, inhibit these methylations thereby disrupting parasite replication. In that regard, carbocyclic adenosine nucleosides are known to be the best SAHase inhibitors;43,44 thus, the synthesis of a series of carbocyclic fleximers was pursued (Figures 11 and 12).
Figure 11.
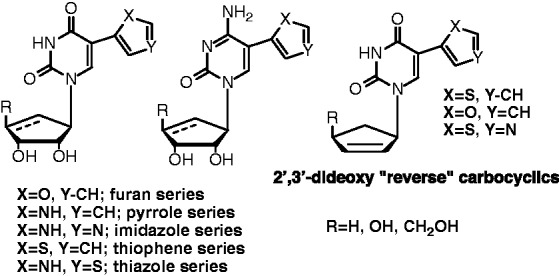
Carbocyclic “reverse” fleximers.
Figure 12.
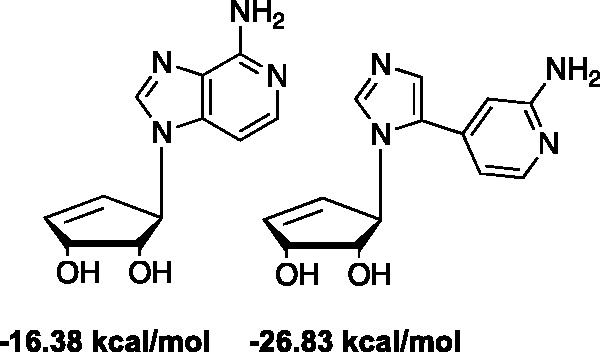
3-Deaza binding energies in SAHase.
The first series we pursued focused on a different connectivity for the base moieties. The connectivity was via the pyrimidine ring with the five-membered ring as the appendage (Figure 11) yielding the “reverse” fleximers.27,45 The inspiration for this change in connectivity came from 2′-deoxy nucleoside analogues first published in 1991 by Herdewijn et al.46–48 Some of these showed impressive activity against herpes simplex virus (HSV-1) and varicella zoster virus (VZV).46–48 Initially, we thought that reversing the connectivity might also lead to inhibition of both purine and pyrimidine metabolizing enzyme since they can be considered 5-subsituted pyrimidines or N-3 glycosylated purines. Interestingly, some of the reverse fleximers proved to be active against adenosine deaminase (ADA),27 by acting as product inhibitors, while the forward analogues failed to show any real activity.
We also synthesized the flex version28 of Borchardt’s truncated 3-deaza-neplanocin49 (NpC) shown in Figure 12, which was computationally predicted to be a better binder in SAHase than the parent 3-deazaNpc (unpublished data). As shown in Figure 13, the ability to sample more of the binding site may lead to better binding and biological activity. The amino groups and the 3′-OH groups were both positioned identically in the binding site; however, the imidazole ring on the flex-analogue was able to explore new areas in the binding site, interacting with His353. The truncated 3-deazaNpC was originally pursued due to its activity against Ebola, but unfortunately never made it past preliminary animal studies.50–52
Figure 13.

Flex-3-deazaNpC and 3-deaza-NpC.
At this point, it was clear that it was time to move to the next level to really test the limits of the fleximer concept. Thanks to a grant from the National Institutes of Health, we were able to pursue application of the fleximer approach to the modified sugars found in several FDA-approved nucleoside therapeutics. The first series that was designed (Figure 14) was based on the 2′-methyl modifications found in sofosbuvir (Me/F) and IDX-184 (Me/OH), given the excitement over their potent activity against HCV.53,54 Unfortunately, the route to realize these analogues was extremely tedious—21 steps with an overall yield of 0.09%—i.e. no future as a therapeutic! Despite the low yields, we were able to obtain enough to make the McGuigan ProTide55,56 and have both tested. The results (unpublished) in the HCV replicon assay were only moderate (Figure 14). Notably, however, no toxicity was noted at all levels tested and the presence of the ProTide moiety did increase the activity, albeit only slightly.
Figure 14.

2′ Modified fleximers in the HCV replicon assay.
After the tedious and low-yielding routes to the previous series, the second series was selected based on the facile synthetic routes available. Applying the acyclic sugar found in acyclovir with the flex-base approach, a series of acyclic fleximers were made (Figure 15).29 Acyclovir is an FDA-approved drug to treat HSV,57,58 but had also recently shown some activity against HIV.59 After broad screen testing, the compounds showed activity against human coronaviruses, including severe acute respiratory syndrome (SARS) and middle east respiratory syndrome (MERS).29 This was quite unexpected due to the lack of any nucleosides to that point demonstrating anti-coronavirus activity. The previous lack of active nucleoside analogues has been attributed to a unique exonuclease utilized by the coronaviruses, which recognizes nucleoside analogues and excises them.60,61 Importantly, acyclovir itself exhibits no activity at all against SARS and MERS;29 thus, it is clear that the flex approach is responsible for the potent activity observed.
Figure 15.

Acyclovir and Flex-Acyclovir analogues.
We anticipated these compounds would act as chain terminators given the known MOA for acyclovir,58 so the compounds, including the triphosphate (2MR04, Figure 15), were screened against the MERS and SARS polymerases. Interestingly, the triphosphate 2MR04 was not incorporated into the nascent RNA strand but did inhibit MERS RNA-dependent RNA polymerase (RdRp) but not the corresponding SARS RdRp (unpublished data). Additional studies are currently underway to fully characterize these differences as well as to elucidate just exactly how these compounds are working in coronaviruses.
In addition to the polymerase studies, the flex-nucleosides/tides were also screened against a panel of viral and human MTases. Surprisingly, some of the fleximers exhibited potent activity as low as 18 μM against the ZIKV, DENV, RSV, and MERS N7- and 2′O-methyltransferases, but not the corresponding human MTases (manuscript in preparation). This selectivity is of course, critical for ensuring a safe but effective therapeutic, so these results are highly promising. More recent investigations for these compounds also revealed potent activity against Ebola (EBOV) and Sudan viruses,30 as well as Dengue (DENV), Yellow Fever (YFV), and Zika (ZIKV) viruses (unpublished data); however, those results will be reported elsewhere once we complete the series.
The pursuit of fleximers has not been limited to our group—after our initial papers, several other researchers have adopted the fleximer approach. Recently Pochet et al. utilized an enzymatic transglycosylation in their synthetic route to realize a series of 2′-deoxyribose imidazole-substituted fleximers (Scheme 1).62 Using purine nucleoside phosphorylase (PNP) or nucleoside N-deoxyribosyltransferases (NDT), the base of thymidine was readily switched with a wide variety of aryl-substituted imidazoles which are prepared using Suzuki-Miyaura cross-coupling methodology.62 Their findings revealed that most of the reactions went in good yield, with both enzymes recognizing the fleximers, although NDT proved to be the most accepting of the two enzymes.62
Scheme 1.

Pochet’s transglycosylation route to fleximers.
Hudson et al. published a paper on click fleximers, where they utilized the popular copper-catalyzed azide-alkyne Huisgen cycloaddition reaction shown in Scheme 2.63 By employing a sugar with a 1′-azide group, they were able to couple to a 5-ethynyl cytosine which generated a series of 1,2,3-triazole-pyrimidine fleximers (Scheme 2).63 They also studied the compounds conformationally and spectroscopically and were able to show that the diamino analogue displayed a characteristic deep purple fluorescence, something that could be exploited for various studies.63 In addition, they confirmed the previous studies by Wetmore64 discussed below to show that the fleximers prefer a co-planar arrangement.63
Scheme 2.
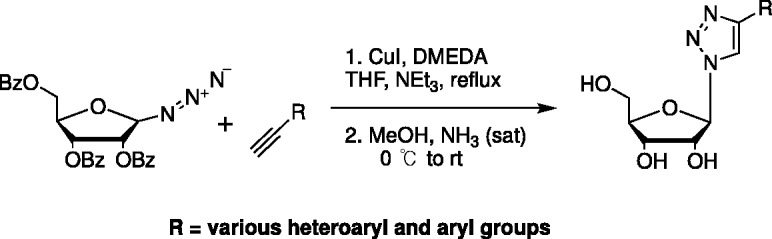
Hudson’s click fleximers.
Koszytkowska-Stawinska et al. also synthesized triazole fleximers utilizing the Bannert cascade reaction,65 which involves a [3,3]-sigmatropic rearrangement (Scheme 3); however, these analogues contained a methylene group between the azide and the pyrimidine moieties (Scheme 3). They synthesized several ribose analogues, as well as some acyclic phosphonate analogues; however, no biological activity was included in their report.65
Scheme 3.
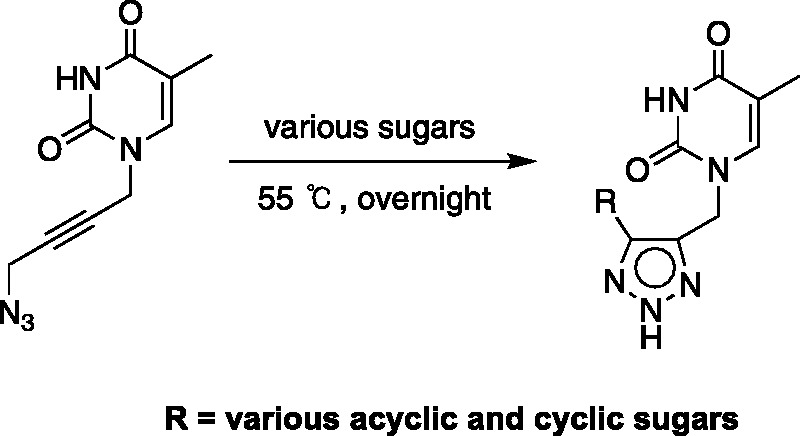
Fleximer analogues via the Bannert Cascade reaction.
Finally, Peyrottes et al. incorporated the triazole in their β-hydroxyphosphonate fleximers (Scheme 4).66 In place of the pyrimidines found in the natural purine bases and the fleximers from our group, they employed a wide range of substituted benzenes.66 Interestingly, some modest activity was noted for a few of the compounds against cytosolic 5′-nucleotidase II, an enzyme involved in regulating purine nucleotide pools that has also been linked to causing resistance to some cancer drugs.66
Scheme 4.

Peyrottes’ azido-fleximers.
In more of a serendipitous approach, Robins et al. discovered a unique route to realizing some oxadiazole-substituted imidazole fleximers by means of an unexpected ring opening of an adenosine N-oxide nucleoside occurred when treated with various anhydrides (Scheme 5).67 While rearrangements are not uncommon with N-oxide nucleosides, they typically result in 2-substituted adenosines, not in the split base found in the fleximers.67 Although interesting, the resulting oxadizaole ring lacks the critical hydrogen bonding elements found in the purine ring system needed for recognition and base pairing, thereby limiting their utility.
Scheme 5.
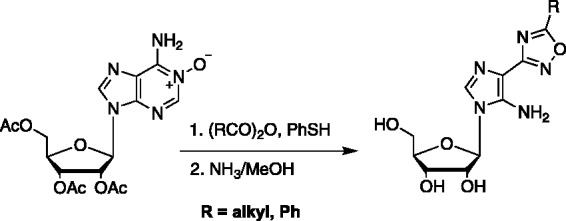
Robins’ ring opening route to an oxadiazole fleximer.
As part of study from Gundersen et al., a series of fleximers employing benzene rings instead of the typical ribose, 2′-deoxyribose sugar were constructed (Figure 16).68 Previous studies from this group with purines substituted with benzene rings led to some interesting activity against mycobacteria,68–70 thus provided a precedent to pursue other analogues. One major difference for these analogues is the connectivity of the benzene ring to the flex base involves a methylene group, not a typical C-N glycosidic bond (Figure 16). Notably, one of their compounds exhibited excellent activity against Mycobacterium tuberculosis with an IC50 of 14 μM.68
Figure 16.
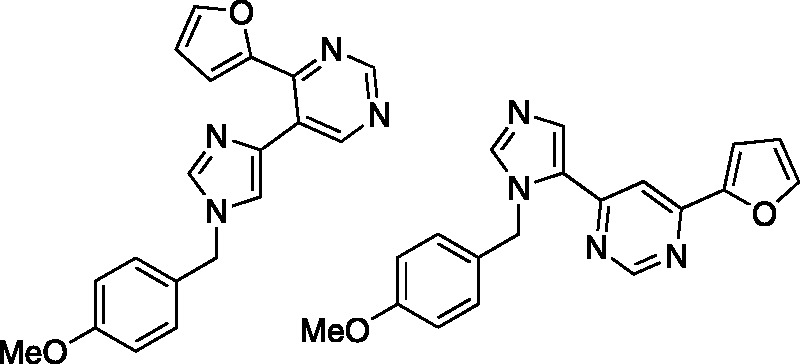
Gunderson’s fleximers.
Finally, Bardon and Wetmore carried out an extensive computational study using density functional theory in an effort to investigate a number of factors related to the fleximers’ structure and function.64 Their studies included assessing barriers to rotation, base pairing properties as well as their various connectivities. Their results showed that there was very little barrier of rotation, with computational barriers less than 40 kJ/mol−1 and that inclusion of a ribose sugar lowered that even further.64 In terms of base pairing, the fleximers all paired correctly and maintained normal purine hydrogen bonding patterns. The preferred connectivity varied based on the presence of the sugar ring (Figure 17). Without a sugar present, the distal conformation possesses the lowest energy conformation, while with a sugar, the proximal was preferred.64 Moreover, the two base components preferred to stay planar in most cases, particularly the proximal analogues; however, the distals tended to prefer a more perpendicular conformation based on the specific base.64
Figure 17.

Various connectivities for Flex-G.
In summary, the results outlined herein underline the growing importance of the flex approach for targeting viruses and other biologically significant enzymes. It is important to keep in mind that the various fleximers described herein were not recognized by every enzyme they were investigated against, nor did they work in the same way in the various enzymes they did inhibit or were recognized by. This speaks to our theory that the fleximer and enzyme are working as “dance partners,” which leads to their selectivity rather than indiscriminant activity (and potential toxicity). As each new series is pursued and studied, the results have increased our information about the endless possibilities for these unique nucleosides. As a result, we look forward to exploring new analogues and reporting on their properties and activity in the future.
Acknowledgements
The author would also like to thank her graduate students not only for actually doing great science but also for their help editing this paper.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author gratefully acknowledges the National Institutes of Health for funding much of this work (R21 AI097685 and R21 AI094639), as well as UMBC’s Technology Catalyst Fund.
ORCID iD
Katherine Seley-Radtke http://orcid.org/0000-0002-0154-3459.
References
- 1.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol 2004; 2: 704–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perigaud C Gosselin G andImbach JL. Nucleoside analogues as chemotherapeutic agents: a review. Nucleoside Nucleotides 1992; 11: 903–945. [Google Scholar]
- 3.Jordheim LP, Durantel D, Zoulim F, et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 2013; 12: 447–464. [DOI] [PubMed] [Google Scholar]
- 4.De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov 2002; 1: 13–25. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E. Antiviral drug discovery and development: where chemistry meets with biomedicine. Antiviral Res 2005; 67: 56–75. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin Microbiol Rev 2003; 16: 569–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Clercq E andHolý A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov 2005; 4: 928–940. [DOI] [PubMed] [Google Scholar]
- 8.Holý A. Antiviral acyclic nucleoside phosphonates structure activity studies. Antiviral Res 2006; 71: 248–253. [DOI] [PubMed] [Google Scholar]
- 9.Elion GB, Furman PA, Fyfe JA, et al. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A 1977; 74: 5716–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faulds D andHeel RC. Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs 1990; 39: 597–638. [DOI] [PubMed] [Google Scholar]
- 11.Boyd MR, Bacon TH, Sutton D, et al. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob Agents Chemother 1987; 31: 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lalezari JP, Stagg RJ, Kuppermann BD, et al. Intravenous cidofovir for peripheral cytomegalovirus retinitis in patients with AIDS. A randomized, controlled trial. Ann Intern Med 1997; 126: 257–263. [DOI] [PubMed] [Google Scholar]
- 13.Dando T andPlosker G. Adefovir dipivoxil: a review of its use in chronic hepatitis B. Drugs 2003; 63: 2215–2234. [DOI] [PubMed] [Google Scholar]
- 14.De Clercq E. Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem Pharmacol 2007; 73: 911–922. [DOI] [PubMed] [Google Scholar]
- 15.Ray AS Fordyce MW andHitchcock MJ. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Res 2016; 125: 63–70. [DOI] [PubMed] [Google Scholar]
- 16.De Clercq E. Selective anti-herpesvirus agents. Antivir Chem Chemother 2013; 23: 93–101. [DOI] [PubMed] [Google Scholar]
- 17.Deval J. Antimicrobial strategies: inhibition of viral polymerases by 3'-hydroxyl nucleosides. Drugs 2009; 69: 151–166. [DOI] [PubMed] [Google Scholar]
- 18.Fung HB Stone EA andPiacenti FJ. Tenofovir disoproxil fumarate: a nucleotide reverse transcriptase inhibitor for the treatment of HIV infection. Clin Ther 2002; 24: 1515–1548. [DOI] [PubMed] [Google Scholar]
- 19.Seley KL Zhang L andHagos A. “Fleximers”. Design and synthesis of two novel split nucleosides. Org Lett 2001; 3: 3209–3210. [DOI] [PubMed] [Google Scholar]
- 20.Seley KL, Zhang L, Hagos A, et al. “Fleximers”. Design and synthesis of a new class of novel shape-modified nucleosides. J Org Chem 2002; 67: 3365–3373. [DOI] [PubMed] [Google Scholar]
- 21.Seley KL Salim S andZhang L. “Molecular chameleons”. Design and synthesis of C-4-substituted imidazole fleximers. Org Lett 2005; 7: 63–66. [DOI] [PubMed] [Google Scholar]
- 22.Leonard NJ Sprecker MA andMorrice AG. Defined dimensional changes in enzyme substrates and cofactors. Synthesis of lin-benzoadenosine and enzymatic evaluation of derivatives of the benzopurines. J Am Chem Soc 1976; 98: 3987–3994. [DOI] [PubMed] [Google Scholar]
- 23.Wauchope OR Velasquez M andSeley-Radtke K. Synthetic Routes to a Series of Proximal and Distal 2'-Deoxy Fleximers. Synthesis (Stuttg) 2012; 44: 3496–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wauchope OR, Johnson C, Krishnamoorthy P, et al. Synthesis and biological evaluation of a series of thieno-expanded tricyclic purine 2'-deoxy nucleoside analogues. Bioorg Med Chem 2012; 20: 3009–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Jochmans D, Ku T, et al. Bicyclic and Tricyclic “Expanded” Nucleobase Analogues of Sofosbuvir: New Scaffolds for Hepatitis C Therapies. ACS Infect Dis 2015; 1: 357–366. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z Ku TC andSeley-Radtke KL. Thiophene-expanded guanosine analogues of Gemcitabine. Bioorg Med Chem Lett 2015; 25: 4274–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmermann SC, Sadler JM, O'Daniel PI, et al. “Reverse” carbocyclic fleximers: synthesis of a new class of adenosine deaminase inhibitors. Nucleosides Nucleotides Nucleic Acids 2013; 32: 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann SC, O'Neill E, Ebiloma GU, et al. Design and synthesis of a series of truncated neplanocin fleximers. Molecules 2014; 19: 21200–21214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters HL, Jochmans D, de Wilde AH, et al. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg Med Chem Lett 2015; 25: 2923–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yates MK, Raje MR, Chatterjee P, et al. Flex-nucleoside analogues - Novel therapeutics against filoviruses. Bioorg Med Chem Lett 2017; 27: 2800–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang PK, Gordon RK, Tal J, et al. S-Adenosylmethionine and methylation. Faseb J 1996; 10: 471–480. [PubMed] [Google Scholar]
- 32.Hausmann S Vivarès CP andShuman S. Characterization of the mRNA capping apparatus of the microsporidian parasite Encephalitozoon cuniculi. J Biol Chem 2002; 277: 96–103. [DOI] [PubMed] [Google Scholar]
- 33.Fabrega C, Hausmann S, Shen V, et al. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell 2004; 13: 77–89. [DOI] [PubMed] [Google Scholar]
- 34.Bujnicki JM, Prigge ST, Caridha D, et al. Structure, evolution, and inhibitor interaction of S-adenosyl-L-homocysteine hydrolase from Plasmodium falciparum. Proteins 2003; 52: 624–632. [DOI] [PubMed] [Google Scholar]
- 35.Seley KL, Quirk S, Salim S, et al. Unexpected inhibition of S-adenosyl-L-homocysteine hydrolase by a guanosine nucleoside. Bioorg Med Chem Lett 2003; 13: 1985–1988. [DOI] [PubMed] [Google Scholar]
- 36.Blackburn GM. Nucleic acids in chemistry and biology. Royal Society of Chemistry: Cambridge, UK, 2006. [Google Scholar]
- 37.Polak M Seley KL andPlavec J. Conformational properties of shape modified nucleosides–fleximers. J Am Chem Soc 2004; 126: 8159–8166. [DOI] [PubMed] [Google Scholar]
- 38.Pastuszak I, Ketchum C, Hermanson G, et al. GDP-L-fucose pyrophosphorylase. Purification, cDNA cloning, and properties of the enzyme. J Biol Chem 1998; 273: 30165–30174. [DOI] [PubMed] [Google Scholar]
- 39.Quirk S andSeley KL. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry 2005; 44: 10854–10863. [DOI] [PubMed] [Google Scholar]
- 40.Quirk S andSeley KL. Identification of catalytic amino acids in the human GTP fucose pyrophosphorylase active site. Biochemistry 2005; 44: 13172–13178. [DOI] [PubMed] [Google Scholar]
- 41.Mair G Ullu E andTschudi C. Cotranscriptional cap 4 formation on the Trypanosoma brucei spliced leader RNA. J Biol Chem 2000; 275: 28994–28999. [DOI] [PubMed] [Google Scholar]
- 42.Hausmann S, Zheng S, Fabrega C, et al. Encephalitozoon cuniculi mRNA cap (guanine N-7) methyltransferase: methyl acceptor specificity, inhibition BY S-adenosylmethionine analogs, and structure-guided mutational analysis. J Biol Chem 2005; 280: 20404–20412. [DOI] [PubMed] [Google Scholar]
- 43.De Clercq E. S-adenosylhomocysteine hydrolase inhibitors as broad-spectrum antiviral agents. Biochem Pharmacol 1987; 36: 2567–2575. [DOI] [PubMed] [Google Scholar]
- 44.De Clercq E. Carbocyclic adenosine analogues as S-adenosylhomocysteine hydrolase inhibitors and antiviral agents: recent advances. Nucleosides Nucleotides 1998; 17: 625–634. [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann SC, Sadler JM, Andrei G, et al. Carbocyclic 5'-nor “reverse” fleximers. Design, synthesis, and preliminary biological activity. Med Chem Commun 2011; 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wigerinck P, Snoeck R, Claes P, et al. Synthesis and antiviral activity of 5-heteroaryl-substituted 2'-deoxyuridines. J Med Chem 1991; 34: 1767–1772. [DOI] [PubMed] [Google Scholar]
- 47.Wigerinck P, Pannecouque C, Snoeck R, et al. 5-(5-Bromothien-2-yl)-2'-deoxyuridine and 5-(5-chlorothien-2-yl)-2'-deoxyuridine are equipotent to (E)-5-(2-bromovinyl)-2'-deoxyuridine in the inhibition of herpes simplex virus type I replication. J Med Chem 1991; 34: 2383–2389. [DOI] [PubMed] [Google Scholar]
- 48.Wigerinck P, Kerremans L, Claes P, et al. Synthesis and antiviral activity of 5-thien-2-yl-2'-deoxyuridine analogues. J Med Chem 1993; 36: 538–543. [DOI] [PubMed] [Google Scholar]
- 49.Hasobe M, McKee JG, Borcherding DR, et al. 9-(trans-2',trans-3'-Dihydroxycyclopent-4'-enyl)-adenine and -3-deazaadenine: analogs of neplanocin A which retain potent antiviral activity but exhibit reduced cytotoxicity. Antimicrob Agents Chemother 1987; 31: 1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bray M, Raymond JL, Geisbert T, et al. 3-deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res 2002; 55: 151–159. [DOI] [PubMed] [Google Scholar]
- 51.De Clercq E. Ebola virus (EBOV) infection: Therapeutic strategies. Biochem Pharmacol 2015; 93: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bray M Driscoll J andHuggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res 2000; 45: 135–147. [DOI] [PubMed] [Google Scholar]
- 53.Stedman C. Sofosbuvir, a NS5B polymerase inhibitor in the treatment of hepatitis C: a review of its clinical potential. Therap Adv Gastroenterol 2014; 7: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lalezari J, Asmuth D, Casiró A, et al. Short-term monotherapy with IDX184, a liver-targeted nucleotide polymerase inhibitor, in patients with chronic hepatitis C virus infection. Antimicrob Agents Chemother 2012; 56: 6372–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGuigan C, Cahard D, Sheeka HM, et al. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J Med Chem 1996; 39: 1748–1753. [DOI] [PubMed] [Google Scholar]
- 56.McGuigan C, Hassan-Abdallah A, Srinivasan S, et al. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J Med Chem 2006; 49: 7215–7226. [DOI] [PubMed] [Google Scholar]
- 57.Elion GB. Acyclovir: discovery, mechanism of action, and selectivity. J Med Virol 1993; Suppl 1: 2–6. [DOI] [PubMed] [Google Scholar]
- 58.Reardon JE andSpector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J Biol Chem 1989. 264: 7405–7411. [PubMed] [Google Scholar]
- 59.McMahon MA, Siliciano JD, Lai J, et al. The antiherpetic drug acyclovir inhibits HIV replication and selects the V75I reverse transcriptase multidrug resistance mutation. J Biol Chem 2008; 283: 31289–31293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subissi L, Posthuma CC, Collet A, et al. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc Natl Acad Sci U S A 2014; 111: E3900–E3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Y, Wu L, Shaw N, et al. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc Natl Acad Sci U S A 2015; 112: 9436–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vichier-Guerre S, Dugué L, Bonhomme F, et al. Expedient and generic synthesis of imidazole nucleosides by enzymatic transglycosylation. Org Biomol Chem 2016; 14: 3638–3653. [DOI] [PubMed] [Google Scholar]
- 63.St Amant AH, Bean LA, Guthrie JP, et al. Click fleximers: a modular approach to purine base-expanded ribonucleoside analogues. Org Biomol Chem 2012; 10: 6521–6525. [DOI] [PubMed] [Google Scholar]
- 64.Bardon AB andWetmore SD. How flexible are fleximer nucleobases? A computational study. J Phys Chem A 2005; 109: 262–272. [DOI] [PubMed] [Google Scholar]
- 65.Koszytkowska-Stawinska M andSas W. Synthesis of novel NH-1,2,3-triazolo-nucleosides by the Banert cascade reaction. Tetrahedron 2013; 69: 2619–2627. [Google Scholar]
- 66.Nguyen Van T, Hospital A, Lionne C, et al. Beta-hydroxyphosphonate ribonucleoside analogues derived from 4-substituted-1,2,3-triazoles as IMP/GMP mimics: synthesis and biological evaluation. Beilstein J Org Chem 2016; 12: 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowak I Cannon JF andRobins MJ. N-oxides of adenosine-type nucleosides undergo pyrimidine ring opening and closure to give 5-amino-4-(1,2,4-oxadiazol-3-yl)imidazole derivatives. Org Lett 2006; 8: 4565–4568. [DOI] [PubMed] [Google Scholar]
- 68.Read ML, Braendvang M, Miranda PO, et al. Synthesis and biological evaluation of pyrimidine analogs of antimycobacterial purines. Bioorg Med Chem 2010; 18: 3885–3897. [DOI] [PubMed] [Google Scholar]
- 69.Bakkestuen AK, Gundersen L-L, Langli G, et al. 9-Benzylpurines with inhibitory activity against Mycobacterium tuberculosis. Bioorg Med Chem Lett 2000; 10: 1207–1210. [DOI] [PubMed] [Google Scholar]
- 70.Andresen G, Gundersen L-L, Nissen-Meyer J, et al. Cytotoxic and Antibacterial Activity of 2-Oxopurine Derivatives. Bioorg Med Chem Lett 2002; 12: 567–569. [DOI] [PubMed] [Google Scholar]