

Abstract
Menkes disease (MD) is a rare infantile onset neurodegenerative disorder due to mutations in the X linked ATP7A gene. These patients can present with failure to thrive, severe psychomotor retardation, seizures and hypopigmented hair, which is characteristic of this condition. A number of neuro-radiological findings have been reported in this condition. We report the spectrum of neuro-radiological findings in three affected boys being treated at our centre. We suggest that magnetic resonance imaging (MRI) and, in particular magnetic resonance angiography (MRA) when taken in the context of the clinical presentation may be helpful in making an early diagnosis of this devastating condition.
Keywords: Magnetic resonance angiography, Menkes disease, neuroradiology
INTRODUCTION
Menkes disease (MD) is a rare infantile-onset neurodegenerative disorder caused by mutations in the X-linked ATP7A gene.[1] This gene encodes a copper-transporting ATPase, which has the dual role of ATP-driven copper efflux from cells and the intracellular transport of copper to the copper-requiring enzymes.[2] These cuproenzymes catalyze a diverse range of essential biochemical reactions.[3] Affected individuals have unusually low copper levels in serum, brain, hair, and skin. The clinical features of this condition, namely failure to thrive, severe psychomotor retardation, seizures, and the characteristic hypopigmented “kinky” hair, occur as a consequence of dysfunction of these enzyme systems.[2,4] The prognosis is poor with relentless neurological deterioration and death within the first few years for most patients.
A number of neuroradiological findings have been reported in this condition. These include cerebral and cerebellar atrophy, delayed myelination, tortuosity of the intra- and extracranial vessels, involvement of the deep gray nuclei, white matter lesions of the temporal lobe, and subdural fluid collections.[5,6,7,8,9]
We describe the spectrum of neuroradiological findings in three affected boys with MD, being treated at our center.
PATIENTS AND METHODS
We reviewed patients with a diagnosis of MD who presented to our tertiary neurology center. All three boys were born at term and in each case pregnancy, labor, and the neonatal period were unremarkable. The patients underwent cerebral magnetic resonance imaging (MRI) and magnetic resonance angiogram (MRA).
RESULTS
Patient 1
A 12-month-old infant of mixed African Caribbean/white origin presented with developmental delay, generalized convulsive seizures, and recurrent urinary tract infections. Developmental concerns were first noted at 4 months of age. He was hypotonic on examination and had thin, sparse, hypopigmented hair.
The serum copper value was <4.1 μmol/L (reference range, 11–22 μmol/L) and ceruloplasmin value was <0.1 μmol/L (reference range, 1.2–2.4 μmol/L). The diagnosis of MD was confirmed through molecular genetic studies. Cerebral MRA and MRI demonstrated excessive cerebral vessel tortuosity, generalized cerebral and cerebellar atrophy, corpus callosum atrophy, and increased extra-axial spaces [Figure 1]. Copper-replacement therapy was commenced with copper histidine starting at 50 μg/kg/day in two divided doses, checking copper levels a week to 10 days later, and altering doses, thereafter, depending on the response. The maximum total dose used was 1 mg/day. There was slight improvement in his neurological status. From the age of 3 years until his death at the age of 6 years, he had recurrent severe upper gastrointestinal hemorrhages secondary to gastric and small bowel ulcers. He developed other complications in the form of bony abnormalities and bladder diverticula.
Figure 1.
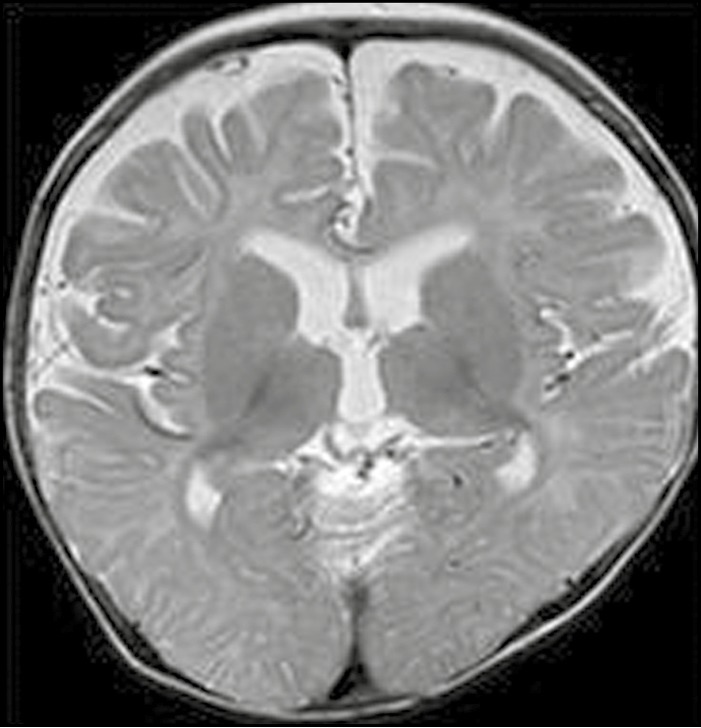
Axial T2-weighted image from an 11-month-old boy illustrating cerebral atrophy with expansion of the extra-axial CSF spaces and ventricles. Cerebellar atrophy (not shown) was also present
Patient 2
A 6-month-old infant was referred for investigation of extreme hypotonia, developmental delay, and generalized convulsive seizures. He had previously been treated for unilateral hip dislocation. On examination, the child had cherubic features, coarse hair, and extreme hypotonia.
Electron microscopy of the hair showed changes consistent with pili torti. Serum copper value was <2 μmol/L, ceruloplasmin value was 0.07 μmol/L, and plasma and cerebrospinal fluid (CSF) lactate levels were elevated (4.1 and 2.8 mmol/L). The diagnosis was confirmed through molecular genetic studies, which revealed a mutation variant in intron 9 in ATP7A gene. MRI and MRA revealed bilateral temporal lobe swelling, which was more marked on the right, bilateral basal ganglia lesions, demyelination, and mild vessel tortuosity [Figures 2 and 3].
Figure 2.
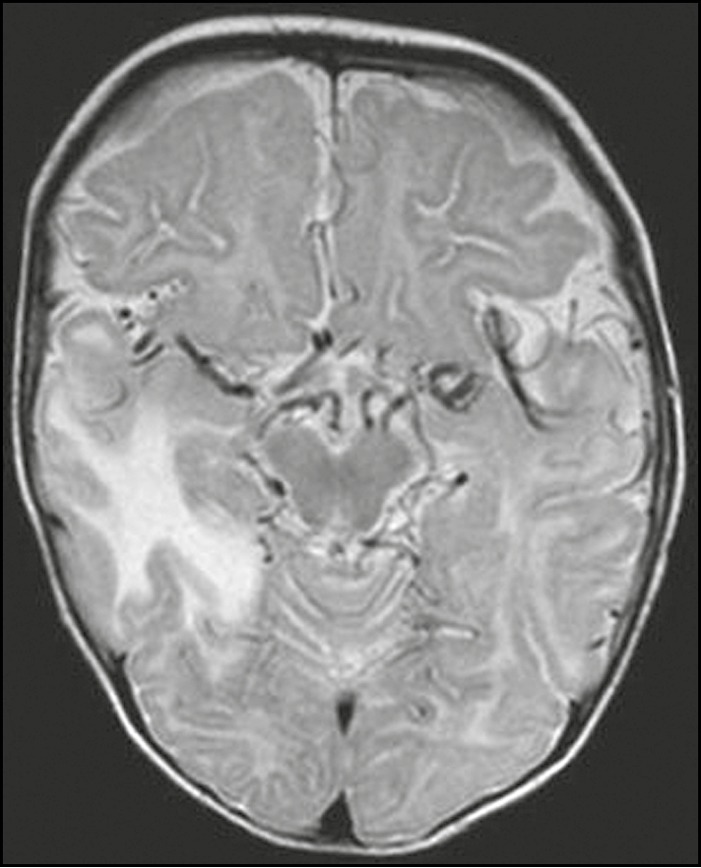
Axial T2-weighted image highlighting marked tortuosity of vessels within the basal cisterns. The child also had swollen right temporal lobe, a recognized feature of Menkes disease
Figure 3.
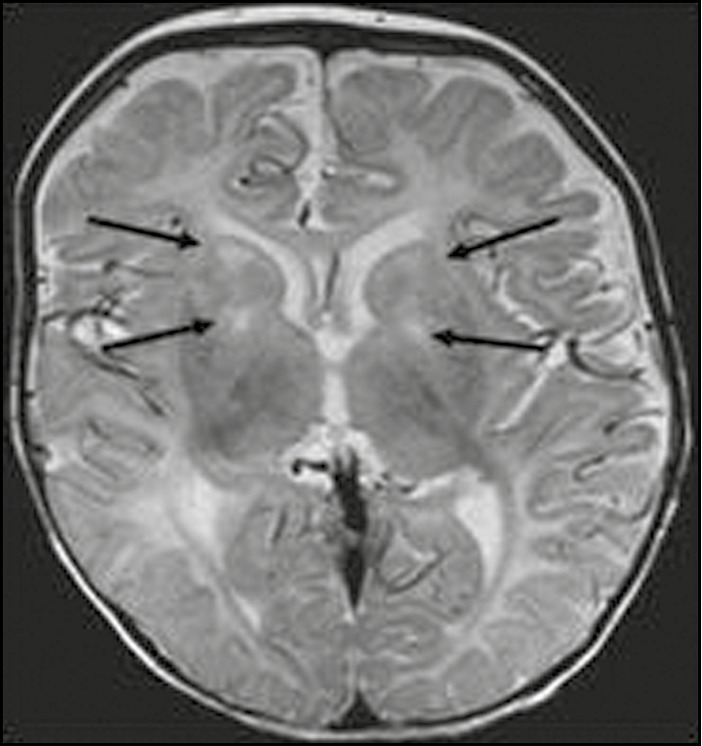
Axial T2-weighted image from a 7-month-old boy. Abnormal hyperintense signal (as denoted by arrows) in bilateral basal ganglia (corpus striatum and globus pallidus)
He was treated with copper soon after presentation, and this was associated with normalization of serum and CSF lactate levels. However, he made less neurodevelopmental progress. He had frequent daily seizures despite being administered with three anticonvulsants. At the age of 3 years, he developed recurrent episodes of urinary retention and was found to have bladder diverticula.
Patient 3
A 16-month-old male infant was admitted with severe developmental delay and a history of infantile spasms since 3 months of age. He was markedly hypotonic with abnormally coarse hypopigmented curly hair. Serum copper and ceruloplasmin values were 2.5 μmol/L and 6.05 μ/L, respectively. Electron microscopy of the hair showed typical changes of pili torti. MRI and MRA at 14 months of age showed excessive vessel tortuosity with cerebellar atrophy and some periventricular deep white matter lesions [Figures 4 and 5]. He remained stable on two anticonvulsants. Developmental progress was limited. He developed bladder diverticula at 2 years of age. He is currently 4 years old and has recurrent admissions for lung aspiration and seizures.
Figure 4.
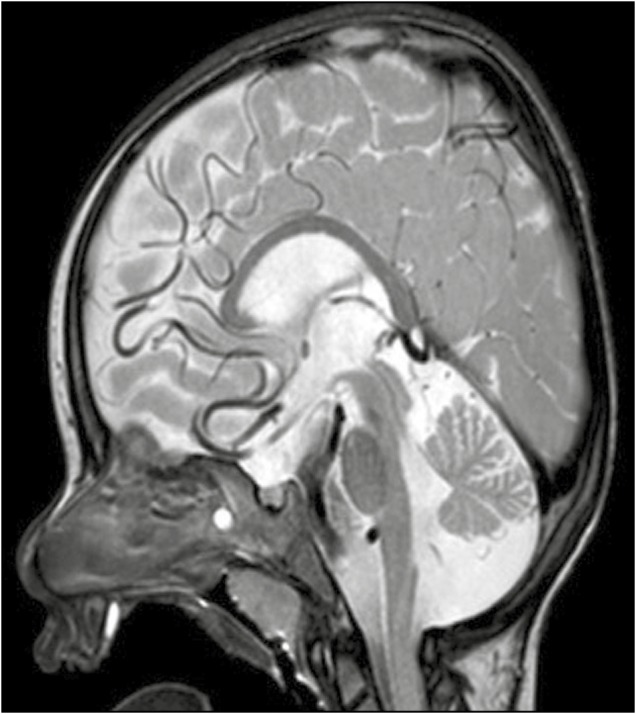
Midline sagittal T2-weighted image from a 14 month old boy with tortuosity of the anterior cerebral arteries and cerebellar atrophy
Figure 5.
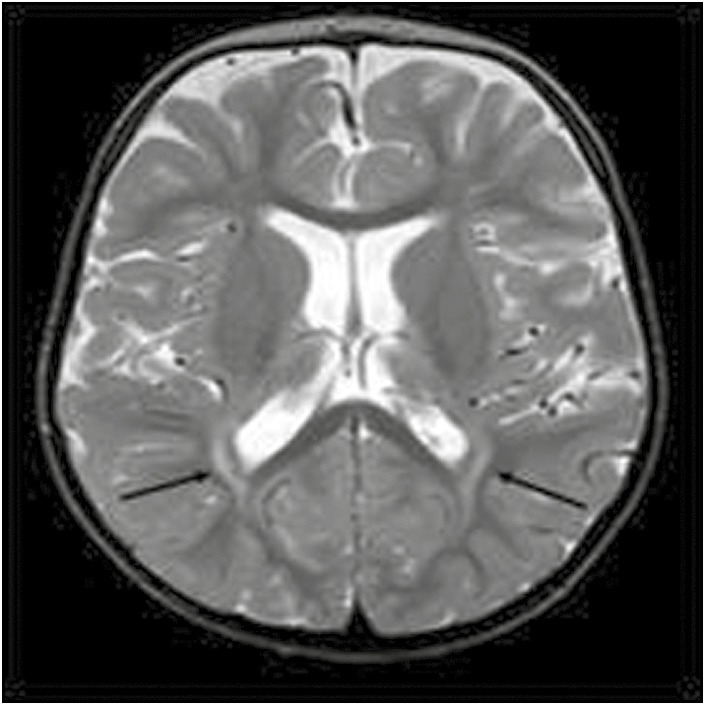
Axial T2-weighted image showing abnormal signal in the deep parieto-occipital white matter bilaterally (as denoted by arrows)
DISCUSSION
The incidence of MD is estimated to be between 1 in 40,000 and 250,000 live births with up to a third of cases arising as a result of de novo mutations.[2,4,10] Infants commonly present at 2–3 months of age with severe hypotonia, developmental regression, failure to thrive, and seizures. The typical coarse “steely” hair, which appears normal at birth, is often the first feature to suggest the diagnosis. Electron microscopy of hair reveals twisting of the hair shaft (pili torti), narrowing, and fragmentation. Bladder diverticula and bony abnormalities resembling scurvy are additional features that are seen later.[4]
Significant secondary mitochondrial dysfunction is also noted to be common in untreated individuals. This was exemplified in two of our patients who had elevated serum lactate values (and in one child with raised CSF lactate) at presentation, which subsequently, normalized following correction of serum copper levels in response to replacement therapy.
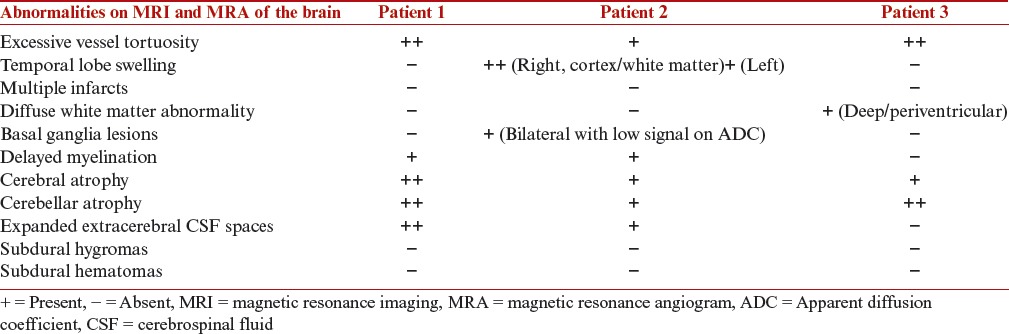
The neuroradiological changes in MD have been well described. Our patients demonstrated the whole spectrum of changes with the exception of subdural collections. Tortuosity of intracranial vessels, and cerebral and cerebellar atrophy were consistent features seen in each child. Table 1 gives an overview of the MRI and MRA findings in patients with MD.
Table 1.
MRI and MRA findings in patients with Menkes disease
Takahashi et al. [7] first described the use of MRA in MD to demonstrate the cerebral vascular abnormalities in the form of excessive coiling and tortuosity. These findings have been reported in almost all patients with MD in the literature and may be visible as early as 5 weeks of age, often predating the appearance of cerebral atrophy.[11,12] Husain et al. [13] found that MD was the condition with the highest incidence of MRA abnormalities and concluded that this was where MRA was most likely to provide additional valuable information.
One child (patient 2) had involvement of the temporal lobes. Some authors[2,14] noted that the temporal lobe changes in patients with MD are reminiscent of stroke-like lesions seen in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), a mitochondrial cytopathy. Others have reported the association between temporal lobe changes and seizures.[15] The temporal lobe abnormalities in MD would seem to represent a combination of vasogenic and cytotoxic edema arising from several factors, namely mitochondrial dysfunction, impaired blood flow through distorted vessels, and increased metabolic demand as a result of seizure activity.
Patient 2 was the only child to display basal ganglia lesions. His pretreatment CSF and serum lactate values were elevated and this, in conjunction with deep gray matter involvement, initially suggested the diagnosis of a mitochondrial cytopathy. Basal ganglia lesions are associated with restricted diffusion on diffusion-weighted MRI, decreased apparent diffusion coefficient (ADC) values, and a lactate peak on magnetic resonance spectroscopy (MRS) in keeping with cytotoxic edema, also a feature of the mitochondrial cytopathies.[16]
The natural history of MD is progressive and extensive degeneration of gray matter with resultant neuronal loss, and gliosis visible radiologically as cerebral and cerebellar atrophy and subdural collections represent the end stage of these processes.[11] Treatment with subcutaneous copper supplementation is available, but for maximum efficacy, this needs to be started as soon as possible, ideally during the neonatal period. Unfortunately, even when instituted in this time frame, therapy is not universally successful because those patients with severe mutations lack sufficient functional protein to transport copper across the blood–brain barrier.[4]
Early diagnosis of MD can be challenging, as there is an overlap between the normally low copper values of healthy neonates and those affected with the disorder.[4] Genetic testing, although available, may be time-consuming, if there is no affected family member as a result of the large size of the ATP7A gene, where over 350 mutations have been reported to date.[10] However, affected infants do have a specific serum and urine catecholamine profile, which is present from early life and shows promise as an early screening tool in the presymptomatic stages of the disease.[4]
CONCLUSION
To summarize the findings from our case series, MRI changes in MD include excessive tortuosity of intracerebral vessels, cerebral and cerebellar atrophy, temporal lobe changes, delayed myelination, and expanded extracerebral CSF spaces. MRA is a valuable adjunct to MRI in the delineation of vessel tortuosity, which is the hallmark of this condition.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
REFERENCES
- 1.Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 2.Kodama H, Fujisawa C, Bhadhprasit W. Pathology, clinical features and treatments of congenital copper metabolic disorders—Focus on neurologic aspects. Brain Dev. 2011;33:243–51. doi: 10.1016/j.braindev.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Lutsenko S, Gupta A, Burkhead JL, Zuzel V. Cellular multitasking: The dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Biochem Biophys. 2008;476:22–32. doi: 10.1016/j.abb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605–14. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faerber EN, Grover WD, DeFilipp GJ, Capitanio MA, Liu TH, Swartz JD. Cerebral MR of Menkes kinky-hair disease. AJNR Am J Neuroradiol. 1989;10:190–2. [PMC free article] [PubMed] [Google Scholar]
- 6.Ichihashi K, Yano S, Kobayashi S, Miyao M, Yanagisawa M. Serial imaging of Menkes disease. Neuroradiology. 1990;32:56–9. doi: 10.1007/BF00593944. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi S, Ishii K, Matsumoto K, Higano S, Ishibashi T, Zuguchi M, et al. Cranial MRI and MR angiography in Menkes’ syndrome. Neuroradiology. 1993;35:556–8. doi: 10.1007/BF00588724. [DOI] [PubMed] [Google Scholar]
- 8.Ozawa H, Kodama H, Murata Y, Takashima S, Noma S. Transient temporal lobe changes and a novel mutation in a patient with Menkes disease. Pediatr Int. 2001;43:437–40. doi: 10.1046/j.1442-200x.2001.01402.x. [DOI] [PubMed] [Google Scholar]
- 9.Ito H, Mori K, Sakata M, Naito E, Harada M, Kagami S. Transient left temporal lobe lesion in Menkes disease may influence the generation of tonic spasms. Brain Dev. 2011;33:345–8. doi: 10.1016/j.braindev.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Møller LB, Mogensen M, Horn N. Molecular diagnosis of Menkes disease: Genotype-phenotype correlation. Biochimie. 2009;91:1273–7. doi: 10.1016/j.biochi.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Leventer RJ, Kornberg AJ, Phelan EM, Kean MJ. Early magnetic resonance imaging findings in Menkes’ disease. J Child Neurol. 1997;12:222–4. doi: 10.1177/088307389701200314. [DOI] [PubMed] [Google Scholar]
- 12.Cosimo QC, Daniela L, Elsa B, Carlo DV, Giuseppe F. Kinky hair, kinky vessels, and bladder diverticula in Menkes disease. J Neuroimaging. 2011;21:e114–6. doi: 10.1111/j.1552-6569.2010.00476.x. [DOI] [PubMed] [Google Scholar]
- 13.Husain AM, Smergel E, Legido A, Faerber EN, Foley CM, Miles DK, et al. Comparison of MRI and MRA findings in children with a variety of neurologic conditions. Pediatr Neurol. 2000;23:307–11. doi: 10.1016/s0887-8994(00)00187-9. [DOI] [PubMed] [Google Scholar]
- 14.Ito H, Mori K, Sakata M, Naito E, Harada M, Minato M, et al. Pathophysiology of the transient temporal lobe lesion in a patient with Menkes disease. Pediatr Int. 2008;50:825–7. doi: 10.1111/j.1442-200X.2008.02750.x. [DOI] [PubMed] [Google Scholar]
- 15.Ekici B, Calışkan M, Tatlı B. Reversible temporal lobe edema: An early MRI finding in Menkes disease. J Pediatr Neurosci. 2012;7:160–1. doi: 10.4103/1817-1745.102597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnerias C, Boddaert N, Guiraud P, Desguerre I, Hertz Pannier L, Dulac O, et al. Unusual magnetic resonance imaging features in Menkes disease. Brain Dev. 2008;30:489–92. doi: 10.1016/j.braindev.2007.12.014. [DOI] [PubMed] [Google Scholar]