

Short abstract
Nucleoside analogs represent the largest class of small molecule-based antivirals, which currently form the backbone of chemotherapy of chronic infections caused by HIV, hepatitis B or C viruses, and herpes viruses. High antiviral potency and favorable pharmacokinetics parameters make some nucleoside analogs suitable also for the treatment of acute infections caused by other medically important RNA and DNA viruses. This review summarizes available information on antiviral research of nucleoside analogs against arthropod-borne members of the genus Flavivirus within the family Flaviviridae, being primarily focused on description of nucleoside inhibitors of flaviviral RNA-dependent RNA polymerase, methyltransferase, and helicase/NTPase. Inhibitors of intracellular nucleoside synthesis and newly discovered nucleoside derivatives with high antiflavivirus potency, whose modes of action are currently not completely understood, have drawn attention. Moreover, this review highlights important challenges and complications in nucleoside analog development and suggests possible strategies to overcome these limitations.
Keywords: Nucleoside analog, antiviral agent, arthropod-borne flavivirus, antiviral therapy, inhibitor
Introduction
The genus Flavivirus belongs to the Flaviviridae family and includes more than 70 single-stranded plus-sense RNA viral species. Flaviviruses of human medical importance are tick- or mosquito-transmitted viruses with typical representatives being tick-borne encephalitis virus (TBEV), Omsk hemorrhagic fever virus (OHFV), Kyasanur Forest disease virus (KFDV), Alkhurma hemorrhagic fever virus (AHFV), Powassan virus (POWV), West Nile virus (WNV), dengue virus (DENV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), or Zika virus (ZIKV).1,2 The Flaviviridae family also includes some less known or neglected viruses, such as louping ill virus (LIV), Usutu virus, Langat virus, or Wesselsbron virus.3–6 The flaviviral genome is a single-stranded, plus-sense RNA of about 11 kb in length that encodes a single polyprotein, which is co- and posttranslationally processed into three structural (capsid, premembrane or membrane, and envelope) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5).7 Both NS3 and NS5 proteins possess enzymatic activities reported to be important targets for antiviral development. Whereas NS3 acts as a serine protease, a 5′-RNA triphosphatase, a nucleoside triphosphatase (NTPase), and a helicase,8,9 NS5 consists of a complex containing the RNA-dependent RNA polymerase (RdRp) and the methyltransferase (MTase) activities.10,11
Flaviviral infections are accompanied by a wide spectrum of distinct clinical manifestations, ranging from relatively mild fevers and arthralgia to severe viscerotropic symptoms (YFV and DENV), hemorrhagic fevers (KFDV and OHFV), encephalitis/myelitis (JEV, WNV, and TBEV), and neuropathic or teratogenic manifestations (ZIKV). More than 200 million clinical cases of flaviviral infections, including numerous deaths, are reported annually worldwide.12 Currently no specific antiviral therapies are available to treat patients with flaviviral infections, thus the search for safe and effective small-molecule inhibitors that would be active against these viruses represents a high research priority.13
Nucleoside analog inhibitors have figured prominently in the search for effective antiviral agents.14 Nucleoside analogs are synthetic, chemically modified nucleosides that mimic their physiological counterparts (endogenous nucleosides) and block cellular division or viral replication by impairment DNA/RNA synthesis or by inhibition of cellular or viral enzymes involved in nucleoside/tide metabolism (Figure 1).15 The first antiviral analogs were developed in the late 1960s and currently there are over 25 approved therapeutic nucleosides used for the therapy of viral infections of high medical importance, such as HIV/AIDS (tenofovir),16,17 hepatitis B (lamivudine/entecavir),18,19 hepatitis C (sofosbuvir),20 or herpes infections (acyclovir).21 So far, numerous nucleoside analogs have been described to inhibit arthropod-transmitted flaviviruses. Since these viruses are closely related to the hepatitis C virus (HCV), for which many potent inhibitors are being currently developed, anti-HCV nucleoside analogs represent promising tools to be repurposed against other viruses within the Flaviviridae family.12
Figure 1.

Intracellular uptake and metabolism of nucleoside analogs and nucleoside analog prodrugs. Nucleoside analogs enter cells through specific plasma membrane nucleoside transporters. Inside the cell, the compounds are phosphorylated by cellular nucleoside kinases resulting in formation of nucleoside mono-, di-, and triphosphates. The first kinase phosphorylation is the rate-limiting step of the triphosphate conversion, which can be overcome by the monophosphate prodrug approach based on the introduction of a phosphorylated group into the 5′ nucleoside position. The phosphorylated group includes protecting moieties to increase hydrophobicity and facilitate the cellular uptake of the prodrug. Monophosphate prodrugs enter cells independently of membrane transporters and the protecting groups are removed by intracellular esterases or phosphoramidases after cell penetration. The triphosphates of nucleoside species represent the active forms of nucleoside analogs that act by inhibiting cellular or viral enzymes, such as DNA/RNA polymerases. During DNA/RNA replication, nucleoside analogs are incorporated into nascent DNA or RNA chains resulting in termination of nucleic acid synthesis or in accumulation of mutations in viral genomes to suppress viral replication due to error catastrophe. At normal physiological conditions, intracellular nucleoside concentrations are maintained at low levels due to nucleoside/nucleotide catabolic pathways, such as deamination (oxidation) of heterocyclic base, hydrolysis or phosphorolysis of heterocyclic base, and hydrolysis of phosphomonoester bonds. These catabolic reactions also concern most nucleoside analogs containing the natural N-glycosidic bond and/or the degradable functional groups of the heterocyclic base. Figure created using Servier Medical Art available on www.servier.com.
The aim of this review is to provide an overview of known antiviral agents targeting selected arthropod-borne flaviviruses and to discuss their characteristic properties, modes of action, and advantages or limitations of their therapeutic use. Moreover, the important challenges and complications in antiflavivirus nucleoside analog development are highlighted and possible strategies to overcome their shortcomings are suggested.
Major classes of antiflavivirus nucleosides
Nucleoside analogs active against arthropod-borne flaviviruses are usually classified according to their targets in the viral life cycle. Such antiviral molecules act as inhibitors of flaviviral RdRp,22–26 MTase,27,28 and helicase/NTPase.29,30 Other nucleoside scaffolds suppress host cell enzymes involved particularly in nucleoside biosynthetic pathways31–34 or in some cases, exhibit multiple modes of action.32,35 In vitro antiviral effects and cytotoxicity profiles of the most important antiflavivirus nucleoside analogs are summarized in Table 1. An overview of in vivo antiflaviviral activities of selected nucleosides as evaluated in different animal models is presented in Table 2.
Table 1.
In vitro antiviral activities and cytotoxicity profiles of selected nucleoside inhibitors of flaviviral replication.
| Structure | Virus | Strain | Cell line | Assay | EC50 (µM) | CC50 (µM) | References |
|---|---|---|---|---|---|---|---|
| Inhibitors of flaviviral RdRp | |||||||
GS-441524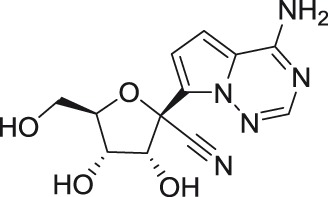
|
AHFV | 200300001 | Vero | CPE | 49.9 | ND | Lo et al.45 |
| KFDV | P9605 | Vero | CPE | 46.3 | ND | Lo et al.45 | |
| TBEV | Hypr | Vero | CPE | 51.2 | ND | Lo et al.45 | |
| OHFV | Bogoluvovska | Vero | CPE | 50.6 | ND | Lo et al.45 | |
| YFV | ND | ND | Cell based | 11 | >30 | Cho et al.43 | |
| DENV-2 | ND | ND | Cell based | 9.46 | >30 | Cho et al.43 | |
| WNV | ND | ND | Cell based | >30 | >30 | Cho et al.43 | |
GS-5734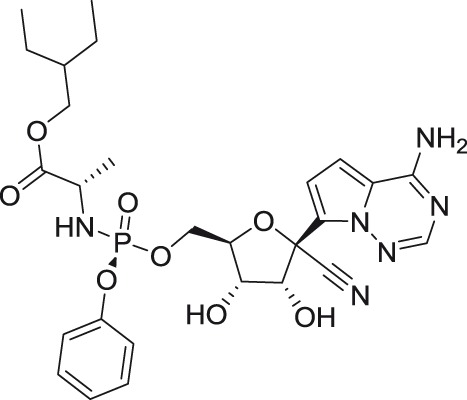
|
AHFV | 200300001 | Vero | CPE | 4.2 | ND | Lo et al.45 |
| KFDV | P9605 | Vero | CPE | 1.8 | ND | Lo et al.45 | |
| TBEV | Hypr | Vero | CPE | 2.1 | ND | Lo et al.45 | |
| OHFV | Bogoluvovska | Vero | CPE | 1.2 | ND | Lo et al.45 | |
2′-C-methyladenosine
|
TBEV | Hypr | PS | VTR | 1.4 | >50 | Eyer et al.54 |
| WNV | New York isol. | Vero | CPE | 5.1 | 25 | Migliaccio et al.49 | |
| DENV-2 | New Guinea C | Vero | CPE | 4 | 18 | Migliaccio et al.49 | |
| YFV | 17-D | Vero | CPE | 3.2 | 13 | Migliaccio et al.49 | |
| ZIKV | MR766 | Vero | VTR | 5.26 | >100 | Eyer et al.23 | |
7-Deaza-2′-C-methyl-adenosine
|
TBEV | Hypr | PS | VTR | 1.1 | >50 | Eyer et al.54 |
| WNV | New York isol. | Vero | CPE | 4.5 | 250 | Olsen et al.50 | |
| DENV-2 | New Guinea C | Vero | CPE | 15 | >320 | Olsen et al.50 | |
| YFV | 17-D | Vero | CPE | 15 | >320 | Olsen et al.50 | |
| ZIKV | MR766 | Vero | CPE | 20 | >357 | Zmurko et al.62 | |
| ZIKV | MR766 | Vero | VYR | 9.6 | >357 | Zmurko et al.62 | |
| ZIKV | MR766 | Vero | PA | 1.3 | >357 | Zmurko et al.62 | |
| ZIKV | MR766 | Vero | IFA | 5.7 | >357 | Zmurko et al.62 | |
2′-C-methylguanosine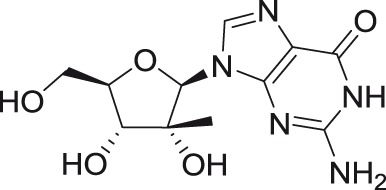
|
TBEV | Hypr | PS | VTR | 1.4 | >50 | Eyer et al.54 |
| WNV | New York isol. | Vero | CPE | 30 | >100 | Migliaccio et al.49 | |
| DENV-2 | New Guinea C | Vero | CPE | 13.6 | >60 | Migliaccio et al.49 | |
| YFV | 17-D | Vero | CPE | 17 | >50 | Migliaccio et al.49 | |
| ZIKV | MR766 | Vero | VTR | 22.25 | >100 | Eyer et al.23 | |
INX-08189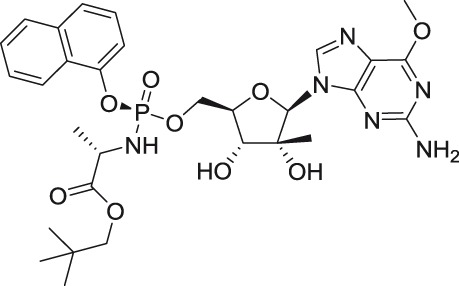
|
DENV-2 | ND | Huh-7 | Replicon | 0.0142 | >1 | Yeo et al.58 |
2′-C-methylcytidine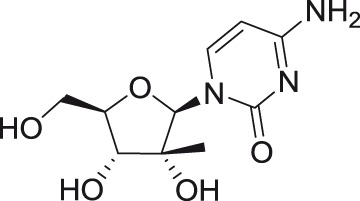
|
TBEV | Hypr | PS | VTR | 1.8 | >50 | Eyer et al.54 |
| AHFV | 200300001 | A549 | qRT-PCR | 2.5 | Flint et al.55 | ||
| AHFV | 200300001 | A549 | CPE | 15.3 | |||
| KDFV | P9605 | A549 | CPE | 7.2 | |||
| OHFV | Bogoluvovska | A549 | CPE | 3.2 | |||
| POWV | Byers | A549 | CPE | 5.5 | |||
| DENV | ND | Huh-7 | Replicon | 11.2 | – | Lee et al.56 | |
| YFV | 17-D | Vero | Visual inspection | 2.5a | 22a | Julander et al.63 | |
| YFV | 17-D | Vero | Neutral red uptake | 2.1a | 22a | Julander et al.63 | |
| YFV | 17-D | Vero | VYR | 0.7a | 22a | Julander et al.63 | |
| ZIKV | MR766 | Vero | VTR | 10.51 | >100 | Eyer et al.23 | |
2′-C-methyluridine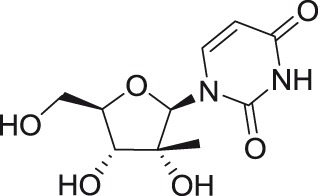
|
TBEV | Hypr | PS | VTR | 11.1 | >50 | Eyer et al.54 |
| ZIKV | MR776 | Vero | VTR | 45.45 | >100 | Eyer et al.23 | |
Sofosbuvir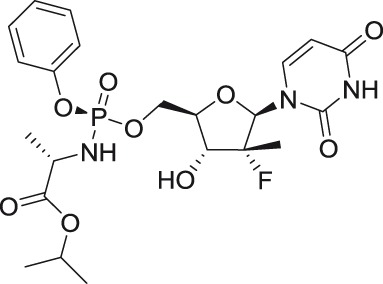
|
ZIKV | ND | BHK-21, SH-sy5y, Huh-7 | CPE | 0.12–1.9 | >300 | Sacramento et al.25 |
| ZIKV | ND | - | RdRp inhibition | 0.38 | ND | Sacramento et al.25 | |
| ZIKV ZIKV ZIKV |
PRVABC59 Paraiba Dakar 41519 |
Huh-7, Jan Huh-7, Jan Huh-7, Jan |
PA qRT-PCR |
1–5 1–5 1–5 |
>200 >200 >200 |
Bullard-Feibelman et al.69 | |
2′-C-ethynyladenosine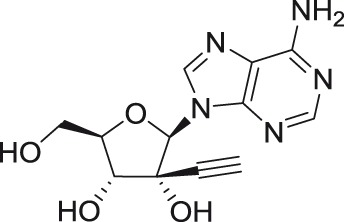
|
DENV-2 | ND | A549 | CFI | 1.41 | 40 | Chen et al.22 |
NITD008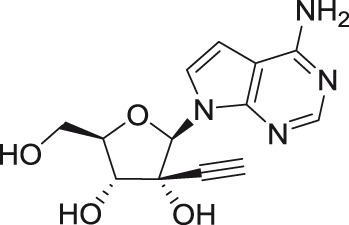
|
DENV-2 DENV-2 DENV-2 DENV-2 DENV-2 |
New Guinea C MY10245 MY10340 MY22366 MY22713 |
A549 PBMC PBMC PBMC PBMC |
CFI CFI CFI CFI CFI |
0.46–2.61 0.58 0.58 0.58 0.58 |
>100 >100 >100 >100 >100 |
Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 |
| ZIKV | GZ01/2016 | Vero | VTR | 0.241 | ND | Deng et al.76 | |
| ZIKV | GZ01/2016 | Vero | qRT-PCR | 0.137 | ND | Deng et al.76 | |
| ZIKV | FSS13025/2010 | Vero | VTR | 0.950 | ND | Deng et al.76 | |
| ZIKV | FSS13025/2010 | Vero | qRT-PCR | 0.283 | ND | Deng et al.76 | |
| TBEV | Hypr | A549 | CPE, CFI, VTR | 0.9–2.99 | >100 | Lo et al.77 | |
| AHFV | 200300001 | A549 | CPE, CFI, VTR | 1.51–9.29 | >100 | Lo et al.77 | |
| KDFV | P9605 | A549 | CPE, CFI, VTR | 1.42–4.01 | >100 | Lo et al.77 | |
| OHFV | Bogoluvovska | A549 | CPE, CFI, VTR | 0.61–3.04 | >100 | Lo et al.77 | |
NITD449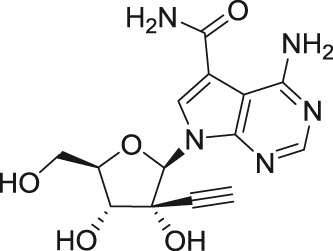
|
DENV (1–4) |
New Guinea C MY10245 MY10340 MY22366 MY22713 |
A549 | CFI CFI CFI CFI CFI |
1.62–6.99 | ND ND ND ND ND |
Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 |
| PBMC PBMC PBMC PBMC |
5 5 5 5 |
||||||
NITD203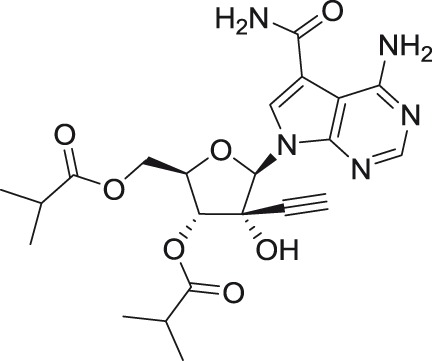
|
DENV (1–4) |
New Guinea C MY10245 MY10340 MY22366 MY22713 |
A549 | CFI CFI CFI CFI CFI |
0.54–0.71 | ND ND ND ND ND |
Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 Chen et al.72 |
| PBMC PBMC PBMC PBMC |
<0.1 <0.1 <0.1 <0.1 |
||||||
4′-C-azidocytidine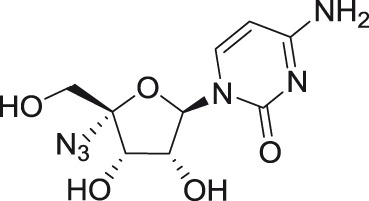
|
TBEV | Hypr | PS | VTR | 2.7 | >50 | Eyer et al.54 |
Balapiravir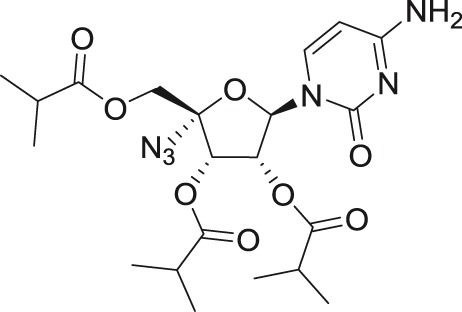
|
DENV (1–4) |
Various | DC | qRT-PCR qRT-PCR qRT-PCR |
5.2–6 | ND ND ND |
Nguyen et al.83 Nguyen et al.83 Nguyen et al.83 |
| Huh-7 | 1.9–11 | ||||||
| PHM | 1.3–3.2 | ||||||
RO-9187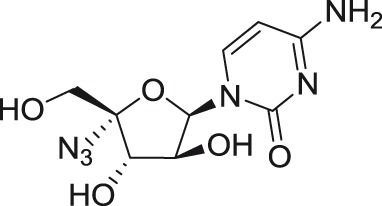
|
TBEV | Hypr | PS | VTR | 0.3 | >50 | Eyer et al.54 |
BCX4430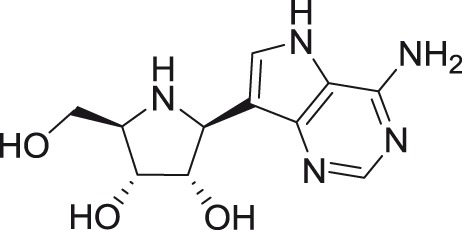
|
TBEV | Hypr | PS PS PS PS |
VTR VTR VTR VTR |
1.48 | >100 >100 >100 >100 |
Eyer et al.90 Eyer et al.90 Eyer et al.90 Eyer et al.90 |
| LIV | LI/31 | 12.33 | |||||
| KFDV | W-377 | 11.37 | |||||
| WNV | Eg101 | 2.33 | |||||
| YFV | 17-D | ND | ND | 14.1 | >100 >100 |
Warren et al.89 Warren et al.89 Warren et al.89 |
|
| JEV | SA14 | 43.6 | |||||
| DENV-2 | New Guinea C | 32.8 | >296 | ||||
| ZIKV ZIKV |
Various Various |
Vero, Huh-7, RD Vero, Huh-7, RD |
CPE | 3.8–11.7a | >100a >100a |
Julander et al.91 Julander et al.91 |
|
| VYR | 5.4–18.2b | ||||||
T-1106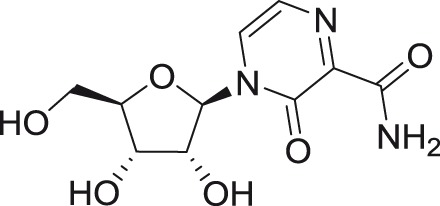
|
YFV YFV YFV |
17-D 17-D 17-D |
Vero Vero Vero |
Neutral red uptake | 1800 | >4000 >4000 >4000 |
Julander et al.94 Julander et al.94 Julander et al.94 |
| CPE | 2630 | ||||||
| Luciferase based | 1080 | ||||||
6-Methyl-7-deazaadenosine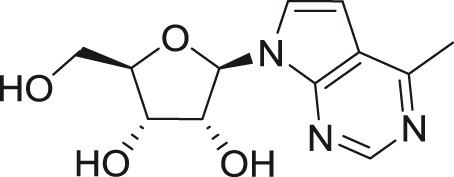
|
DENV-2 DENV-2 |
ND ND |
Vero Vero |
PA | 0.062 | ND ND |
Wu et al.96 Wu et al.96 |
| qRT-PCR | 0.039 | ||||||
N6-(9-antranylmethyl) adenosine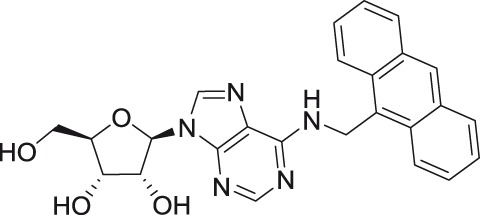
|
TBEV | Absettarov | PEK | PA | 15 | >50c | Orlov et al.35 |
N6-(1-pyrenylmethyl) adenosine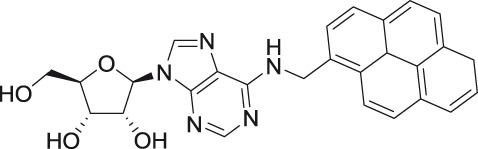
|
TBEV | Absettarov | PEK | PA | 6 | >50c | |
| N6-benzyl-5′-O-triisopropylsilyl adenosine 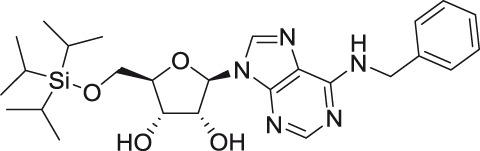
|
TBEV | Absettarov | PEK | PA | 5 | >50c | |
| N6-benzyl-5′-O-trityl adenosine 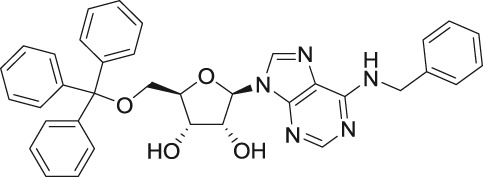
|
TBEV | Absettarov | PEK | PA | 2 | >50c | |
N6-benzyl-5′-O-tert-butyldimethylsilyl-adenosine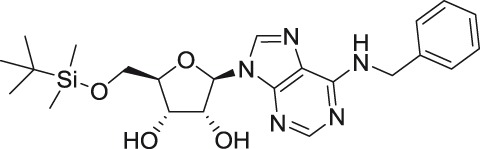
|
TBEV | Absettarov | PEK | PA | 20 | >50c | |
2′,5′Di-O-trityluridine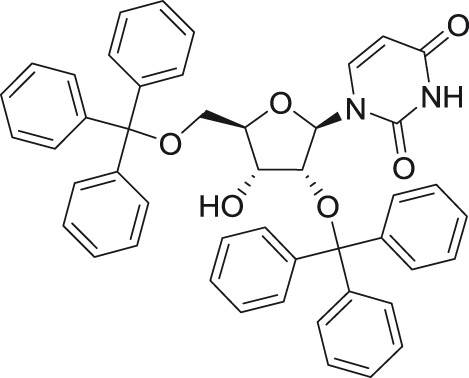
|
DENV-2 | New Guinea C | Vero Vero |
CPE CPE |
30a | >100a >100a |
Saudi et al.104 Saudi et al.104 |
| YFV | 17-D | 1.2a | |||||
3′,5′Di-O-trityluridine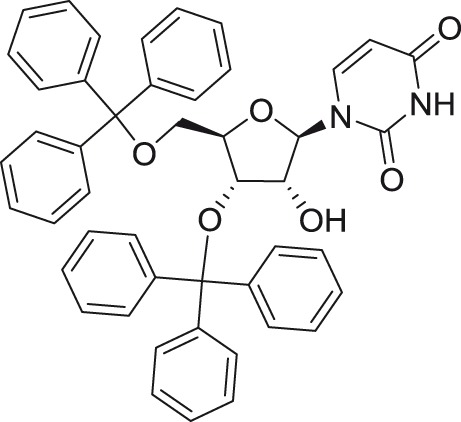
|
DENV-2 | New Guinea C | Vero Vero |
CPE CPE |
1.75a | >10a | |
| YFV | 17-D | 1a | >85a | ||||
| Inhibitors of flaviviral methyltransferase | |||||||
GRL-002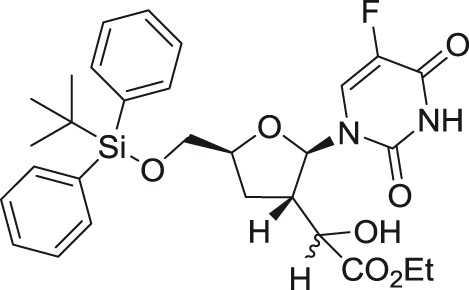
|
WNV | NY99 | – | N-7 methylation inhibition | 33.9d | – | Chen et al.111 |
| WMV | NY99 | – | 2′-O-methylation inhibition | 5.5d | – | Chen et al.111 | |
| WMV | NY99 | A549 | VTR | 52 | 48 | Chen et al.111 | |
GRL-003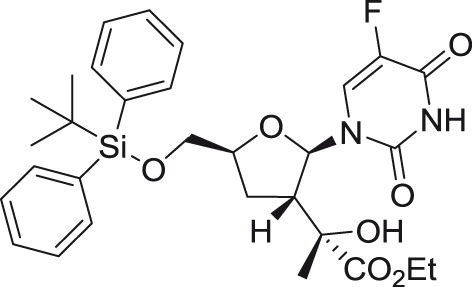
|
WMV | NY99 | – | N-7 methylation inhibitin | 17.3d | – | Chen et al.111 |
| WMV | NY99 | – | 2′-O-methylation inhibition | 19.8d | – | Chen et al.111 | |
| WMV | NY99 | A549 | VTR | 27 | 236 | Chen et al.111 | |
| Flex 1, R=H, Ac, Flex 1-TP, R=triphosphate 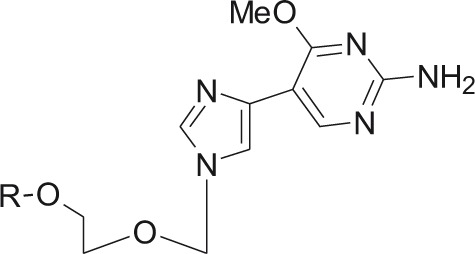
|
DENV-3 | ND | – | 2′-O-methylation inhibition | 22d | – | K. Seley-Radtke, manuscript in preparation |
| ZIKV | French Polynesisa (2013/PF KJ776791.2) | – | 2′-O-methylation inhibition | 22d | – | ||
Flex 2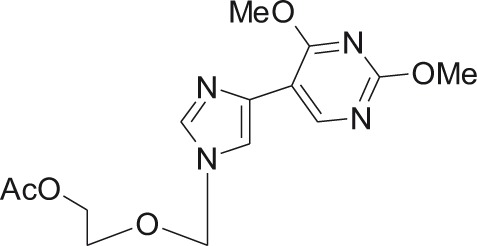
|
DENV-3 | ND | – | 2′-O-methylation inhibition | 3.2d | – | unpublished results, Smee laboratory, Utah |
| Ribavirin and other nucleoside synthesis inhibitors | |||||||
Ribavirin
|
DENV (1–4) |
Various | Vero | CPE | 19.8–41.9a | >100a,e | Crance et al.31 |
| JEV | Nakayama | Vero | CPE | 134.1a | >100a,e | Crance et al.31 | |
| WNV | E101 | Vero | CPE | 71.2a | >100a,e | Crance et al.31 | |
| USUV | DakArD 19848 | Vero | CPE | 62.6a | >100a,e | Crance et al.31 | |
| LGTV | ND | Vero | CPE | 33.9a | >100a,e | Crance et al.31 | |
| YFV | 17D and FNV | Vero | CPE | 42.4; 48.2a | >100a,e | Crance et al.31 | |
| WESSV | ND | Vero | CPE | 91.7a | >100a,e | Crance et al.31 | |
| ZIKV | Various | Vero, Huh-7, RD | CPE | 3.8–142.9a | >100a,e | Crance et al.31 and Julander et al.91 | |
| ZIKV | Various | Vero, Huh-7, RD | VYR | 9.52–281b | >100a,e | ||
ETAR
|
DENV-2 | ND | Vero | ND | 9.5 | >1000 | McDowell et al.135 |
IM18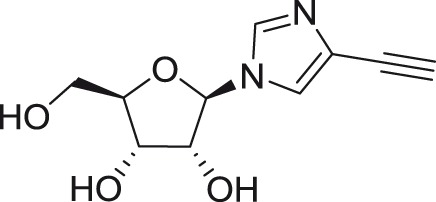
|
DENV-2 | ND | Vero | ND | 106.1 | ND | McDowell et al.135 |
6-Azauridine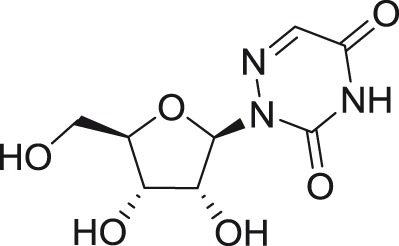
|
DENV (1–4) |
Various | Vero | CPE | 0.1–0.5a | >100a,e | Crance et al.31 |
| JEV | Nakayama | Vero | CPE | 0.5a | >100a,e | Crance et al.31 | |
| WNV | E101 | Vero | CPE | 0.2a | >100a,e | Crance et al.31 | |
| USUV | DakArD 19848 | Vero | CPE | 0.1a | >100a,e | Crance et al.31 | |
| LGTV | ND | Vero | CPE | 0.2a | >100a,e | Crance et al.31 | |
| YFV | 17D and FNV | Vero | CPE | 0.2; 0.2a | >100a,e | Crance et al.31 | |
| WESSV | ND | Vero | CPE | 1.3a | >100a,e | Crance et al.31 | |
| ZIKV | DakArB 11514 | Vero | CPE | 1.5a | >100a,e | Crance et al.31 | |
| Rigid amphipathic nucleosides | |||||||
5-(Perylen-3-yl)ethynyl-arabino-uridine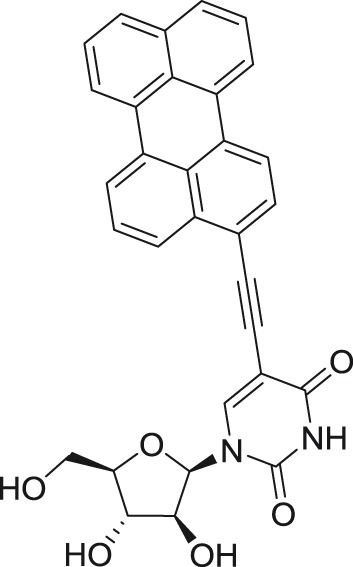
|
TBEV | Absettarov | PEK | PA | 0.018f | >50c | Orlov et al.101 |
5-(Perylen-3-yl)ethynyl-2′-deoxy-uridine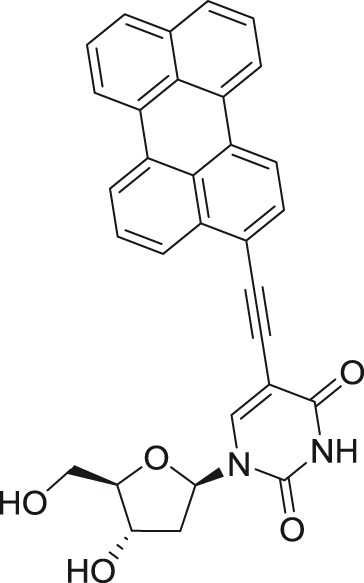
|
TBEV | Absettarov | PEK | PA | 0.024f | >50c | Orlov et al.101 |
5-(Pyren-1-yl)ethynyl-2′-deoxy-uridine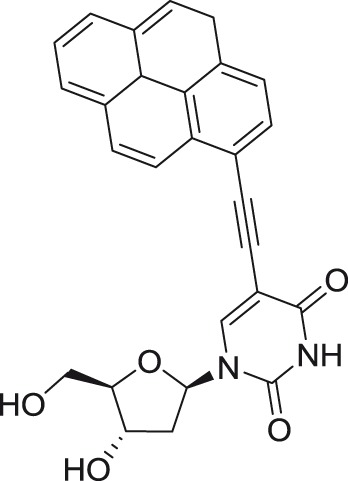
|
TBEV | Absettarov | PEK | PA | 0.98f | >50c | Orlov et al.101 |
AHFV: Alkhurma hemorrhagic fever virus; CFI: cellular flavivirus immunodetection; CPE: cytopathic effect reduction assay; DC: dendritic cells; DENV: dengue virus; JEV: Japanese encephalitis virus; KFDV: Kyasanur Forest disease virus; LGTV: Langat virus; LIV: louping ill virus; ND: not determined; OHFV: Omsk hemorrhagic fever virus; PA: plaque reduction assay; PBMC: peripheral blood mononuclear cells; PHM: primary human macrophages; POWV: Powassan virus; TBEV: tick-borne encephalitis virus; USUV: Usutu virus; VTR: viral titer reduction assay; VYR: viral yield reduction assay; WESSV: Wesselsbron virus; WNV: West Nile virus; YFV: yellow fever virus; ZIKV: Zika virus.
aEC50 and CC50 values are expressed as µg/ml.
bEC90 values, expressed as µg/ml.
cCC50 (24 h): the cell culture was treated for 24 h with the appropriate compound to obtain the cytotoxicity data.
dIC50 values obtained from enzyme-based inhibition assays.
eCC50 values for confluent compound-treated cells.
fEC50 (sim): the cell culture was TBEV infected and simultaneously treated with the appropriate compound.
Table 2.
Examples of in vivo antiflaviviral activities of selected nucleoside analogs.
| Compound | Animal model | Admin. route | Treatment start | Treatment length | Dose | Virus | Infection route | Survival rate (%) | References |
|---|---|---|---|---|---|---|---|---|---|
| 7-Deza-2′-C-methyladenosine | BALB/c mouse | i.p. | 0 dpi | 17 days | 25 mg/kg/2× day | TBEV | s.c. | 60 | Eyer et al.61 |
| 5–15 mg/kg/2× day | 35–50 | ||||||||
| AG129 mouse | p.o. | −1 h | 10 days | 50 mg/kg/day | ZIKV | i.p. | 25 | Zmurko et al.62 | |
| 2′-C-methylcytidine | ICR suckling mouse | i.c. | 1 dpi | 5 days | 15 or 30 mg/kg/day | DENV | i.c. | 60 | Lee et al.56 |
| Syrian golden hamster | i.p. | −4 h | 4–7 days | 120 mg/kg/day | YFV | i.p. | 90 | Julander et al.63 | |
| 3 dpi | 80 | ||||||||
| Sofosbuvir | C57BL/6J mouse | p.o. | 1 dpi | 7 days | 33 mg/kg/day | ZIKV | s.c. | 50 | Bullard-Feibelman et al.69 |
| Suckling Swiss mouse | i.p. | −24 h | 7 days | 20 mg/kg/day | ZIKV | i.p. | 40 | Ferreira et al.71 | |
| 2 dpi | 25 | ||||||||
| NITD008 | AG129 mouse | p.o. | 0 h | ND | 25–50 mg/kg/day | DENV | i.v. | 100 | Yin et al.26 |
| 1 dpi | 25 mg/kg/day | 70 | |||||||
| A129 mouse | p.o. | ND | 5 days | 50 mg/kg/day | ZIKV | i.p. | 50 | Deng et al.76 | |
| T-1106 | Syrian golden hamster | i.p. | −4 h | 8 days | 100 mg/kg/2× day | YFV | i.p. | 100 | Julander et al.93 |
| 3 dpi | 100 | ||||||||
| 5 dpi | 20 | ||||||||
| BCX4430 | Syrian golden hamster | i.p. | −4 h | 7 days | 12.5–125 mg/kg/day | YFV | i.p. | 100 | Julander et al.92 |
| 4 dpi | 200 mg/kg/day | 80 | |||||||
| AG129 mouse | i.m. | −4 h | 7–8 days | 300 mg/kg/day | ZIKV | s.c. | 90 | Julander et al.91 | |
| 1 dpi | 85 | ||||||||
| 3 dpi | 10 |
DENV: dengue virus; i.c.: intracerebral administration; i.m.: intramuscular administration; i.p.: intraperitoneal administration; i.v.: intravenous administration; ND: not determined; p.o.: per os (oral administration); s.c.: subcutaneous administration; TBEV: tick-borne encephalitis virus; YFV: yellow fever virus; ZIKV: Zika virus.
Inhibitors of flaviviral NS5 RdRp
The flavivirus NS5 protein is approximately 900 amino acids in length and consists of the NH2-terminal MTase domain required for the 5′-RNA capping process and the COOH-terminal RdRp domain responsible for the replication of the viral RNA genome.10,36 Flaviviral RdRp is a right hand-shaped structure with fingers, palm, and thumb domains; the palm domain is the catalytic domain carrying the polymerase active site that coordinates two Mg2+ ions essential for catalyzing the polymerization reaction.37 Nucleoside inhibitors of flaviviral RdRp are the most attractive targets for antiviral drug design; as human replication/transcription enzymes lack RdRp activity, such compounds are expected to show fewer deleterious side effects and favorable safety profiles.12,15,38,39
The mode of action for nucleoside RdRp inhibitors is based on the premature termination of viral nucleic acid synthesis.40 Following the intracellular phosphorylation, the 5′-triphosphate metabolites are competitively incorporated into the flaviviral RNA nascent chains (Figure 1). This prevents further extension of the incorporated analog by addition of the next nucleoside triphosphate resulting in formation of incomplete (nonfunctional) viral RNA chains.41 Nucleoside inhibitors of flaviviral RdRp act as “nonobligate chain terminators,” as their 3′-hydroxyl group is conformationally constrained or sterically/electronically hindered, thus decreasing the potency to form a phosphodiester linkage with the incoming nucleoside triphosphate.40 The nonobligate terminators differ from “obligate chain terminators,” in which the 3′-hydroxy group is completely missing, as exemplified by numerous nucleoside reverse transcriptase inhibitors for the treatment of HIV infections.42 Chemical modifications of the heterobase moiety, different types of glycosidic bonds, and substitutions at different positions of the sugar ring, which are typical modifications for nucleoside inhibitors of flaviviral RdRps, are shown in Table 3.
Table 3.
Heterobase substitutions and ribose modifications of selected flaviviral RdRp nucleoside inhibitors.
| Heterobase identity/modification | Type of the glycosidic bond | Ribose substitution | Ribose position | Example |
|---|---|---|---|---|
| 4-Aza-7,9-dideazaadenie 4-Aza-7,9-dideazaadenie |
C-glycosidic C-glycosidic |
-CN (α) -CN (α) |
C1′ C1′ |
GS-441524 GS-5734 |
| Adenine 7-Deazaadenine Guanine Cytosine Uracil 6-O-methylguanine |
N-glycosidic N-glycosidic N-glycosidic N-glycosidic N-glycosidic N-glycosidic |
-CH3 (β) -CH3 (β) -CH3 (β) -CH3 (β) -CH3 (β) -CH3 (β) |
C2′ C2′ C2′ C2′ C2′ C2′ |
2′-C-methyladenosine 7-Deaza-2′-C-methyladenosine 2′-C-methylguanosine 2′-C-methylcytidine 2′-C-methyluridine INX-08189 |
| Uracil | N-glycosidic | -F (α), CH3 (β) | C2′ | Sofosbuvir |
| Adenine 7-Deazaadenine 7-Deaza-7-carbamoyladenine |
N-glycosidic N-glycosidic N-glycosidic |
-ethynyl (β) -ethynyl (β) -ethynyl (β) |
C2′ C2′ C2′ |
2′-C-ethynyladenosine NITD008 NITD449 |
| Cytosine Cytosine |
N-glycosidic N-glycosidic |
-H (α), OH (β) + N3 (α) –N3 (α) |
C2′+ C4′ C4′ |
4′-Azido 4′-Azido-aracytidine |
| 9-Deazaadenine | C-glycosidic | O exchanged for N | – | BCX4430 |
| 3-Oxopyrazine-2-carboxamide | N-glycosidic | No substitution | – | T-1106 |
| 6-Methyl-7-deazaadenine | N-glycosidic | No substitution | – | 6-Methyl-7-deazadenosine |
| Uracil Uracil |
N-glycosidic N-glycosidic |
Trityl Trityl |
C2′ and C5′ C3′ and C5′ |
2′,5′di-O-trityluridine 3′,5′di-O-trityluridine |
1′-Cyano substituted nucleosides
GS-441524, a 1′-cyano substituted C-nucleoside derived from 4-aza-7,9-dideazaadenosine,43 was developed by Gilead Sciences, Inc. as a treatment for filovirus infections also showing reasonable antiviral activity against paramyxo- and pneumoviruses.44 Within the Flaviviridae family, this compound exerted micromolar inhibitory activity against YFV and DENV-2 (EC50 values of 11 and 9.46 µM, respectively) in various cell-based screening systems.43 Surprisingly, considerably less favorable in vitro activities (>30–51.2 µM) were reported for WNV and tick-borne flaviviruses, such as AHFV, KFDV, TBEV, and OHFV.43,45
A phosphoramidate prodrug of GS-441524, referred to as GS-5734, recently entered Phase II clinical trials for treatment of Ebola infections, displayed 10- to 40-fold higher antiviral effect against members of the TBEV serocomplex when compared with its parental analog GS-441524. The increased antiviral potency could be related to an improved conversion of the prodrug to the biologically active form; however, the reported SI values (2.4–8.3) indicate a low therapeutic potential for this nucleoside to treat flaviviral infections.45 Further substitutions of GS-441524 molecule at the C1′ position with methyl, vinyl, or methyl–ethynyl moieties yielded compounds with considerably reduced potency and a narrower spectrum of antiviral activity.43
2′-C-methyl substituted nucleosides
2′-C-Methyl-nucleoside scaffolds represent the initial major class of therapeutic nucleosides developed by Merck Research Laboratories to demonstrate potent inhibition of HCV replication.46–50 Antiviral activity of 2′-C-methylated nucleosides beyond the Flaviviridae family was reported for representatives of Picornaviridae and Caliciviridae families,51–53 indicating the potential for broad-spectrum inhibitory activity for these compounds within the positive single-stranded RNA viruses.
The 2′-C-methyl substituent introduced at the nucleoside β-face appears to be an important structural element for highly selective micromolar inhibition of tick-borne flaviviruses, particularly for TBEV, AHFV, KDFV, OHFV, and POWV, when assayed in porcine stable kidney cells (PS), human neuroblastoma cells UKF-NB4, or adenocarcinomic human alveolar basal epithelial cells A549.24,54,55 From mosquito-transmitted flaviviruses, 2′-C-methylated nucleosides inhibited WNV, DENV, and YFV in cell-based or cell-free reporter assay systems, showing low micromolar antiviral activities.50,56,57 A phosphoramidate prodrug of 6-O-methyl-2′-C-methylguanosine, denoted as INX-08189, exerted nanomolar inhibitory activity against DENV-2, and the combination of INX-08189 with ribavirin resulted in significant synergistic anti-DENV activity in vitro.58 7-Deaza-2′-C-methyladenosine together with other 2′-C-methylated species was the first described nucleoside-based inhibitors of ZIKV, after its epidemiological outbreaks in Oceania and Latin America.23,59 7-Deaza-2′-C-methyladenosine showed anti-ZIKV potency not only on immortalized cell lines, but also on induced pluripotent stem cell-derived neuronal cell types, such as cortical neurons, motor neurons, and astrocytes.60 The triphosphate analogs of 2′-C-methylated nucleosides exhibited strong inhibitory activity in a polymerase-based in vitro assay using an active recombinant ZIKV RdRp.61
Strong antiflaviviral activity for several 2′-C-methyl modified nucleosides was also demonstrated using numerous in vivo efficacy models. For example, 7-deaza-2′-C-methyladenosine substantially improved disease outcome, increased survival, and reduced signs of neuroinfection and viral titers in the brains of BALB/c mice infected with a lethal dose of TBEV.62 This compound also reduced viremia in AG129 mice infected with the African strain of ZIKV.59 Moreover, 2′-C-methylcytidine protected suckling mice challenged with DENV56 and hamsters infected with a lethal dose of YFV, even when administered up to three days postinfection.63
2′-Fluoro-2′-C-methyl substituted nucleosides
Similar to the 2′-C-methylated nucleosides, analogs possessing the 2′-α-fluoro-2′-β-C-methyl modification were initially identified as promising inhibitors of HCV polymerase activity.64 Sofosbuvir, a phosphoramidate prodrug of 2′-fluoro-2′-C-methyluridine developed by Gilead Sciences, Inc., is one of the most potent and selective inhibitors in this series and was approved by the Food and Drug Administration (FDA) for the treatment of chronic HCV infection.65 Sofosbuvir is nontoxic for most human cell lines and is a very poor substrate for human mitochondrial RdRp, resulting in an acceptable safety profile and negligible mitochondrial toxicity.66,67
Sofosbuvir was demonstrated to inhibit the ZIKV RdRp in a recombinant polymerase assay68 and to suppress ZIKV replication in different cell-based systems using U87 glioblastoma cells, baby hamster kidney fibroblasts (BHK-21), SH-sy5y neuroblasts, hepatocarcinoma Huh-7 cells, Jar human placental choriocarcinoma cells, neural stem cells, and brain organoids with nanomolar or low micromolar inhibitory activity.25,69 Interestingly, no inhibition of ZIKV replication with sofosbuvir even at the 50 µM level was observed in Vero cells,23 indicating that the sofosbuvir-mediated anti-ZIKV effect is strongly cell-type dependent.70 Sofosbuvir protected inbred C57BL/6J mice, which were previously immunosuppressed with a single dose of anti-Ifnar1 blocking monoclonal antibody, against ZIKV-induced mortality.69 Moreover, this compound reduced viral titer in blood plasma, spleen, kidney, and brain in suckling Swiss albino mice and prevented virus-induced neuromotor impairment in ZIKV-infected animals.71
Surprisingly however, sofosbuvir was inactive against TBEV, when screened on both PS and UKF-NB4 cells.54 Another 2′-α-fluoro-2′-β-C-methyl modified nucleoside, PSI-6206, and its 3′,5′-diester prodrug mericitabine, also displayed no activity against TBEV replication. The lack of anti-TBEV activity for the 2′-α-fluoro-2′-β-C-methyl modified nucleosides could be ascribed to their inefficient intracellular conversion to their corresponding triphosphates and, moreover, to extensive deamination/demethylation in the tested cell cultures resulting in their conversion to uridine.46,54
2′-C-ethynyl substituted nucleosides
2′-C-Ethynyl substituted nucleosides have been primarily identified as inhibitors of DENV.22,72–75 2′-C-Ethynyladenosine represents the lead compound in this series, which inhibited DENV-2 replication with an EC50 of 1.41 µM in cell-based assays and with a CC50 value of 40 µM.22 The 7-deaza derivative of 2′-C-ethynyladenosine, denoted as NITD008, inhibited DENV of different serotypes at submicromolar or low micromolar concentrations when tested in BHK-21 cells, A549 cells, Huh-7 hepatocarcinoma cells, and human peripheral blood mononuclear cells (PBMCs), showing a significantly improved cytotoxicity profile (CC50 of >100 µM) compared with that of 2′-C-ethynyladenosine.26 NITD008 was also assayed against WNV, YFV, and ZIKV and exhibited excellent in vitro inhibitory parameters and a protective effect against WNV and ZIKV infections in mouse efficacy models.26,76 NITD008 was also reported to effectively inhibit the in vitro replication of TBEV, AHFV, KDFV, OHFV, and POWV, with nanomolar or low micromolar antiviral levels observed in various cell-based screening systems.77
Introduction of the C7 carbamoyl moiety to NITD008 molecule provided another 2′-ethynyl modified derivative, referred to as NITD449. Despite its low micromolar anti-DENV efficacy, this nucleoside disappointingly exhibited only low levels in plasma when dosed orally.72 To increase the oral bioavailability, the isobutyryl ester prodrug of NITD449, designed as NITD203, was synthesized. NITD203 successfully exhibited both nanomolar anti-DENV activity as well as improved pharmacokinetic parameters.72 Although NITD008 and NITD203 showed anti-DENV potency in rodent models, even when the treatment was delayed up to 48 h after infection, both nucleosides failed in preclinical toxicity studies in rats and dogs due to their insufficient safety profiles.26,72 NITD008 failed to achieve no-observed-adverse-effect levels (NOAEL) when rats (10 mg/kg/day) and dogs (1 mg/kg/day) were dosed daily for two weeks.26 Similarly, NOAEL was not achieved for NITD203 in the two-week toxicity test when rats were dosed at 30 and 75 mg/kg/day.72
Further substitutions of the C7 position of NITD008 with fluoro or cyano moieties provided compounds with nano- or low micromolar anti-DENV activities and acceptable cytotoxicity profiles. In contrast, 2′-C-ethyl, -vinyl, or -methylethynyl substituted derivatives of 2′-C-ethynyladenosine were completely inactive when tested against DENV. Similarly, the exchange of the adenine base for cytosine or guanine also yielded inactive compounds.22,72
2′-O-substituted nucleosides
2′-O-Methyl substituted adenosine, guanosine, cytidine, and uridine were evaluated for their potential anti-TBEV activity; however, no or negligible antiviral effects were observed when tested in both PS and UKF-NB4 cells.54 Such abrogation of the nucleoside inhibitory activity could be related to the elimination of the 2′-α-hydroxy hydrogen bond donor/acceptor properties when the methyl moiety is introduced at the nucleoside O2′ position.46,54 The ability of flaviviral RdRp to discriminate among nucleosides modified at the 2′-position on the nucleoside α-face is likely related to the need of the polymerase to avoid incorporation of 2′-α-deoxynucleoside monophosphates into the viral nascent RNA chain.46
3′-C- and 3′-O-substituted nucleosides
Methylation of the C3′ or O3′ position to generate the corresponding 3′-C-methyl or 3′-O-methyl modified structures resulted in a complete loss of anti-TBEV activity, regardless of the purine/pyrimidine heterobase identity. These nucleosides exerted no cytotoxic effects and caused no morphological changes in PS or porcine embryo kidney (PEK) cell cultures.35,54 Similarly, 3′-deoxynucleosides exhibited no detectable inhibitory effect on TBEV replication.54 In contrast, several nucleosides with a trityl group at the C3′ position showed micromolar inhibitory activity against DENV and YFV (see below).78 The observed inactivity of 3′-O-methylated and 3′-dehydroxylated nucleosides could be related to either a strict requirement of the TBEV RdRp active site for a 3′-hydroxyl group to form the appropriate hydrogen bonding interactions with the nucleoside triphosphate molecule, or, to inefficient cellular uptake and metabolism to convert the nucleoside molecule into the corresponding triphosphate form.46,54
4′-Azido substituted nucleosides
Using high-throughput screening of large nucleoside libraries in combination with a rational drug design approach by investigators at Roche, several cytidine analogs with an azido group at the C4′ position were identified as potent inhibitors of HCV replication in subgenomic replicon assays.79–81 It was later shown that these compounds were also highly active against henipaviruses and other paramyxoviruses.82 Two 4′-azido modified nucleoside analogs, 4′-azidocytidine (R-1479) and 4′-azido-aracytidine (RO-9187), showed nano- or micromolar in vitro antiviral activity against TBEV.54 Moreover, both compounds were found to be active also against WNV (L. Eyer, manuscript in preparation).
The anti-TBEV activity of RO-9187 (the arabino-counterpart of 4′-azidocytidine) was unexpected, as this compound lacks the 2′-α-hydroxy moiety, a determinant which was generally considered to be crucial for specific hydrogen-bonding interactions with RdRps during the RNA replication process.54,80 Some additional interactions of the polymerase active site with both the 2′-β-hydroxy substituent and the 4′-azido substituent are thought to compensate for the loss of the 2′-α-hydroxy interaction, resulting in the strong and selective anti-TBEV activity of RO-9187. Interestingly, the anti-TBEV efficacy of both 4′-azido modified nucleosides was cell-type dependent; the compounds were active only in PS cells, but not in UKF-NB4 cells.54
An ester prodrug of 4′-azidocytidine, denoted as balapiravir, was completely inactive against TBEV in vitro, probably because of its poor intracellular uptake or insufficient kinase phosphorylation in the tested host cell lines.54 In contrast, balapiravir was reported to show strong in vitro antiviral activity against DENV of various serotypes; it was the first direct antiviral agent tested clinically for DENV infection. Unfortunately, this compound failed to achieve antiviral efficacy in DENV patients, which was reflected in a negligible reduction of DENV viremia and persistence of clinical symptoms, even though the plasma concentration of the compound was higher than the 50% effective concentration.83 One of the possible explanations is that DENV infection stimulates PBMCs to produce cytokines, which are responsible for the decreased efficiency of the conversion of balapiravir to its triphosphate form.84
Interestingly, nucleoside analogs combining the 4′-azido moiety with the 2′-C-methyl group in one molecule (e.g. 2′-C-methyl-4′-azidocytidine) did not exhibit any antiviral activity when tested against HCV; however, the corresponding 5′-monophosphate prodrugs displayed considerably improved virus inhibitory effects (EC50 values in the micromolar ranges) without apparent cytotoxicity.85 Other interesting compounds, 4′-azido-2′-deoxy-2′-C-methylcytidine and its ester prodrugs, were found to show nano- or low micromolar antiviral efficacy in vitro.86 Unfortunately, such nucleoside scaffolds have not been evaluated against arthropod-borne flaviviruses; the reported results originate from HCV replicon-based assays.85,86
Imino-C-nucleoside analog BCX4430
BCX4430, developed by BioCryst Pharmaceuticals Inc., is an adenosine analog with the furanose oxygen on the ribose ring replaced by nitrogen and the heterobase nitrogen 9 replaced by carbon.87 This interesting nucleoside is classified as imino-C-nucleoside.88 BCX4430 was initially described as an inhibitor of filovirus infections, exerting antiviral activity against a broad spectrum of single-stranded RNA viruses, particularly against members of the Bunyaviridae, Arenaviridae, Picornaviridae, Orthomyxoviridae, Paramyxoviridae, Coronaviridae, and Flaviviridae families.89 Currently, this compound has entered Phase I clinical trials for Ebola virus disease treatment focused on intramuscular administration of BCX4430 in healthy volunteers and to date has shown promising pharmacokinetics properties and good tolerability.87
BCX4430 is active against numerous mosquito-transmitted flaviviruses, such as WNV (EC50 of 2.33 µM)90 and representatives of both the African and Asian lineages of ZIKV (3.8–11.7 µg/ml).91 For YFV, JEV, and DENV-2, micromolar EC50 values were reported.89 In vivo efficacy of BCX4430 was also demonstrated in a lethal hamster model of YFV infection and in a mouse model of ZIKV infection.91,92 Low micromolar antiviral activity of BCX4430 was also described for some of the medically important tick-borne flaviviruses, such as TBEV, LIV, and KFDV.90
Heterocyclic base-modified nucleosides
Heterocyclic base-modified nucleosides with demonstrated antiflaviviral activities include T-1106,93–95 6-methyl-7-deazaadenosine,96 and numerous N6-alkyl or aryl substituted nucleosides.35,97 T-1106 is a ribosylated analog of the pyrazine derivative T-705 (6-fluoro-3-hydroxy-2-pyrazinecarboxamide, favipiravir),98 which was described to inhibit the HCV RdRp in enzyme assays.93 Nucleoside inhibitor T-1106 displayed a negligible in vitro activity against YFV in Vero cells, as well as in luciferase-based assays.94 In contrast, this compound exerted favorable efficacy, bioavailability, and low toxicity in a hamster model of YFV infection, using a hamster-adapted Jimenez YFV strain. After intraperitoneal application of 100 mg/kg/day, T-1106 improved survival rates, serum parameters, weight gain, and mean day to death when administered up to four days after virus challenge.93,94 In this model, the combination of T-1106 with ribavirin gave superior effects compared to monotherapy (with either T-1106 or ribavirin).95
6-Methyl-7-deazaadenosine is a hydrophobic mimic of adenosine showing nanomolar antiviral activity against DENV-2 in a Vero cell-based screening system, as well as in luciferase-driven DENV-2 replicon assay, with no cytotoxic effects noted after 7 h of treatment.96 Related compounds such as 6-methyl-1-deazaadenosine and 6-methyl-4-deazaadenosine were completely inactive when tested against DENV-2. Mechanistic studies of the 5′-triphosphate of 6-methyl-7-deazaadenosine revealed that this nucleotide is an efficient substrate for viral RdRp (screened against polio RdRp) and is incorporated into nascent viral RNA strains mimicking both ATP and GTP.96
N6-Alkyl or aryl substituted nucleosides, originally identified as inhibitors of Lassa fever virus, Marburg virus, and enterovirus A71, showed interesting bioactivity profiles when tested against TBEV.35,99,100 Whereas N6-methyladenosine and N6-benzyladenosine were completely inactive, nucleosides with bulky substituents, such as N6-(9-anthracenylmethyl)adenosine and N6-(1-pyrenylmethyl)adenosine, exerted a micromolar anti-TBEV effect. In contrast, N2- and N4-substituted analogs showed no antiviral activity. Moderate anti-YFV and anti-DENV activities were also observed in N6-subsitituted analogs of 5′,3′-O- and 5′,2′-O-tert-butyldiphenylsilyl-modified adenosine.97 The mechanism of action of these nucleosides is poorly understood; however, they likely interact with the RdRp or MTase domain of the flaviviral NS5 protein, as demonstrated by docking studies.35 Bulky aromatic substituents could also play a role in the interaction with the viral membrane resulting in cell entry inhibition.101
Tritylated nucleosides
In a large-scale cell-based screening campaign of alkylated, silylated, or acylated pyrimidine nucleosides, 2′,5′di-O-trityluridine and 3′,5′di-O-trytiluridine were identified as inhibitors of DENV-2 and YFV replication, showing high antiviral potency and favorable cytotoxicity profiles in Vero cells.78,102,103 Substantial antiviral effect against YFV was observed also in 2′-deoxy-3′,5′-di-O-trityluridine and in several 5-halogenated bis-tritylated pyrimidine nucleosides; however, their anti-DENV activity was proven to be weaker.104 Thymidine or 2′-deoxyuridine congeners of 2′,5′- and 3′,5′-tritylated nucleosides led to the loss of antiflavivirus activity or to increased compound cytotoxicity.
The mechanisms of action of tritylated nucleosides are not completely understood. Based on the observed inhibition of DENV replication in subgenomic replicon assays, it is assumed that these compounds may be acting as inhibitors of intracellular viral replication events rather than suppressing either early or late processes of viral infection, such as entry or assembly.103 Although the presence of large, hydrophobic trityl moieties does not make these structures ideal candidates for further drug development, their chemical structures may provide valuable information for advanced mechanistic studies and for further development of related nucleoside scaffolds with improved biological parameters.104
Nucleoside inhibitors of flaviviral MTase
The NH2 domain of flaviviral NS5 protein is associated with the virus’s MTase activity, which is involved in methylation of the 5′-cap structure of genomic RNA.8,105 The flaviviral cap structure is formed by the conserved dinucleotide sequence AG (m7GpppAm) and is crucial for mRNA stability and efficient translation.106 Two topologically distinct methylation reactions are mediated by the flaviviral NS5 MTase: the N7 of guanine is methylated by the (guanine-N7)-MTase and the first nucleotide of RNA transcript is further methylated at the ribose 2′-hydroxyl by (nucleoside-2′-O)-MTase. The resulting product of the methyl donation for both methylation reactions by S-adenosyl-l-methionine is the nucleoside analog S-adenosyl-l-homocysteine (SAH).107
SAH and the nucleoside antibiotic sinefungin are natural nonselective inhibitors of many eukaryotic and viral MTases, including those of DENV108,109 or ZIKV.61 Chemical derivatization of SAH at the N6 position provided inhibitors with nano- or low micromolar activity against DENV MTase, which did not inhibit the corresponding human enzymes.110 Other rationally designed nucleosides with potent inhibitory activity against MTase contain a thymine base with a hydrophobic methyl tert-butyl substituent at the 5′ position of the sugar moiety.111 Two of these nucleosides, GRL-002 and GRL-003, inhibited the N7 and 2′-O MTase activity of WNV in enzyme-based assays and the observed MTase inhibition was in agreement with the micromolar in vitro anti-WNV efficacy.111 Another class of promising selective antiflavivirus compounds is represented by 5′-silylated 3′-1,2,3-triazole-substituted nucleoside scaffolds derived from 3′-azidothymidine. Similar structures were originally developed for HIV-1 inhibition.27 Both the 5′-silyl protecting group and the 3′ bulky triazole substituent appeared to be crucial structural elements for low micromolar inhibition of flaviviral MTase in enzyme-based assays, as well as for inhibition of WNV and DENV replication in cell culture. These nucleosides inhibit the methylation reactions through competitive interactions with the substrate binding site and also with the GTP-binding pocket of the flaviviral NS5 MTase.28
Recently, a novel series of flexible nucleoside analogs known as “fleximers” have exhibited activity against several hard to treat viruses, including filoviruses such as Ebola and Sudan,112 coronaviruses including Severe Acute Respiratory Virus and Middle East Respiratory Virus,113 as well as most recently, flaviviruses including ZIKV and DENV. The fleximers feature a “split” purine nucleobase that has been shown to impart significant activity to the nucleoside scaffold as well as to allow it to overcome resistance related to point mutations.114–116 The most recent series combined the fleximer approach with the acyclic nucleoside acyclovir, an FDA approved drug for herpes virus. While these analogs inhibited the aforementioned viruses, acyclovir shows no activity against any of those viruses, thereby underscoring the importance of the fleximer approach. Since those initial findings, these compounds have also demonstrated potent activity against DENV and ZIKV (K. Seley-Radtke, manuscript in preparation). Preliminary results indicate that these compounds target the DENV and ZIKV NS5 in at least its cap-MTase activity, with negligible effects on the cognate human N7-MTase. In that regard, initial screening revealed promising levels of MTase inhibition, particularly for the triphosphate of the compound (Flex 1-TP), with an IC50 of 22 µM for both the DENV and ZIKV 2′-O-MTase (K. Seley-Radtke, manuscript in preparation). As a result, Flex 1-TP was further tested against DENV NS5, and while it was not incorporated, it successfully inhibited further incorporations of additional nucleotides, thereby halting replication, however not as a typical chain terminator. In addition, the acetate-protected dimethoxy analog (Flex 2) is a potent DENV inhibitor (3.2 µM, unpublished results, Smee laboratory, Utah); however, more work needs to be done to fully elucidate these novel compounds’ mechanism of action. It may well be that these nucleotide analogs target the NS5 protein at both the RdRp and MTase regions, which would make them highly effective viral inhibitors, with low probability of viral resistance developing.
Nucleoside inhibitors of flaviviral NTPase/helicase
Flaviviral NTPase and helicase activities are associated with the COOH-proximal domain of the NS3 protein.117 Flaviviral helicases are capable of unwinding duplex RNA structures during viral replication by disrupting the hydrogen bonds keeping the two strands together.118,119 The helicase activity is strictly associated with NTP hydrolysis (NTPase activity); the released chemical energy is used for the translocation of the enzyme along the double-helix structures, capturing the exposed single strand regions or for a direct disruption of the hydrogen bonds between the two RNA strains.120
Specific nucleoside inhibitors of flaviviral NTPase/helicase can interact with dsRNA or DNA resulting in the weakened stability of double-helix structures or by steric hindrance of translocation of the enzyme along the polynucleotide chain. Such a mechanism could modulate the efficacy of the unwinding reaction or NTPase activity of the enzyme and therefore, affect the viral replication process.117–121 Weak inhibitory effects on flaviviral NTPase/helicases were described for ribavirin triphosphate,124,125 5′-O-fluorosulfonylbenzoyl esters of purine nucleosides,126,127 or halogenated benzotriazole-modified nucleosides.30,122 These compounds were primarily evaluated in enzyme-based assays for their putative anti-HCV activity; however, some of them have been found to inhibit also NTPase/helicases of WNV, JEV, or DENV.117–127
Ribavirin and other nucleoside synthesis inhibitors
Ribavirin, a nucleoside analog featuring a [1,2,4]triazole ring for a nucleobase, is a licensed drug against various RNA viruses. The predominant inhibitory mechanism for ribavirin against flaviviral replication is the suppression of de novo biosynthesis of guanine nucleotides through direct inhibition of inosine monophosphate dehydrogenase, an enzyme converting inosine monophosphate to xanthosine monophosphate, a precursor in GTP biosynthesis.32 Speculation over other modes of action for ribavirin includes specific inhibition of the viral RdRp,128 accumulation of mutations in viral genomes resulting in error catastrophe,129,130 interference with mRNA capping guanylation,131 and immuno-modulation promoting the Th1 antiviral response.32,132,133
Ribavirin was shown to exert a moderate inhibitory effect for multiple mosquito-borne flaviviruses in various cell cultures,31,134,135 often being used as a positive control in many in vitro25,59,92 and in vivo antiviral studies.91–94 Ribavirin administered to YVF-infected hamsters challenged intraperitoneally, resulted in significant improvement in survival rates, even if the therapy was started two days post-YFV infection.93,94 In contrast however, in primates, only a weak prophylactic effect on viremia in rhesus monkeys challenged with DENV was observed.136 Several ribavirin derivatives were recently synthesized and showed interesting bioactivity profiles: ETAR (1-β-d-ribofuranosyl-3-ethynyl-[1,2,4]triazole) and IM18 (1-β-d-ribofuranosyl-4-ethynyl-[1,3]imidazole) inhibited DENV-2 replication in Vero cells by more than 10-fold compared with ribavirin and showed no detectable cytotoxic effects up to 1000 µM.135 Another derivative, EICAR (1-β-d-ribofuranosyl-5-ethynyl-imidazole-4-carboxamide), was reported to possess a similar in vitro spectrum of antiviral activity, but lower selectivity compared with those of ribavirin.137
Two nucleosides whose antiviral activity is based on the depletion of the intracellular nucleoside pool are 6-azauridine and 5-aza-7-deazaguanosine. 6-Azauridine and its derivatives are inhibitors of orotidine monophosphate decarboxylase blocking cellular de novo pyrimidine biosynthesis.31,33 6-Azauridine proved to be active against numerous arthropod-borne flaviviruses,55 however exhibited slight cytotoxicity with an inhibitory effect on the growth of host cells.24 A triacetate prodrug of 6-azauridine showed low micromolar activity against AHFV and WNV in vitro55 and very low toxicity in both animal and human studies.138 Another derivative, 2-thio-6-azauridine, exerted a moderate inhibitory effect on WNV.33 Similar results were achieved using 5-aza-7-deazaguanosine (ZX-2401)139; this compound exhibited synergistic in vitro anti-YFV activity in combination with interferon. The mechanism of action for 5-aza-7-deazaguanosine is currently unknown; however, it is conceivable that it likely resembles that of ribavirin.34
Rigid amphipathic nucleosides
Nucleoside derivatives containing bulky aromatic substituents (perylene or pyrene moieties) attached to the heterocyclic base were originally synthesized as fluorescent nucleoside probes140–142; later these structures were identified as inhibitors of herpes simplex virus, type 1 and 2 (HSV-1 and HSV-2), vesicular stomatitis virus, and Sindbis virus replication.143 Further studies demonstrated their broad-spectrum antiviral activity against other enveloped viruses, such as influenza virus, murine cytomegalovirus, and HCV.144 The mechanism of action of these rigid amphipathic nucleosides is based on their incorporation into the viral or cellular membranes, preventing fusion.143,144 Alternatively, these nucleosides may also function by photosensitization of lipid membranes, resulting in irreversible damage of enveloped virion particles.145
5-(Perylen-3-yl)ethynyl-arabinouridine and 5-(perylen-3-yl)ethynyl-2′-deoxyuridine were shown to act as strong inhibitors of TBEV in PEK cell culture.101 The perylene moiety as well as the rigid ethynyl linker appeared as crucial structural elements for nanomolar anti-TBEV activity and low cytotoxicity (>50 µM). Interestingly, uracil nucleosides bearing the pyrene moiety, such as 5-[(pyren-3-yl)methoxypropyn-1-yl]-2′-deoxyuridine and 5-(pyren-1-yl)ethynyl-2′-deoxyuridine, showed almost a 10-fold lower anti-TBEV potency compared to their perylene-substituted counterparts. Such compounds, if not used as therapeutic agents, could still contribute to a better understanding of different modes of action of various nucleoside scaffolds.101
Challenges and complications of antiflavivirus nucleoside analog development
Introduction of various chemical substituents into different positions of the nucleoside scaffold can dramatically affect the physicochemical properties of the compound. Such modifications can also significantly influence the compound’s biological/pharmacokinetic parameters, such as cellular uptake,15,146 the ability of the compound to be activated (phosphorylated) by cellular kinases,147 degradation by nucleoside catabolic enzymes,148 or cellular toxicity.149,150 Use of nucleoside analogs can also result in the undesirable emergence of drug-resistant virus mutants.151–153 This section highlights the most important challenges and complications toward the development of nucleoside inhibitors of arthropod-transmitted flaviviruses and suggests possible strategies to surmount these difficulties.
For most nucleoside analogs, the first kinase phosphorylation is the rate-limiting step for the conversion to the nucleoside triphosphates. This limitation has a major influence on nucleoside analog antiviral activity147,154 but can be bypassed by the use of a monophosphate prodrug approach based on the introduction of the phosphorylated group into the 5′ nucleoside position. The phosphorylated group includes masking moieties on the charged phosphate leading to a neutral and eventually hydrophobic entity able to deliver the nucleoside 5′-monophosphate into the cells (Figure 1).15 The monophosphate prodrug approach has been shown to convert some inactive nucleosides into strong inhibitors, or, has improved the kinetics parameters of intracellular nucleoside triphosphate formation.85 This strategy, together with enantioselective purification, led to the development of the phosphoramidate prodrug sofosbuvir, which exhibited considerably increased phosphorylation efficacy compared to the parent nucleoside, 2′-C-fluoro-2′-C-methyluridine.155,156
Rapid degradation of nucleoside analogs by nucleoside catabolic pathways (Figure 1) is another undesirable phenomenon, which can adversely affect the antiviral potency of some nucleoside analogs.148 To address this problem, appropriate structural changes can be introduced into the nucleoside scaffolds to protect the nucleosides from metabolic deactivation.157 Such a strategy was successful for 2′-C-methyladenosine, which was found to be rapidly deaminated by cellular adenosine deaminase to the inactive inosine derivative and/or degraded by purine nucleoside phosphorylase, resulting in poor bioavailability and rapid clearance of the nucleoside in plasma.49,50 Substitution of the adenine N7 nitrogen for a carbon provided metabolically stable 7-deaza-2′-C-methyladenosine, which is a poor substrate for both nucleoside catabolic enzymes. This compound was characterized by excellent bioavailability and half-life in beagle dogs and rhesus monkeys.50 Another possible strategy to increase the metabolic stability of therapeutic nucleosides is based on the introduction of a C-glycosidic bond into the nucleoside scaffold to generate C-nucleoside analogs, such as BCX443089 or GS-5734.43 The major advantage of C-nucleosides over the canonical N-nucleosides lies in their resistance to unwanted phosphorolysis by intracellular phosphorylases, which otherwise would cleave the N-glycosidic linkage.88
Individual host cellular types can display differences in expression levels of nucleoside kinases and other enzymes/proteins involved in nucleoside metabolism and transport. This can then result in cell-type dependent antiviral activity as manifested by different EC50 values for the same inhibitor when assayed on different cell lines.158,159 Strong anti-TBEV activity for 2′-C-methylguanosine, 4′-azidocytidine, and 4′-azido-aracytidine in PS cells was associated with rapid and efficient nucleoside conversion to the corresponding triphosphates. On the other hand, no anti-TBEV effect or phosphorylation products were observed when both compounds were tested in UKF-NB4.54 Similarly, the loss of anti-ZIKV activity in Vero cells for sofosbuvir is likely related to the increased expression of the multidrug resistance ABC transporter in this cell line, resulting in the efflux of the compound from the cells.23,25 Clearly, cell-type dependent antiviral activity of some nucleosides can considerably affect the results of antiviral screens and therefore, using multiple clinically relevant cell lines for evaluation of compounds for antiviral activity is important.70
Another possible complication in nucleoside drug development is an undesirable toxicity profile for the nucleoside inhibitor, which can be related to poor selectivity between viral and human enzymes.39,149,150 A typical example of a nonselective nucleoside analog is 7-deazaadenosine (tubercidin), which exhibits high in vitro cytotoxicity. This has been attributed to the incorporation of tubercidin monophosphate into cellular DNA/RNA by human polymerases.50 In contrast, two derivatives of tubercidin, 7-deaza-2′-C-methyladenosine and 6-methyl-7-deazaadenosine, are selectively recognized by flaviviral RdRp and are nontoxic in most mammalian cell lines.50,96 Some nucleoside analogs inhibit the mitochondrial DNA or RNA polymerase γ, resulting in mitochondrial toxicity.149,160 This has been the primary reason for the failure of several promising nucleosides/prodrugs in clinical trials, as has been shown for some 2′-C-methyl- and 4′-azido modified nucleosides.67 Newly developed compounds should also be evaluated for their genotoxicity and mutagenicity, as well as for renal, cardiovascular, or liver toxicity using various biochemical in vitro assays.161–163 Nevertheless, even if a compound successfully passes through numerous in vitro tests, it can still exhibit harmful side effects when tested in animals, as observed with the adenosine analog NITD008.26
In that regard, the availability of suitable animal models is crucial for the successful evaluation of a nucleoside’s therapeutic in vivo antiviral efficacy. Some flaviviruses, particularly DEVN and ZIKV, do not readily replicate or cause pathology in immunocompetent mice and, therefore, the use of these rodents as animal infection models is substantially limited.164–166 To overcome this problem, suckling or young mice,167 AG129 mice lacking INF-α/β and INF-γ receptors,168 IFNAR−/− mice lacking only the IFN-α/β receptor,169 or immunosuppressed mice170 can be used as appropriate models to evaluate nucleosides in vivo. However, as these animals are defective in an immune response, this model may also underestimate the real efficacy of the test compounds. Rodent-adapted flavivirus strains, such as the hamster-adapted YFV strain Jimenez,171 mice-adapted DENV strain D2S10,26 or ZIKV African strain Dakar 41519,172 represent other possible options for in vivo antiviral studies; the biological properties of such viruses can be, however, considerably different compared with those of the parent human-adapted strains.169
Another issue is related to the length of therapeutic treatment. Most tick- and mosquito-borne flaviviruses cause acute infections, in which short-time treatment duration is expected, ranging between several days to weeks.173 This is in contrast to chronic diseases, such as HCV, HBV, and HIV infections, which require long-lasting, and sometimes lifelong, treatment regimens.72,173 The differences between the acute and chronic diseases should be considered during preclinical development of antiflaviviral inhibitors. Thus, some compounds that show insufficient safety profiles when tested for treatment of chronic infections can be still suitable and safe for short-term therapy of acute flaviviral diseases.72
Antiviral therapies based on chemical inhibitors of viral replication can be accompanied with a rapid emergence of drug-resistant mutants which substantially complicates the course of infection treatment, as seen in HIV, HBV, or HCV infections.152,153 In flaviviruses, a rapid evolution of resistance to 2′-C-methylated nucleoside inhibitors was observed; this resistance was associated with a signature mutation S603T (in AHVF and TBEV)55,62 or S604T (in ZIKV)68 within the active site of the viral RdRp. Interestingly, the biological properties of the TBEV mutant viruses were dramatically affected, which was manifested by resistance-associated loss of viral replication efficacy in cell culture and a highly attenuated virulence phenotype in mice. This resulted in an unusually low mortality rate when mice were infected with the mutant strain.62 As TBEV mutants resistant to 2′-C-methylated nucleosides are highly sensitive to 4′-azido substituted nucleosides,62 a combination treatment based on two or more inhibitors could be a possible strategy in order to minimize the risks for the emergence of viral drug resistance.174–176
Conclusions
More than 200 million clinical cases caused by arthropod-borne flaviviruses, including numerous deaths, are reported worldwide annually. So many cases of infection indicate the importance for the pursuit of new small molecule-based therapeutics to combat emerging viral pathogens. Among these, inhibitors of flaviviruses represent a critical unmet medical need. In that regard, nucleoside inhibitors of flaviviral RdRps are the most attractive targets for antiviral drug design. Nucleosides with the methyl- or ethynyl- modification at the C2′ position and their 2′-fluoro derivatives are the best understood antiflavivirus nucleoside analogs, many of which were initially developed for treatment of HCV infections and later reemployed to suppress replication of other non-HCV flaviviruses. Other important antiflavivirus nucleosides are represented by inhibitors of nucleoside biosynthesis, whose mode of action is predominantly based on depletion of the intracellular nucleoside pool. Nucleoside inhibitors of flaviviral MTase and NTPase/helicase, as well as some newly discovered flavivirus inhibitors, such as tritylated nucleosides, rigid amphipathic nucleosides, or N6-aryl-substituted adenosine derivatives, whose mechanisms of action are still poorly understood, can be used as initial structures or starting points for further developments of new generations of nucleoside scaffolds with improved biological parameters. Taken together, specific nucleoside analog-based antiviral therapy in combination with effective vaccination strategies could provide potent prophylactic and curative tools to treat human infections caused by flaviviruses.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Ministry of Health of the Czech Republic (grant no. 16–34238A), Czech Science Foundation (grant no. 16–20054S) (to DR and RN), and by Project “FIT” (Pharmacology, Immunotherapy, nanotoxicology; CZ.02.1.01/0.0/0.0/15_003/0000495), which was funded by the European Regional Development Fund (to DR).
References
- 1.Baier A. Flaviviral infections and potential targets for antiviral therapy In: Ruzek D. (ed) Flavivirus encephalitis. 1st ed Rijeka: InTech, 2011, pp.89–104. [Google Scholar]
- 2.Lazear HM Stringer EM andde Silva AM.. The emerging zika virus epidemic in the Americas research priorities. J Am Med Assoc 2016; 315: 1945–1946. [DOI] [PubMed] [Google Scholar]
- 3.Jeffries CL, Mansfield KL, Phipps LP, et al. Louping ill virus: an endemic tick-borne disease of Great Britain. J Gen Virol 2014; 95: 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashraf U, Ye J, Ruan XD, et al. Usutu virus: an emerging flavivirus in Europe. Viruses 2015; 7: 219–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rumyantsev AA Murphy BR andPletnev AG.. A tick-borne Langat virus mutant that is temperature sensitive and host range restricted in neuroblastoma cells and lacks neuroinvasiveness for immunodeficient mice. J Virol 2006; 80: 1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackburn NK andSwanepoel R.. An investigation of flavivirus infections of cattle in Zimbabwe-Rhodesia with particular reference to Wesselsbron virus. J Hyg 1980; 85: 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers TJ, Hahn CS, Galler R, et al. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 1990; 44: 649–688. [DOI] [PubMed] [Google Scholar]
- 8.Koonin EV. Computer-assisted identification of a putative methyltransferase domain in Ns5 protein of flaviviruses and lambda-2 protein of reovirus. J Gen Virol 1993; 74: 733–740. [DOI] [PubMed] [Google Scholar]
- 9.Zhou YS, Ray D, Zhao YW, et al. Structure and function of flavivirus NS5 methyltransferase. J Virol 2007; 81: 3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu JQ Liu WC andGong P.. A structural overview of RNA-dependent RNA polymerases from the Flaviviridae family. Int J Mol Sci 2015; 16: 12943–12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou G, Chen YL, Dong H, et al. Functional analysis of two cavities in flavivirus NS5 polymerase. J Biol Chem 2011; 286: 14362–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deval J Symons JA andBeigelman L.. Inhibition of viral RNA polymerases by nucleoside and nucleotide analogs: therapeutic applications against positive-strand RNA viruses beyond hepatitis C virus. Curr Opin Virol 2014; 9: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puig-Basagoiti F, Tilgner M, Forshey BM, et al. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob Agents Chemother 2006; 50: 1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Clercq E. A 40-year journey in search of selective antiviral chemotherapy. Annu Rev Pharmacol Toxicol 2011; 51: 1–24. [DOI] [PubMed] [Google Scholar]
- 15.Jordheim LP, Durantel D, Zoulim F, et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 2013; 12: 447–464. [DOI] [PubMed] [Google Scholar]
- 16.Benhamou Y Tubiana R andThibault V.. Tenofovir disoproxil fumarate in patients with HIV and lamivudine-resistant hepatitis B virus. N Engl J Med 2003; 348: 177–178. [DOI] [PubMed] [Google Scholar]
- 17.Ray AS Fordyce MW andHitchcock MJM.. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res 2016; 125: 63–70. [DOI] [PubMed] [Google Scholar]
- 18.Huang YS, Chang SY, Sheng WH, et al. Virological response to tenofovir disoproxil fumarate in HIV-positive patients with lamivudine-resistant hepatitis B virus coinfection in an area hyperendemic for hepatitis B virus infection. PLoS One 2016; 11: e0169228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam YF, Seto WK, Wong D, et al. Seven-year treatment outcome of entecavir in a real-world cohort: effects on clinical parameters, HBsAg and HBcrAg levels. Clin Trans Gastroenterol 2017; 8: e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stedman C. Sofosbuvir, a NS5B polymerase inhibitor in the treatment of hepatitis C: a review of its clinical potential. Ther Adv Gastroenterol 2014; 7: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Clercq E andHolý A.. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov 2005; 4: 928–940. [DOI] [PubMed] [Google Scholar]
- 22.Chen YL, Yin Z, Duraiswamy J, et al. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob Agents Chemother 2010; 54: 2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eyer L, Nencka R, Huvarova I, et al. Nucleoside inhibitors of zika virus. J Infect Dis 2016; 214: 707–711. [DOI] [PubMed] [Google Scholar]
- 24.Eyer L, Valdes JJ, Gil VA, et al. Nucleoside inhibitors of tick-borne encephalitis virus. Antimicrob Agents Chemother 2015; 59: 5483–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacramento CQ, de Melo GR, de Freitas CS, et al. The clinically approved antiviral drug sofosbuvir inhibits zika virus replication. Sci Rep 2017; 7: 40920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin Z, Chen YL, Schul W, et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci USA 2009; 106: 20435–20439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vernekar SK Qiu L andZacharias J.. Synthesis and antiviral evaluation of 4′-(1,2,3-triazol-1-yl)thymidines. MedChemComm 2014; 5: 603–608. [Google Scholar]
- 28.Vernekar SK, Qiu L, Zhang J, et al. 5′-Silylated 3′-1,2,3-triazolyl thymidine analogues as inhibitors of West Nile Virus and Dengue Virus. J Med Chem 2015; 58: 4016–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borowski P, Lang M, Haag A, et al. Characterization of imidazo[4,5-d]pyridazine nucleosides as modulators of unwinding reaction mediated by West Nile virus nucleoside triphosphatase/helicase: evidence for activity on the level of substrate and/or enzyme. Antimicrob Agents Chemother 2002; 46: 1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borowski P, Deinert J, Schalinski S, et al. Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses. Eur J Biochem 2003; 270: 1645–1653. [DOI] [PubMed] [Google Scholar]
- 31.Crance JM, Scaramozzino N, Jouan A, et al. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res 2003; 58: 73–79. [DOI] [PubMed] [Google Scholar]
- 32.Leyssen P, Balzarini J, De Clercq E, et al. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of inosine monophosphate dehydrogenase. J Virol 2005; 79: 1943–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrey JD, Smee DF, Sidwell RW, et al. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antiviral Res 2002; 55: 107–116. [DOI] [PubMed] [Google Scholar]
- 34.Ojwang JO, Ali S, Smee DF, et al. Broad-spectrum inhibitor of viruses in the Flaviviridae family. Antiviral Res 2005; 68: 49–55. [DOI] [PubMed] [Google Scholar]
- 35.Orlov AA, Drenichev MS, Oslovsky VE, et al. New tools in nucleoside toolbox of tick-borne encephalitis virus reproduction inhibitors. Bioorg Med Chem Lett 2017; 27: 1267–1273. [DOI] [PubMed] [Google Scholar]
- 36.Lescar J andCanard B.. RNA-dependent RNA polymerases from flaviviruses and Picornaviridae. Curr Opin Struct Biol 2009; 19: 759–767. [DOI] [PubMed] [Google Scholar]
- 37.Choi KH andRossmann MG.. RNA-dependent RNA polymerases from Flaviviridae. Curr Opin Struct Biol 2009; 19: 746–751. [DOI] [PubMed] [Google Scholar]
- 38.Behnam MAM, Nitsche C, Boldescu V, et al. The medicinal chemistry of Dengue virus. J Med Chem 2016; 59: 5622–5649. [DOI] [PubMed] [Google Scholar]
- 39.Boldescu V, Behnam MAM, Vasilakis N, et al. Broad-spectrum agents for flaviviral infections: Dengue, Zika and beyond. Nat Rev Drug Discov 2017; 16: 565–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Clercq E andNeyts J.. Antiviral agents acting as DNA or RNA chain terminators. Handb Exp Pharmacol 2009; 189: 53–84. [DOI] [PubMed] [Google Scholar]
- 41.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol 2004; 2: 704–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cihlar T andRay AS.. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res 2010; 85: 39–58. [DOI] [PubMed] [Google Scholar]
- 43.Cho A, Saunders OL, Butler T, et al. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg Med Chem Lett 2012; 22: 2705–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016; 531: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo MK, Jordan R, Arvey A, et al. GS-5734 and its parent nucleoside analog inhibit filo-, pneumo-, and paramyxoviruses. Sci Rep 2017; 7: 43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eldrup AB, Allerson CR, Bennett CF, et al. Structure-activity relationship of purine ribonucleosides for inhibition of hepatitis C virus RNA-dependent RNA polymerase. J Med Chem 2004; 47: 2283–2295. [DOI] [PubMed] [Google Scholar]
- 47.Carroll SS, Tomassini JE, Bosserman M, et al. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J Biol Chem 2003; 278: 11979–11984. [DOI] [PubMed] [Google Scholar]
- 48.Carroll SS, Koeplinger K, Vavrek M, et al. Antiviral efficacy upon administration of a HepDirect prodrug of 2′-C-methylcytidine to hepatitis C virus-infected chimpanzees. Antimicrob Agents Chemother 2011; 55: 3854–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Migliaccio G, Tomassini JE, Carroll SS, et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem 2003; 278: 49164–49170. [DOI] [PubMed] [Google Scholar]
- 50.Olsen DB, Eldrup AB, Bartholomew L, et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob Agents Chemother 2004; 48: 3944–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lefebvre DJ, De Vleeschauwer AR, Goris N, et al. Proof of concept for the inhibition of foot-and-mouth disease virus replication by the anti-viral drug 2′-C-methylcytidine in severe combined immunodeficient mice. Transbound Emerg Dis 2014; 61: E89–E91. [DOI] [PubMed] [Google Scholar]
- 52.Rocha-Pereira J, Jochmans D, Dallmeier K, et al. Inhibition of norovirus replication by the nucleoside analogue 2′-C-methylcytidine. Biochem Biophys Res Commun 2012; 427: 796–800. [DOI] [PubMed] [Google Scholar]
- 53.Rocha-Pereira J, Jochmans D, Debing Y, et al. The viral polymerase inhibitor 2′-C-methylcytidine inhibits Norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J Virol 2013; 87: 11798–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eyer L, Smidkova M, Nencka R, et al. Structure-activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res 2016; 133: 119–129. [DOI] [PubMed] [Google Scholar]
- 55.Flint M, McMullan LK, Dodd KA, et al. Inhibitors of the tick-borne, hemorrhagic fever-associated flaviviruses. Antimicrob Agents Chemother 2014; 58: 3206–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JC, Tseng CK, Wu YH, et al. Characterization of the activity of 2′-C-methylcytidine against dengue virus replication. Antiviral Res 2015; 116: 1–9. [DOI] [PubMed] [Google Scholar]
- 57.Mateo R Nagamine CM andKirkegaard K.. Suppression of drug resistance in Dengue virus. MBio 2015; 6: e01960–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeo KL, Chen YL, Xu HY, et al. Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob Agents Chemother 2015; 59: 2086–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanko K, Eggermont K, Patel A, et al. Replication of the Zika virus in different iPSC-derived neuronal cells and implications to assess efficacy of antivirals. Antiviral Res 2017; 145: 82–86. [DOI] [PubMed] [Google Scholar]
- 60.Hercik K, Brynda J, Nencka R, et al. Structural basis of Zika virus methyltransferase inhibition by sinefungin. Arch Virol 2017; 162: 2091–2096. [DOI] [PubMed] [Google Scholar]
- 61.Eyer L, Kondo H, Zouharova D.et al. Escape of tick-borne flavivirus from 2′ -C-methylated nucleoside antivirals is mediated by a single conservative mutation in NS5 that has a dramatic effect on viral fitness. J Virol 2017; e01028–17. [DOI] [PMC free article] [PubMed]
- 62.Zmurko J, Marques RE, Schols D, et al. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl Trop Dis 2016; 10: e0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Julander JG, Jha AK, Choi JA, et al. Efficacy of 2′-C-methylcytidine against yellow fever virus in cell culture and in a hamster model. Antiviral Res 2010; 86: 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sofia MJ, Chang W, Furman PA, et al. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J Med Chem 2012; 55: 2481–2531. [DOI] [PubMed] [Google Scholar]
- 65.Keating GM andVaidya A.. Sofosbuvir: first global approval. Drugs 2014; 74: 273–282. [DOI] [PubMed] [Google Scholar]
- 66.Arnold JJ, Sharma SD, Feng JY, et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog 2012; 8: e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feng JY, Xu YL, Barauskas O, et al. Role of mitochondrial RNA polymerase in the toxicity of nucleotide inhibitors of hepatitis C virus. Antimicrob Agents Chemother 2016; 60: 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu HT, Hassounah SA, Colby-Germinario SP, et al. Purification of Zika virus RNA-dependent RNA polymerase and its use to identify small-molecule Zika inhibitors. J Antimicrob Chemoth 2017; 72: 727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bullard-Feibelman KM, Govero J, Zhu Z, et al. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res 2017; 137: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mumtaz N, Jimmerson LC, Bushman LR, et al. Cell-line dependent antiviral activity of sofosbuvir against Zika virus. Antiviral Res 2017; 146: 161–163. [DOI] [PubMed] [Google Scholar]
- 71.Ferreira AC, Zaverucha-do-Valle C, Reis PA, et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci Rep 2017; 7: 9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen YL, Yin Z, Lakshminarayana SB, et al. Inhibition of Dengue Virus by an ester prodrug of an adenosine analog. Antimicrob Agents Chemother 2010; 54: 3255–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen YL Yokokawa F andShi PY.. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral Res 2015; 122: 12–19. [DOI] [PubMed] [Google Scholar]
- 74.Latour DR, Jekle A, Javanbakht H, et al. Biochemical characterization of the inhibition of the dengue virus RNA polymerase by beta-D-2′-ethynyl-7-deaza-adenosine triphosphate. Antiviral Res 2010; 87: 213–222. [DOI] [PubMed] [Google Scholar]
- 75.Yin XQ andSchneller SW1.. Deaza-5′-noraisteromycin. Nucleosides Nucleotides Nucleic Acids 2004; 23: 67–76. [DOI] [PubMed] [Google Scholar]
- 76.Deng YQ, Zhang NN, Li CF, et al. Adenosine analog NITD008 is a potent inhibitor of Zika virus. Open Forum Infect Dis 2016; 3: ofw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lo MK, Shi PY, Chen YL, et al. In vitro antiviral activity of adenosine analog NITD008 against tick borne flaviviruses. Antiviral Res 2016; 130: 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McGuigan C, Serpi M, Slusarczyk M, et al. Anti-flavivirus activity of different tritylated pyrimidine and purine nucleoside analogues. Chem Open 2016; 5: 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klumpp K, Leveque V, Le Pogam S, et al. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol Chem 2006; 281: 3793–3799. [DOI] [PubMed] [Google Scholar]
- 80.Klumpp K, Kalayanov G, Ma H, et al. Deoxy-4′-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′-alpha-hydroxyl groups. J Biol Chem 2008; 283: 2167–2175. [DOI] [PubMed] [Google Scholar]
- 81.Smith DB, Kalayanov G, Sund C, et al. The design, synthesis, and antiviral activity of 4′-azidocytidine analogues against hepatitis C virus replication: the discovery of 4′-azidoarabinocytidine. J Med Chem 2009; 52: 219–223. [DOI] [PubMed] [Google Scholar]
- 82.Hotard AL, He B, Nichol ST, et al. 4′-Azidocytidine (R1479) inhibits henipaviruses and other paramyxoviruses with high potency. Antiviral Res 2017; 144: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nguyen NM, Tran CNB, Phung LK, et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis 2013; 207: 1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen YL, Ghafar NA, Karuna R, et al. Activation of peripheral blood mononuclear cells by Dengue virus infection depotentiates balapiravir. J Virol 2014; 88: 1740–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rondla R, Coats SJ, McBrayer TR, et al. Anti-hepatitis C virus activity of novel beta-d-2′-C-methyl-4′ -azido pyrimidine nucleoside phosphoramidate prodrugs. Antivir Chem Chemother 2009; 20: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nilsson M, Kalayanov G, Winqvist A, et al. Discovery of 4′-azido-2′-deoxy-2′-C-methyl cytidine and prodrugs thereof: a potent inhibitor of hepatitis C virus replication. Bioorg Med Chem Lett 2012; 22: 3265–3268. [DOI] [PubMed] [Google Scholar]
- 87.Taylor R, Kotian P, Warren T, et al. BCX4430-A broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J Infect Public Health 2016; 9: 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Clercq E. C-nucleosides to be revisited. J Med Chem 2016; 59: 2301–2311. [DOI] [PubMed] [Google Scholar]
- 89.Warren TK, Wells J, Panchal RG, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014; 508: 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eyer L, Zouharova D, Sirmarova J, et al. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses. Antiviral Res 2017; 142: 63–67. [DOI] [PubMed] [Google Scholar]
- 91.Julander JG, Siddharthan V, Evans J, et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antiviral Res 2017; 137: 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Julander JG, Bantia S, Taubenheim BR, et al. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a hamster model. Antimicrob Agents Chemother 2014; 58: 6607–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Julander JG, Furuta Y, Shafer K, et al. Activity of T-1106 in a hamster model of yellow fever virus infection. Antimicrob Agents Chemother 2007; 51: 1962–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Julander JG, Shafer K, Smee DF, et al. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother 2009; 53: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Julander J, Shafer K, Smee D, et al. Efficacy of T-1106 or T-705, alone or in combination with ribavirin, in the treatment of hamsters infected with yellow fever virus. Antiviral Res 2008; 78: A34. [Google Scholar]
- 96.Wu R, Smidansky ED, Oh HS, et al. Synthesis of a 6-methyl-7-deaza analogue of adenosine that potently inhibits replication of polio and Dengue viruses. J Med Chem 2010; 53: 7958–7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Angusti A, Manfredini S, Durini E, et al. Design, synthesis and anti Flaviviridae activity of N-6-, 5′,3′-O- and 5′,2′-O-substituted adenine nucleoside analogs. Chem Pharm Bull 2008; 56: 423–432. [DOI] [PubMed] [Google Scholar]
- 98.Morrey JD, Taro BS, Siddharthan V, et al. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 2008; 80: 377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Drenichev MS, Oslovsky VE, Sun L, et al. Modification of the length and structure of the linker of N-6-benzyladenosine modulates its selective antiviral activity against enterovirus 71. Eur J Med Chem 2016; 111: 84–94. [DOI] [PubMed] [Google Scholar]
- 100.Tararov VI, Tijsma A, Kolyachkina SV, et al. Chemical modification of the plant isoprenoid cytokinin N-6-isopentenyladenosine yields a selective inhibitor of human enterovirus 71 replication. Eur J Med Chem 2015; 90: 406–413. [DOI] [PubMed] [Google Scholar]
- 101.Orlov AA, Chistov AA, Kozlovskaya LI, et al. Rigid amphipathic nucleosides suppress reproduction of the tick-borne encephalitis virus. Med Chem Commun 2016; 7: 495–499. [Google Scholar]
- 102.Chatelain G, Debing Y, De Burghgraeve T, et al. In search of flavivirus inhibitors: evaluation of different tritylated nucleoside analogues. Eur J Med Chem 2013; 65: 249–255. [DOI] [PubMed] [Google Scholar]
- 103.De Burghgraeve T, Selisko B, Kaptein S, et al. 3′,5′ Di-O-trityluridine inhibits in vitro flavivirus replication. Antiviral Res 2013; 98: 242–247. [DOI] [PubMed] [Google Scholar]
- 104.Saudi M, Zmurko J, Kaptein S, et al. In search of flavivirus inhibitors part 2: tritylated, diphenylmethylated and other alkylated nucleoside analogues. Eur J Med Chem 2014; 76: 98–109. [DOI] [PubMed] [Google Scholar]
- 105.Koonin EV andDolja VV.. Evolution taxonomy of positive-strand RNA viruses – implications of comparative-analysis of amino-acid-sequences. Crit Rev Biochem Mol Biol 1993; 28: 375–430. [DOI] [PubMed] [Google Scholar]
- 106.Cleaves GR andDubin DT.. Methylation status of intracellular dengue type-2 40-S RNA. Virology 1979; 96: 159–165. [DOI] [PubMed] [Google Scholar]
- 107.Dong H, Fink K, Zuest R, et al. Flavivirus RNA methylation. J Gen Virol 2014; 95: 763–778. [DOI] [PubMed] [Google Scholar]
- 108.Chung KY, Dong HP, Chao AT, et al. Higher catalytic efficiency of N-7-methylation is responsible for processive N-7 and 2′-O methyltransferase activity in dengue virus. Virology 2010; 402: 52–60. [DOI] [PubMed] [Google Scholar]
- 109.Lim SP, Wen DY, Yap TL, et al. A scintillation proximity assay for dengue virus NS5 2′-O-methyltransferase-kinetic and inhibition analyses. Antiviral Res 2008; 80: 360–369. [DOI] [PubMed] [Google Scholar]
- 110.Lim SP, Sonntag LS, Noble C, et al. Small molecule inhibitors that selectively block dengue virus methyltransferase. J Biol Chem 2011; 286: 6233–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen H, Liu L, Jones SA, et al. Selective inhibition of the West Nile virus methyltransferase by nucleoside analogs. Antiviral Res 2013; 97: 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yates MK, Raje MR, Chatterjee P, et al. Flex-nucleoside analogues – novel therapeutics against filoviruses. Bioorg Med Chem Lett 2017; 27: 2800–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peters HL, Jochmans D, de Wilde AH, et al. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg Med Chem Lett 2015; 25: 2923–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seley KL, Zhang L, Hagos A, et al. “ Fleximers”. Design and synthesis of a new class of novel shape-modified nucleosides. J Org Chem 2002; 67: 3365–3373. [DOI] [PubMed] [Google Scholar]
- 115.Seley KL, Quirk S, Salim S, et al. Unexpected inhibition of S-adenosyl-L-homocysteine hydrolase by a guanosine nucleoside. Bioorg Med Chem Lett 2003; 13: 1985–1988. [DOI] [PubMed] [Google Scholar]
- 116.Quirk S andSeley KL.. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry 2005; 44: 10854–10863. [DOI] [PubMed] [Google Scholar]
- 117.Gallinari P, Brennan D, Nardi C, et al. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J Virol 1998; 72: 6758–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hodgman TC. A new superfamily of replicative proteins. Nature 1988; 333: 22–23. [DOI] [PubMed] [Google Scholar]
- 119.Kim JL, Morgenstern KA, Griffith JP, et al. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 1998; 6: 89–100. [DOI] [PubMed] [Google Scholar]
- 120.Yao NH, Hesson T, Cable M, et al. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol 1997; 4: 463–467. [DOI] [PubMed] [Google Scholar]
- 121.Tai CL, Chi WK, Chen DS, et al. The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3). J Virol 1996; 70: 8477–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bretner M, Baier A, Kopańska K, et al. Synthesis and biological activity of 1H-benzotriazole and 1H-benzimidazole analogues – inhibitors of the NTpase/helicase of HCV and of some related Flaviviridae. Antivir Chem Chemother 2005; 16: 315–326. [DOI] [PubMed] [Google Scholar]
- 123.Borowski P, Kuehl R, Mueller O, et al. Biochemical properties of a minimal functional domain with ATP-binding activity of the NTPase/helicase of hepatitis C virus. Eur J Biochem 1999; 266: 715–723. [DOI] [PubMed] [Google Scholar]
- 124.Borowski P, Lang M, Niebuhr A, et al. Inhibition of the helicase activity of HCV NTPase/helicase by 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide-5′-triphosphate (ribavirin-TP). Acta Biochim Pol 2001; 48: 739–744. [PubMed] [Google Scholar]
- 125.Borowski P, Mueller O, Niebuhr A, et al. ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochim Pol 2000; 47: 173–180. [PubMed] [Google Scholar]
- 126.Bretner M, Schalinski S, Borowski P, et al. 5′-O-fluorosulfonylbenzoyl esters of purine nucleosides as potential inhibitors of NTPase/helicase and polymerase of Flaviviridae viruses. Nucleosides Nucleotides Nucleic Acids 2003; 22: 1531–1533. [DOI] [PubMed] [Google Scholar]
- 127.Bretner M, Schalinski S, Haag A, et al. Synthesis and evaluation of ATP-binding site directed potential inhibitors of nucleoside triphosphatases/helicases and polymerases of hepatitis C and other selected Flaviviridae viruses. Antivir Chem Chemother 2004; 15: 35–42. [DOI] [PubMed] [Google Scholar]
- 128.Eriksson B, Helgstrand E, Johansson NG, et al. Inhibition of influenza-virus ribonucleic-acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother 1977; 11: 946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crotty S Cameron CE andAndino R.. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA 2001; 98: 6895–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Graci JD andCameron CE.. Quasispecies, error catastrophe, and the antiviral activity of ribavirin. Virology 2002; 298: 175–180. [DOI] [PubMed] [Google Scholar]
- 131.Li CQ, Guillen J, Rabah N, et al. mRNA capping by Venezuelan Equine Encephalitis virus nsP1: functional characterization and implications for antiviral research. J Virol 2015; 89: 8292–8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Graci JD andCameron CE.. Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol 2006; 16: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hultgren C, Milich DR, Weiland O, et al. The antiviral compound ribavirin modulates the T helper (Th)1/Th2 subset balance in hepatitis B and C virus-specific immune responses. J Gen Virol 1998; 79: 2381–2391. [DOI] [PubMed] [Google Scholar]
- 134.Jordan I, Briese T, Fischer N, et al. Ribavirin inhibits West Nile virus replication and cytopathic effect in neural cells. J Infect Dis 2000; 182: 1214–1217. [DOI] [PubMed] [Google Scholar]
- 135.McDowell M, Gonzales SR, Kumarapperuma SC, et al. A novel nucleoside analog, 1-beta-D-ribofuranosyl-3-ethynyl-[1,2,4]triazole (ETAR), exhibits efficacy against a broad range of flaviviruses in vitro. Antiviral Res 2010; 87: 78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Malinoski FJ, Hasty SE, Ussery MA, et al. Prophylactic ribavirin treatment of dengue type-1 infection in rhesus-monkeys. Antiviral Res 1990; 13: 139–150. [DOI] [PubMed] [Google Scholar]
- 137.Gabrielsen B, Phelan MJ, Barthelrosa L, et al. Synthesis and antiviral evaluation of N-carboxamidine-substituted analogs of 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamidine hydrochloride. J Med Chem 1992; 35: 3231–3238. [DOI] [PubMed] [Google Scholar]
- 138.Crutcher WA andMoschella SL.. Double-blind controlled crossover high-dose study of azaribine in psoriasis. Br J Dermatol 1975; 92: 199–205. [DOI] [PubMed] [Google Scholar]
- 139.Dukhan D, Leroy F, Peyronnet J, et al. Synthesis of 5-aza-7-deazaguanine nucleoside derivatives as potential anti-flavivirus agents. Nucleosides Nucleotides Nucleic Acids 2005; 24: 671–674. [DOI] [PubMed] [Google Scholar]
- 140.Korshun VA, Prokhorenko IA, Gontarev SV, et al. New pyrene derivatives for fluorescent labeling of oligonucleotides. Nucleosides Nucleotides 1997; 16: 1461–1464. [Google Scholar]
- 141.Korshun VA, Manasova EV, Balakin KV, et al. New fluorescent nucleoside derivatives – 5-alkynylated 2′-deoxyuridines. Nucleosides Nucleotides 1998; 17: 1809–1812. [Google Scholar]
- 142.Skorobogatyi MV, Malakhov AD, Pchelintseva AA, et al. Fluorescent 5-alkynyl-2′-deoxyuridines: high emission efficiency of a conjugated perylene nucleoside in a DNA duplex. Chembiochem 2006; 7: 810–816. [DOI] [PubMed] [Google Scholar]
- 143.St Vincent MR, Colpitts CC, Ustinov AV, et al. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc Natl Acad Sci USA 2010; 107: 17339–17344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Colpitts CC, Ustinov AV, Epand RF, et al. 5-(Perylen-3-yl) ethynyl-arabino-uridine (aUY11), an arabino-based rigid amphipathic fusion inhibitor, targets virion envelope lipids to inhibit fusion of influenza virus, hepatitis C virus, and other enveloped viruses. J Virol 2013; 87: 3640–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vigant F, Hollmann A, Lee J, et al. The rigid amphipathic fusion inhibitor dUY11 acts through photosensitization of viruses. J Virol 2014; 88: 1849–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li FJ Maag H andAlfredson T.. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci 2008; 97: 1109–1134. [DOI] [PubMed] [Google Scholar]
- 147.Hecker SJ andErion MD.. Prodrugs of phosphates and phosphonates. J Med Chem 2008; 51: 2328–2345. [DOI] [PubMed] [Google Scholar]
- 148.Lane AN andFan TWM.. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 2015; 43: 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Johnson AA, Ray AS, Hanes J, et al. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem 2001; 276: 40847–40857. [DOI] [PubMed] [Google Scholar]
- 150.Lee H Hanes J andJohnson KA.. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 2003; 42: 14711–14719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Locarnini S andWarner N.. Major causes of antiviral drug resistance and implications for treatment of hepatitis B virus monoinfection and coinfection with HIV. Antiviral Ther 2007; 12: H15–H23. [PubMed] [Google Scholar]
- 152.Menendez-Arias L Alvarez M andPacheco B.. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism, of action and resistance. Curr Opin Virol 2014; 8: 1–9. [DOI] [PubMed] [Google Scholar]
- 153.Poveda E, Wyles DL, Mena A, et al. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antiviral Res 2014; 108: 181–191. [DOI] [PubMed] [Google Scholar]
- 154.McGuigan C, Cahard D, Sheeka HM, et al. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J Med Chem 1996; 39: 1748–1753. [DOI] [PubMed] [Google Scholar]
- 155.Lam AM, Espiritu C, Bansal S, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother 2012; 56: 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ross BS, Reddy PG, Zhang HR, et al. Synthesis of diastereomerically pure nucleotide phosphoramidates. J Org Chem 2011; 76: 8311–8319. [DOI] [PubMed] [Google Scholar]
- 157.Peterson LW and, McKenna CE. Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues. Expert Opin Drug Deliv 2009; 6: 405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Becher F, Landman R, Mboup S, et al. Monitoring of didanosine and stavudine intracellular trisphosphorylated anabolite concentrations in HIV-infected patients. AIDS 2004; 18: 181–187. [DOI] [PubMed] [Google Scholar]
- 159.Gao WY, Shirasaka T, Johns DG, et al. Differential phosphorylation of azidothymidine, dideoxycytidine, and dideoxyinosine in resting and activated peripheral-blood mononuclear-cells. J Clin Invest 1993; 91: 2326–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kohler JJ andLewis W.. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ Mol Mutagen 2007; 48: 166–172. [DOI] [PubMed] [Google Scholar]
- 161.Maurer HH. Screening procedures for simultaneous detection of several drug classes used for high throughput toxicological analyses and doping control. A review. Comb Chem High Throughput Screen 2000; 3: 467–480. [DOI] [PubMed] [Google Scholar]
- 162.Mckim JM. Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb Chem High Throughput Screen 2010; 13: 188–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Szymanski P Markowicz M andMikiciuk-Olasik E.. Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int J Mol Sci 2012; 13: 427–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Adachi A andMiura T.. Animal model studies on viral infections. Front Microbiol 2014; 5: 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dowall SD, Graham VA, Rayner E, et al. A susceptible mouse model for Zika virus infection. PLoS Negl Trop Dis 2016; 10: e0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zompi S andHarris E.. Animal models of Dengue virus infection. Viruses 2012; 4: 62–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lee YR, Huang KJ, Lei HY, et al. Suckling mice were used to detect infectious dengue-2 viruses by intracerebral injection of the full-length RNA transcript. Intervirology 2005; 48: 161–166. [DOI] [PubMed] [Google Scholar]
- 168.Sarathy VV, White M, Li L, et al. A lethal murine infection model for Dengue Virus 3 in AG129 mice deficient in type I and II interferon receptors leads to systemic disease. J Virol 2015; 89: 1254–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zellweger RM andShresta S.. Mouse models to study dengue virus immunology and pathogenesis. Front Immunol 2014; 5: 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chan JFW, Zhang AJ, Chan CCS, et al. Zika virus infection in dexamethasone-immunosuppressed mice demonstrating disseminated infection with multi-organ involvement including orchitis effectively treated by recombinant type I interferons. EBioMedicine 2016; 14: 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McArthur MA, Suderman MT, Mutebi JP, et al. Molecular characterization of a hamster viscerotropic strain of yellow fever virus. J Virol 2003; 77: 1462–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Govero J, Esakky P, Scheaffer SM, et al. Zika virus infection damages the testes in mice. Nature 2016; 540: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gubler D Kuno G andMarkoff L.. Flaviviruses In: Knipe DM andHowley PM (eds) Fields virology. 5th ed Philadelphia, PA: Lippincott Williams & Wilkins, 2007, pp.1153–1253. [Google Scholar]
- 174.Carey I andHarrison PM.. Monotherapy versus combination therapy for the treatment of chronic hepatitis B. Expert Opin Investig Drug 2009; 18: 1655–1666. [DOI] [PubMed] [Google Scholar]
- 175.Wong GLH Wong VWS andChan HLY.. Combination therapy of interferon and nucleotide/nucleoside analogues for chronic hepatitis B. J Viral Hepat 2014; 21: 825–834. [DOI] [PubMed] [Google Scholar]
- 176.Su TH andLiu CJ.. Combination therapy for chronic hepatitis B: current updates and perspectives. Gut Liver 2017; 11: 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]