

Abstract
Objective
To evaluate the safety and preliminary pharmacokinetics of a pharmaceutical formulation of purified cannabidiol (CBD) in children with Dravet syndrome.
Methods
Patients aged 4–10 years were randomized 4:1 to CBD (5, 10, or 20 mg/kg/d) or placebo taken twice daily. The double-blind trial comprised 4-week baseline, 3-week treatment (including titration), 10-day taper, and 4-week follow-up periods. Completers could continue in an open-label extension. Multiple pharmacokinetic blood samples were taken on the first day of dosing and at end of treatment for measurement of CBD, its metabolites 6-OH-CBD, 7-OH-CBD, and 7-COOH-CBD, and antiepileptic drugs (AEDs; clobazam and metabolite N-desmethylclobazam [N-CLB], valproate, levetiracetam, topiramate, and stiripentol). Safety assessments were clinical laboratory tests, physical examinations, vital signs, ECGs, adverse events (AEs), seizure frequency, and suicidality.
Results
Thirty-four patients were randomized (10, 8, and 9 to the 5, 10, and 20 mg/kg/d CBD groups, and 7 to placebo); 32 (94%) completed treatment. Exposure to CBD and its metabolites was dose-proportional (AUC0–t). CBD did not affect concomitant AED levels, apart from an increase in N-CLB (except in patients taking stiripentol). The most common AEs on CBD were pyrexia, somnolence, decreased appetite, sedation, vomiting, ataxia, and abnormal behavior. Six patients taking CBD and valproate developed elevated transaminases; none met criteria for drug-induced liver injury and all recovered. No other clinically relevant safety signals were observed.
Conclusions
Exposure to CBD and its metabolites increased proportionally with dose. An interaction with N-CLB was observed, likely related to CBD inhibition of cytochrome P450 subtype 2C19. CBD resulted in more AEs than placebo but was generally well-tolerated.
Classification of evidence
This study provides Class I evidence that for children with Dravet syndrome, CBD resulted in more AEs than placebo but was generally well-tolerated.
Data on the efficacy and safety of cannabidiol (CBD) in various epilepsies are emerging. Randomized controlled trials have suggested that an orally administered pharmaceutical formulation of purified CBD as an add-on to existing antiepileptic drugs (AEDs) has efficacy in treating convulsive seizures associated with Dravet syndrome (DS) and drop seizures associated with Lennox-Gastaut syndrome.1,2 Open-label trials support these findings and suggest there is efficacy in other generalized and focal epilepsies, including tuberous sclerosis complex and febrile infection-related epilepsy syndrome.3–5 Adverse effects of CBD in patients with treatment-resistant epilepsy are well-documented in randomized controlled and open-label trials, and most commonly include somnolence, diarrhea, and decreased appetite.1–5
Data are needed on the pharmacokinetics (PK) of CBD in patients with epilepsy. CBD is metabolized mainly by the cytochrome P450 (CYP) 2C19 and CYP3A4 isoenzymes,6 which are induced by several AEDs (e.g., carbamazepine, topiramate, phenytoin) and are inhibited by others (e.g., valproate).7 CBD can also inhibit the CYP2C family of isozymes (inhibition constant [Ki] = 1–10 μM) and CYP3A4 (Ki = 1 μM).6 Thus, drug–drug interactions could exist between CBD and other AEDs. Several uncontrolled studies found that the level of the active clobazam metabolite, N-desmethylclobazam (N-CLB), is increased by CBD,8–10 and 1 study noted interactions with several other AEDs.9
In preparation for a larger randomized controlled trial of CBD, this trial was conducted to evaluate the dose-ranging safety, tolerability, and preliminary PK data of add-on CBD compared with add-on placebo in children with DS.
Methods
The primary research purpose was to evaluate the safety of multiple doses of CBD compared with placebo (Class I evidence).
Patients
Patients aged 4–10 years with DS, taking 1 or more AEDs and experiencing fewer than 4 convulsive seizures during a 4-week baseline period, were eligible for inclusion. Convulsive seizures were defined as tonic–clonic, tonic, clonic, and atonic. All medications and interventions for epilepsy, including ketogenic diet and vagus nerve stimulation, were stable for 4 weeks prior to screening and remained unchanged throughout the trial.
Standard protocol approvals, registrations, and patient consents
The trial (ClinicalTrials.gov identifier: NCT02091206) was approved by the institutional review board or independent ethics committee at each trial site. All patients or parents/legal representatives provided written informed assent/consent before trial onset.
Trial design
This multisite, randomized, double-blind, placebo-controlled, parallel-group trial was conducted at 12 sites in the United States and United Kingdom between October 2014 and March 2015. The trial comprised 4-week baseline, 3-week treatment, 10-day taper, and 4-week safety follow-up periods. Patients who completed the trial could enter a long-term open-label extension. An independent data safety monitoring committee (DSMC) reviewed safety and PK data to determine the target dose and titration regimen for use in future trials.
Following the 4-week baseline period, patients who satisfied all eligibility criteria were randomly assigned (through central randomization via interactive voice/web response system) at a 4:1 ratio to receive 1 of 3 doses of CBD (5, 10, or 20 mg/kg/d) or placebo of equivalent volume as the treatment doses, added to their stable AED regimens. CBD oral solution (GW Pharmaceuticals Ltd., London, UK) contained 25 or 100 mg/mL CBD; placebo was identical except for the absence of CBD. Study medication was administered twice daily starting at 2.5 mg/kg/d (or equivalent volume for placebo) and increasing by 2.5–5.0 mg/kg every other day to randomized dose, which took 3, 7, and 11 days for the 5, 10, and 20 mg/kg/d doses, respectively. Dose reductions were permitted for adverse events (AEs).
Protocol amendment
The trial protocol was revised in November 2014 to include a review of the diagnosis and classification of seizure types in each patient by an independent panel, appointed by the Epilepsy Study Consortium, using a standard protocol to confirm the diagnosis of DS.1
PK assessments
PK blood samples were taken predose, 2–3 hours postdose, and 4–6 hours postdose on the first day of dosing and again at the same time intervals after 3 weeks of treatment (day 22). The 3 time points (predose, 2.5 hours, and 5 hours) are the minimum required to generate an area under the curve (AUC). Only sparse data were used to derive exposure information; therefore, no other PK parameters were derived. Plasma concentrations (measured as total) were analyzed by ultra-performance liquid chromatography with tandem mass spectrometry using a validated method for the following: CBD, 6-hydroxy-cannabidiol (6-OH-CBD), 7-carboxy-cannabidiol (7-COOH-CBD), clobazam, N-CLB, valproate, levetiracetam, topiramate, and stiripentol. The 7-hydroxy-cannabidiol (7-OH-CBD) metabolite of CBD was also assessed, but due to analytical issues related to the reference material batch used, the data could only be considered qualitative and are not presented.
Safety assessments
Safety assessments included laboratory assessments (hematology, biochemistry, and urinalysis), physical examinations, monitoring of vital signs and ECGs, and assessments of AEs, seizure frequency, and suicidality (Columbia-Suicide Severity Rating Scale, performed by a licensed assessor for the instrument and in children at least 6 years of age).11
Data analyses
All PK and safety data were summarized using descriptive statistics; no formal statistical comparisons were planned or performed. PK exposures were expressed as AUC0–t estimated using a linear trapezoidal linear interpolation method using WinNonlin v6.3. Dose proportionality was assessed by regression analysis.
Results
Patient disposition and baseline demographics
A total of 34 patients were randomized: 30 patients at 8 sites in the United States and 4 patients at 3 sites in the United Kingdom; 32 patients (94%) completed the treatment period, and of these, 24 (75%) entered an open-label extension trial (figure 1). Two patients on CBD were withdrawn during the treatment period. All 34 randomized patients were included in the safety analysis set: 10, 8, and 9 patients in the 5, 10, and 20 mg/kg/d CBD groups, respectively, and 7 patients in the placebo group. All PK analyses were conducted using the safety analysis set.
Figure 1. Disposition of patients.

There was a delay of ≥23 weeks between treatment completion and when the open-label extension trial was available for enrollment. All pharmacokinetic analyses were conducted using the safety analysis set. CBD = cannabidiol.
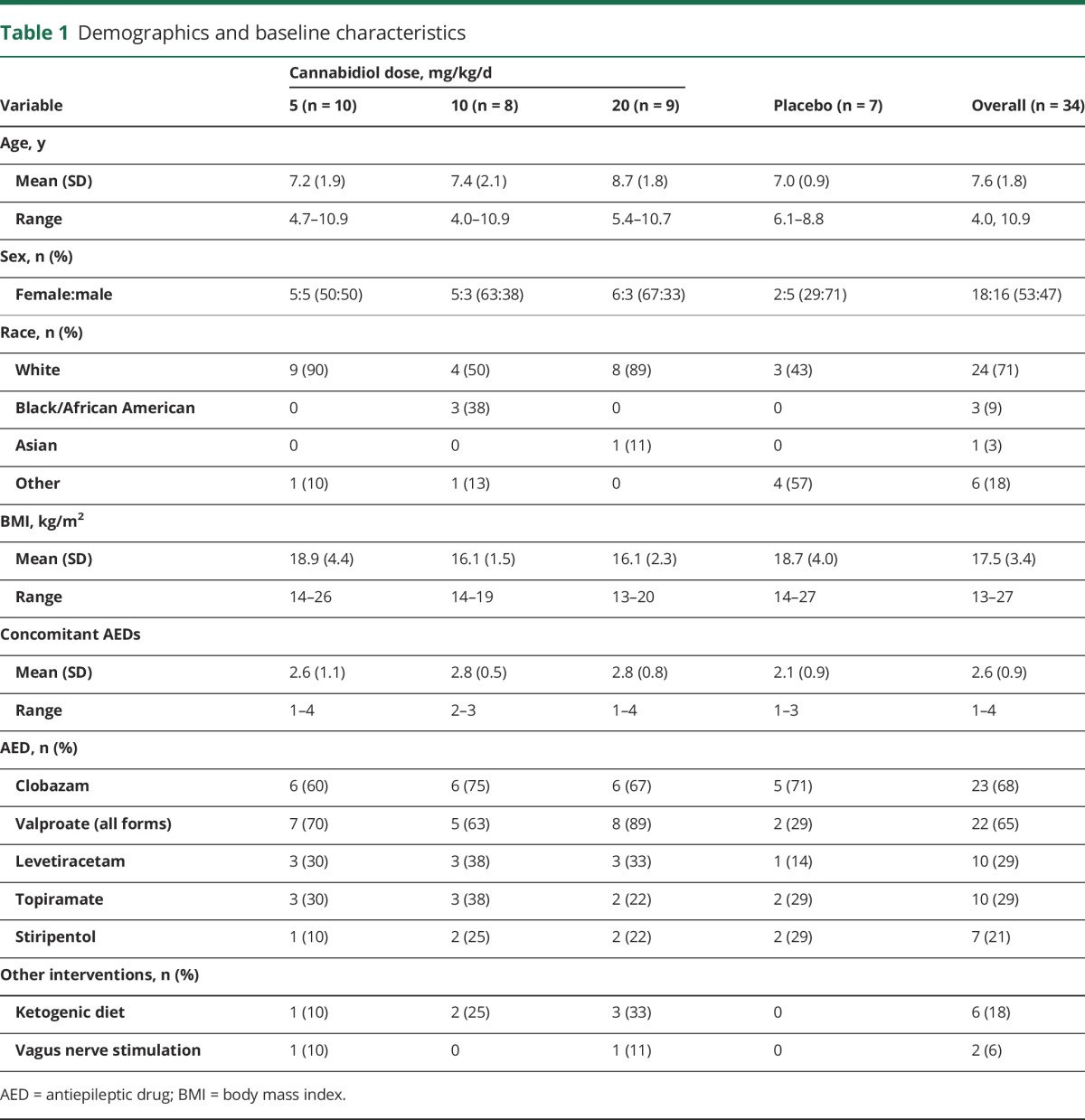
Demographics and baseline characteristics were comparable between groups (table 1). Seventy-one percent of patients were white and the mean age was 7.6 years (range 4.0–10.9 years). The most common concomitant AEDs were clobazam (68%) and valproate (all forms; 65%).
Table 1.
Demographics and baseline characteristics
Pharmacokinetics
Mean plasma concentrations of CBD, 6-OH-CBD, and 7-COOH-CBD over time following single (1.25 mg/kg) or multiple (5, 10, or 20 mg/kg/d) dosing of CBD are shown in figure 2A; data are provided in table e-1 (links.lww.com/WNL/A306). Exposures across the groups on day 1 were consistent with the common starting dose of 1.25 mg/kg. At all doses and timepoints, 7-COOH-CBD was the most abundant circulating metabolite while concentrations of 6-OH-CBD were consistently <10% those of CBD, based on AUC0–t. For each analyte, exposure (based on AUC0–t at end of treatment) increased in a dose-related manner, with no major deviation from dose proportionality (figure 2B). Qualitative data generated for the 7-OH-CBD metabolite also showed a dose-proportional increase, with plasma exposures less than that of CBD. At end of treatment, 7-COOH-CBD levels were 13–17 times those of CBD. Total variability in AUC0–t for CBD was moderate to high (% coefficient of variation [%CV] 20%–121%), and was substantially higher for the metabolites (%CV 57%–1750%) (table e-2). There was no effect of repeated CBD administration on the 6-OH-CBD:CBD ratio for AUC0–t, but there was a marked increase in the 7-COOH-CBD:CBD ratio at end of treatment, suggesting a greater accumulation of this metabolite and that this was a major route of biotransformation (table e-2).
Figure 2. Plasma pharmacokinetics (PK) of cannabidiol (CBD) and metabolites.

(A) Arithmetic mean plasma concentrations of CBD, 6-hydroxy-cannabidiol (6-OH-CBD), and 7-carboxy-cannabidiol (7-COOH-CBD) over time at baseline (day 1) and end of treatment (day 22). Note that the day 1 dose was 1.25 mg/kg for all 3 treatment groups. (B) Dose linearity for CBD, 6-OH-CBD, and 7-COOH-CBD exposures (based on area under the curve [AUC]0–t) at end of treatment (day 22). Qualitative data generated for the 7-OH-CBD metabolite also showed a dose-proportional increase. R2 values relate to geometric mean fitted regression lines.
Following multiple dosing of CBD in patients on regimens containing clobazam (CLB; n = 17 with end of treatment data), there was no relevant change in plasma exposure to CLB; however, there was a notable increase (mean % increase ≥166% in all dose groups) in mean N-CLB concentrations (range −10% to 526%) (figure 3A) and mean N-CLB:CLB ratios (range −43% to 664%) (table e-3, links.lww.com/WNL/A306), with similar mean increases for all 3 CBD doses (5, 10, 20 mg/kg/d). These increases in N-CLB were not observed in patients taking stiripentol, a known potent CYP2C19 inhibitor (n = 4 with end of treatment data) (figure 3B and table e-3). CBD had no effect on systemic exposure to any other AEDs investigated (valproate, levetiracetam, topiramate, or stiripentol), although sample sizes were small (figure e-1, links.lww.com/WNL/A305). Dedicated drug–drug interaction studies with valproate and stiripentol, as well as trials to explore the effects on CYP3A4 and CYP2C19 isozymes as a perpetrator and victim drug, are ongoing.
Figure 3. Percentage change from baseline to end of treatment in plasma clobazam (CLB) and N-desmethylclobazam (N-CLB) concentrations.

(A) By treatment group. Note that 2 patients in the 5 mg/kg/d cannabidiol (CBD) group reduced CLB dose during treatment, but no reason was listed. (B) By stiripentol use in all patients on CBD. Error bars: SEM.
Safety
Treatment-emergent AEs (TEAEs) were reported in all groups: CBD 5 mg/kg/d—8/10 patients (80%); 10 mg/kg/d—5/8 patients (63%); 20 mg/kg/d—7/9 patients (78%); placebo—6/7 patients (86%). TEAEs experienced by >1 patient overall are provided in table 2. TEAEs occurring in 3 or more patients across the combined CBD groups were pyrexia, somnolence, decreased appetite, sedation, vomiting, ataxia, and abnormal behavior. A dose relation was observed for decreased appetite only. Two patients discontinued because of AEs: 1 in the 10 mg/kg/d CBD group due to pyrexia and maculopapular rash, and 1 in the 20 mg/kg/d CBD group due to elevated transaminases meeting a withdrawal criterion (alanine aminotransferase [ALT] or aspartate aminotransferase [AST] >8 × upper limit of normal [ULN]). Transaminases returned to baseline after discontinuation from the trial.
Table 2.
Treatment-emergent adverse events experienced by >1 patient overall
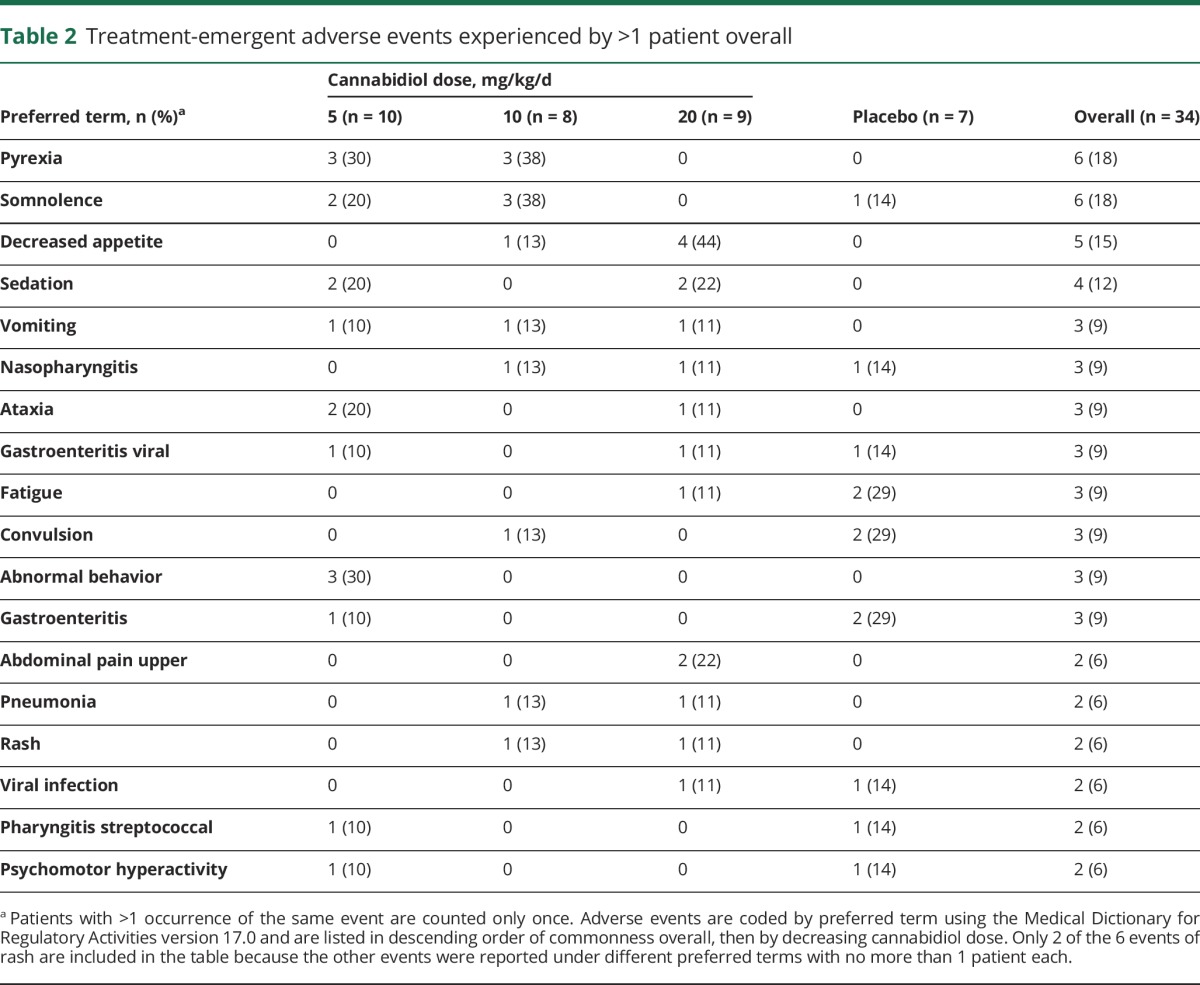
Six patients (18%) experienced a rash that was reported as a TEAE: 5 in the CBD and 1 in the placebo group. The rashes included diffuse maculopapular rash (CBD 10 mg/kg/d), papular rash (CBD 20 mg/kg/d), localized rash on thigh (CBD 20 mg/kg/d), viral rash (CBD 5 mg/kg/d), concomitant rash and hives (CBD 10 mg/kg/d), and diaper rash (placebo). All events of rash resolved. There were no cases showing mucosal involvement and no evidence of Stevens-Johnson syndrome or toxic epidermal necrolysis.
There were no deaths. Five patients experienced serious TEAEs (1, 2, and 1 in the 5, 10, and 20 mg/kg/d CBD groups, respectively, and 1 in the placebo group). Serious TEAEs reported in >1 patient overall were pyrexia (2 in the 10 mg/kg/d CBD group) and convulsion (1 in the 10 mg/kg/d CBD group and 1 in the placebo group). Six patients taking CBD (22%) had elevated ALT or AST >3 × ULN during the trial; none met the criteria for drug-induced liver injury (DILI) as there was no elevation of bilirubin >2 × ULN. These elevations were most common in the 20 mg/kg/d group (4/6 patients). All 6 patients were taking concomitant valproate and 4 of the 6 had concomitant viral/bacterial infections. The dose of valproate was reduced in 1 patient and elevations in all 6 returned to baseline upon either trial completion (n = 5) or withdrawal (n = 1). There were no clinically meaningful observations for clinical laboratory assessments, vital signs, or ECG results. None of the patients on CBD reported TEAEs of worsening of seizures or the appearance of new seizure types during treatment. There were no instances of suicidal ideation on the Columbia-Suicide Severity Rating Scale after day 1 through end of study among the patients for whom the questionnaire was completed.
Discussion
This was a randomized controlled trial of CBD in patients with DS. Patients had reasonably well-controlled seizures (<4 convulsive seizures during a 4-week baseline) obviating the need for concomitant AED dose changes and allowing adequate PK monitoring. This trial showed that exposure to CBD and its metabolites increased in a dose-proportional manner. Between the 2 postdose time points assessed, the highest plasma exposures to CBD were observed during the first time window (2–3 hours). The major circulating metabolite was 7-COOH-CBD and 6-OH-CBD was a relatively minor circulating metabolite. There are no published studies on the anticonvulsant activity of CBD metabolites although such studies are ongoing.
The results show that exposures to CBD and its metabolites are linearly related to dose over a clinically relevant dose range, as assessed by AUC at end of treatment. Total variability in AUC0–t was moderate to high (%CV 20%–121%) and was higher for the metabolites (%CV 57%–1750%). High intersubject variability in cannabinoid PK has been noted previously12 and may reflect various intrinsic and extrinsic factors that have not been fully evaluated in this trial. These could include effects of concomitant medications that induce or inhibit the CYP2C19 and CYP3A4 isoenzymes, genetic variation in the CYP isoenzymes, variability in food effects, or differences in tissue distribution.
The combination of all 3 doses of CBD (5, 10, and 20 mg/kg/d) with clobazam led to a PK interaction with clobazam, resulting in an elevation of N-CLB plasma exposure in patients. This elevation did not occur in the presence of stiripentol and suggests stiripentol had maximally inhibited CYP2C19, although additional evidence is needed as the sample size in the current trial was limited to 4 patients taking both AEDs. Elevated levels of N-CLB could contribute to both beneficial (antiseizure)13 and adverse effects (sedation, fatigue) associated with CBD administration, though additional sedation was not observed in CLB patients in part B of this trial.1 There was no obvious effect on the other AEDs investigated (valproate, topiramate, stiripentol, or levetiracetam). These findings are consistent with those of an open-label trial that found no consistent effect of CBD on levels of clobazam (n = 22), valproate (n = 18), levetiracetam (n = 12), felbamate (n = 12), lamotrigine (n = 10), zonisamide (n = 9), topiramate (n = 5), rufinamide (n = 5), or stiripentol (n = 2).14 Another group reported elevated levels of topiramate, rufinamide, zonisamide, and eslicarbazepine and decreased levels of clobazam following CBD administration.9 However, this trial was an open-label retrospective review with generally small numbers, timing of AED levels relative to dosing may have varied, and the extent of the interaction was not reported; thus, formal interaction studies are required. The discrepancies may reflect differences in methodologies (e.g., consistency of obtaining trough vs random levels at baseline and post-CBD administration, assay techniques), concomitant AEDs and other medications, demographics (e.g., age, sex), and other factors.
In the current trial, CBD resulted in more AEs than placebo. However, it was generally well-tolerated at the 5–20 mg/kg/d dose range and the safety findings were consistent with the profile seen in open-label studies. Elevated transaminases (ALT or AST) were reported without association with DILI. In keeping with a previous trial,3 we observed an increased frequency of elevated transaminases with concomitant use of valproate; all elevations subsequently resolved, 1 after discontinuing CBD and reducing valproate dose, and the remaining 5 after trial completion. The 20 mg/kg/d dose was approved by the DSMC following review of all safety and PK data, and was employed in the subsequent randomized controlled trial (GWPCARE1 part B).
CBD and its metabolites showed dose-proportional PK over a clinically relevant dose range and no interaction with studied AEDs except for an elevation in N-CLB, that was absent if concomitant stiripentol was present, possibly reflecting prior saturation and inhibition of the CYP2C19 isoenzyme. Additional formal PK trials are needed. All 3 doses of CBD were generally well-tolerated and the 20 mg/kg/d dose was chosen by the DSMC as the appropriate dose for further study.
Acknowledgment
The authors thank the patients and the GWPCARE1 part A investigators for their contributions to this study.
Glossary
- 6-OH-CBD
6-hydroxy-cannabidiol
- 7-COOH-CBD
7-carboxy-cannabidiol
- 7-OH-CBD
7-hydroxy-cannabidiol
- AE
adverse event
- AED
antiepileptic drug
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AUC
area under the curve
- CBD
cannabidiol
- CLB
clobazam
- CYP
cytochrome P450
- DILI
drug-induced liver injury
- DS
Dravet syndrome
- DSMC
data safety monitoring committee
- N-CLB
N-desmethylclobazam
- PK
pharmacokinetics
- TEAE
treatment-emergent adverse event
- ULN
upper limit of normal
Footnotes
Class of Evidence: NPub.org/coe
CME Course: NPub.org/cmelist
Contributor Information
Collaborators: GWPCARE1 Part A Study Group, Angus Wilfong, Robert Flamini, Linda Laux, Ian Miller, Helen Cross, Charuta Joshi, Richard Chin, and Sameer Zuberi
Author contributions
Orrin Devinsky: study design, acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Anup D. Patel: study design, acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Elizabeth A. Thiele: study design, acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Matthew H. Wong: study design, acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Richard Appleton: acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Cynthia L. Harden: analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Sam Greenwood: analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Gilmour Morrison: analysis and interpretation of data, drafting or revising the manuscript for intellectual content. Kenneth Sommerville: study design, acquisition of data, analysis and interpretation of data, drafting or revising the manuscript for intellectual content.
Study funding
Study funded by GW Research Ltd.
Disclosure
O. Devinsky has received grant support, paid to his institution, from GW Pharmaceuticals, Novartis, PTC Pharmaceuticals, and Zogenix, and has equity interest in Pairnomix, Tilray, and Egg Rock. A. Patel has received research funding and consulting fees from GW Pharmaceuticals. E. Thiele has received consulting fees from UCB and Eisai, grant support and consulting fees from GW Pharmaceuticals and Zogenix, and is a consultant to Aquestive and Usher Smith. M. Wong received funds from GW Pharmaceuticals associated with conducting this study. R. Appleton received honoraria for professional advice to GW Pharmaceuticals Ltd. in 2014 and 2017. C. Harden has received royalties from Up-to-Date and Springer. S. Greenwood is an employee of GW Research Ltd. G. Morrison is an employee of GW Research Ltd. K. Sommerville is an employee of Greenwich Biosciences and Assistant Professor of Clinical Medicine at Duke University. Go to Neurology.org/N for full disclosures.
References
- 1.Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med 2017;376:2011–2020. [DOI] [PubMed] [Google Scholar]
- 2.O'Connell BK, Gloss D, Devinsky O. Cannabinoids in treatment-resistant epilepsy: a review. Epilepsy Behav 2017;70:341–348. [DOI] [PubMed] [Google Scholar]
- 3.Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 2016;15:270–278. [DOI] [PubMed] [Google Scholar]
- 4.Gofshteyn JS, Wilfong A, Devinsky O, et al. Cannabidiol as a potential treatment for febrile infection-related epilepsy syndrome (FIRES) in the acute and chronic phases. J Child Neurol 2017;32:35–40. [DOI] [PubMed] [Google Scholar]
- 5.Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia 2016;57:1617–1624. [DOI] [PubMed] [Google Scholar]
- 6.Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev 2014;46:86–95. [DOI] [PubMed] [Google Scholar]
- 7.Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: general features and interactions between antiepileptic drugs. Lancet Neurol 2003;2:347–356. [DOI] [PubMed] [Google Scholar]
- 8.Friedman D, Devinsky O. Cannabinoids in the treatment of epilepsy. N Engl J Med 2015;373:1048–1058. [DOI] [PubMed] [Google Scholar]
- 9.Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP; UAB CBD Program. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia 2017;58:1586–1592. [DOI] [PubMed] [Google Scholar]
- 10.Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015;56:1246–1251. [DOI] [PubMed] [Google Scholar]
- 11.Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS): Children's Baseline/Since Last Visit (version 6/23/2010). New York:Research Foundation for Mental Hygiene; 2008. [Google Scholar]
- 12.Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers 2007;4:1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashi S, Yano I, Shibata M, et al. Effect of CYP2C19 polymorphisms on the clinical outcome of low-dose clobazam therapy in Japanese patients with epilepsy. Eur J Clin Pharmacol 2015;71:51–58. [DOI] [PubMed] [Google Scholar]
- 14.Friedman D, Cilio MR, Tilton N, et al. The effect of epidiolex (cannabidiol) on serum levels of concomitant anti-epileptic drugs in children and young adults with treatment-resistant epilepsy in an expanded access program. American Epilepsy Society 2014 annual meeting. Abstract No 2.309. Available at: aesnet.org. Accessed September 29, 2017.