

Abstract
Background
Staphylococcus aureus is a major pathogen causing significant morbidity and mortality worldwide. The emergence of MDR S. aureus strains in the community setting has major implications in disease management. However, data regarding the occurrence and patterns of MDR community-associated S. aureus sub-clones is limited.
Objectives
To use whole-genome sequences to describe the diversity and distribution of resistance mechanisms among community-associated S. aureus isolates.
Methods
S. aureus isolates from skin and soft tissue infections (SSTIs) and nasal colonization were collected from patients within 10 primary care clinics from 2007 to 2015. The Illumina Miseq platform was used to determine the genome sequences for 144 S. aureus isolates. Phylogenetic and bioinformatics analyses were performed using in silico tools. The resistome was assembled and compared with the phenotypically derived antibiogram.
Results
Approximately one-third of S. aureus isolates in the South Texas primary care setting were MDR. A higher proportion of SSTI isolates were MDR in comparison with nasal colonization isolates. Individuals with MDR S. aureus SSTIs were more likely to be African American and obese. Furthermore, S. aureus populations are able to acquire and lose antimicrobial resistance genes. USA300 strains were differentiated by a stable chromosomal mutation in gyrA conferring quinolone resistance. The resistomes were highly predictive of antimicrobial resistance phenotypes.
Conclusions
These findings highlight the high prevalence and epidemiological factors associated with MDR S. aureus strains in the community setting and demonstrate the utility of next-generation sequencing to potentially quicken antimicrobial resistance detection and surveillance for targeted interventions.
Introduction
Since the discovery of Staphylococcus aureus, a major challenge has been its remarkable ability to acquire resistance to antibiotics. The emergence of community-associated MRSA (CA-MRSA) has resulted in an epidemic of skin and soft tissue infections (SSTIs) in the USA.1,2 Whereas MDR has been well described among hospital-associated S. aureus clones, community-associated S. aureus clones have retained susceptibility to non-β-lactam antimicrobials, including macrolides, tetracyclines, fluoroquinolones, lincosamides and trimethoprim/sulfamethoxazole. However, the increased use of these antimicrobials could drive the emergence of new MDR sub-clones of community-associated S. aureus, complicating disease management. The emergence of resistance to these agents among USA300 strains poses a tremendous challenge for treating both community- and healthcare-associated S. aureus infections.3–6
With rapidly advancing technology, microbial WGS is positioned to soon become a viable option in the routine clinical laboratory.7–9 WGS provides valuable information in reconstructing the evolution of antimicrobial resistance and has the potential to substantially increase the speed of antimicrobial resistance detection in the practice setting.10–13 With the ever-increasing rise of bacterial drug resistance, the need for rapid and reliable methods to detect its emergence and predict antimicrobial susceptibility has never been more imperative.
This study aimed to: (i) describe the population structure, diversity and distribution of resistance mechanisms among community-associated S. aureus isolates; and (ii) determine the extent to which the genotype is predictive of resistance.
Materials and methods
Study setting and population
We performed this investigation using clinical and isolate information from a well-described cohort of patients with S. aureus SSTIs or nasal colonization in the South Texas primary care setting. Details of this cohort have been described previously.14,15 Briefly, this study was conducted in collaboration with ten primary care clinics within the South Texas Ambulatory Research Network, a practice-based research network composed of urban, suburban and rural primary care clinics distributed throughout the South Texas region, from 2007 to 2015. Patients were eligible for study enrolment if they provided informed consent, were 18 years of age or older, and presented to one of the participating clinics with a purulent SSTI. Patients were excluded if they were pregnant, incarcerated or had impaired decision-making capacity. Nasal isolates were from patients who presented to these clinics from February to May 2015 to assess for S. aureus nasal colonization. The subsequent analyses conducted in this study were limited to isolates confirmed to be S. aureus and the patients from whom they were obtained.
Ethics
The institutional review board at the UT Health Science Center San Antonio approved the study.
Microbiological analysis
Samples were plated onto blood agar plates and incubated at 35°C for 24 h, then sub-cultured to MRSA-selective agar (MRSASelect chromogenic agar plates; Bio-Rad Laboratories). Latex agglutination tests (StaphAurex®; Thermo Fisher Scientific) and phenotypic screening tests (cefoxitin) were used for the identification and isolation of MRSA. Vitek 2 AST-GP75 cards (bioMérieux) were used to determine the susceptibility of S. aureus study isolates to 14 antibiotics. D-zone tests were performed to identify inducible clindamycin resistance. Mupirocin susceptibility testing was conducted using gradient diffusion testing (Etest, bioMérieux). Antimicrobial MICs were interpreted according to the CLSI document M100-S14 (2014).16 Standard definitions of MDR were used.17
DNA sequencing and analyses
DNA extraction was conducted on the MagNA Pure 96 Instrument for automated DNA extraction (Roche Life Science, Indianapolis, IN, USA). Sequencing libraries were prepared using the NexteraXT DNA sample preparation kit (Illumina Inc.) following the manufacturer’s instructions and sequenced on a MiSeq sequencing instrument (Illumina Inc.).
Sequencing data were imported and analysed using CLC Genomics Workbench 8.1 (Qiagen, Redwood City, CA, USA). Paired-end reads were mapped to reference strain FPR3757 (accession number NC_007793).18 The Fixed Ploidy Variant Detection tool was used to identify SNPs and the Indels and Structural Variants tool was used to identify insertions and deletions.19 SNPs were included in analyses if that position contained at least 15 high-quality reads and ≥ 90% of them supported an alternative allele different from the FPR3757 reference. An in silico SNP validation was performed to assess SNP frequency with varying coverage levels (Table S1, available as Supplementary data at JAC Online). Strain whole-genome sequence FASTQ files have been deposited with NCBI under BioProject PRJNA352260.
For comparative genome evaluation, SNPs were used as a measure of genetic pairwise distances between strains. An SNP matrix was generated using the Reference Sequence Alignment-based Phylogeny builder (REALPHY: http://realphy.unibas.ch/fcgi/realphy). The SNP matrix was uploaded onto MEGA7 and CLCGenomics Workbench 8.5.1 for phylogenetic analyses. Phylogeny was inferred using neighbour joining and the maximum likelihood method based on the Jukes–Cantor model with 500 bootstrap replicates. Consistent with previous literature, clusters of strains were indicated by bootstrap values of >70% for maximum likelihood and neighbour-joining analyses.4
MLST was determined from the sequence data by extracting the sequence at the specific loci of the seven housekeeping genes using the CLCGenomics Workbench 8.5.1 and the S. aureus MLST scheme (www.pubmlst.org; downloaded November 2015). Isolates were considered to be USA300 if they were ST8 and had the Panton–Valentine leucocidin (PVL) genes, as previously validated.20,21
Mobile genetic elements (MGEs) were detected in silico. Sequence reads were assembled de novo into contigs and were used to determine the presence and absence of MGEs. The assembled contigs were screened with PlasmidFinder to identify circular and integrated plasmids using a subset of replicon sequences from 139 fully sequenced plasmids associated with S. aureus.22
Resistome
To assemble the resistome, the presence and absence of known antimicrobial resistance genes was identified using the ARG-ANNOT (Antibiotic Resistance Gene-ANNOTation) database followed by manual searching for chromosomal genes with amino acid variants.23 The genetic elements and accession numbers associated with susceptibility among the S. aureus isolates are listed in Tables S2 and S3.
Discrepancy investigation
Discrepancies were defined as discordances between susceptibility results using Vitek 2 and predicted genotypic susceptibility. Discrepancies were investigated using gradient diffusion testing (Etest, bioMérieux). Concordance was defined as agreement between susceptibility results from Vitek 2 or gradient diffusion with genotype. Major errors (MEs) occur when the phenotypic result (Vitek 2) is susceptible and the genotypic prediction is resistant. Very major errors (VMEs) occur when the phenotypic result (Vitek 2) is resistant but the genotypic prediction is susceptible. The initial phenotypic and genotypic analysis of resistance was conducted blind by two investigators.
Statistical analyses
Statistical analyses were performed using SPSS 23.0® (IBM Corp, Armonk, NY, USA). The χ2 test or Fisher’s exact test was used for dichotomous or categorical variables. Student’s t-test was used for continuous variables. P < 0.05 indicated statistical significance.
Results
MLST of S. aureus
We sequenced 144 S. aureus isolates (112 from SSTIs and 32 from nasal colonization) recovered from 144 patients. Seventy-one strains were MRSA and 73 were MSSA. All MRSA isolates belonged to the ST8 clonal group (Figure S1). Although, the majority of MSSA belonged to ST8 and ST5, we identified a variety of unique isolates that belonged to other ST types; these included livestock-associated ST97 and ST59 clones, and multiple novel MLST clonal types with single-locus variants of ST8, ST5 and ST12 (Table S4).
To estimate the population structure, sample reads were mapped onto a single core reference genome, FPR3757. One hundred and twenty-three isolates had sufficient coverage for the analyses. We identified a total of 53 840 SNP sites compared with the reference sequence FPR3757. After excluding MGEs, the strains differed by an average of 3548 SNPs (range 15–47 574). Based on the neighbour-joining method, the isolates fell into four clades (Figure 1). The first group comprised only ST8 isolates that grouped very tightly with FPR3757, differing by an average of 160 SNPs (range 15–1215). This magnitude of inter-strain distance is similar to previous reports.24 Approximately 70% of the S. aureus isolates included in this study clustered in Group 1. Of note, methicillin-susceptible and -resistant strains were intermingled, suggesting a common lineage. Group 2 encompassed two branches that were both similarly distant from the reference strain: one group was composed mainly of ST5 strains, which are known to be predominantly hospital-acquired (HA)-MRSA strains; the other group was more heterogeneous. The third group comprised ST59 MSSA strains. The fourth group was composed mainly of ST45 and ST30 MSSA strains.
Figure 1.

SNP-based phylogenetic analyses of S. aureus strains. (a) Radial phylogenetic tree mapping the isolates based on the concatenated SNP distances. Group 1 (green ring) comprised primarily ST8 clustered tightly with reference FPR3757. Group 2 (blue rings) comprised two groups similarly distant from FPR3757. Group 3 (purple ring) comprised three ST59 isolates. Group 4 (orange ring) comprised ST30 and ST45 isolates. (b) Zoomed-in view of Group 1 cluster composed of ST8 isolates.
To evaluate the concordance of traditional MLST typing and WGS data, the MLST strain types were mapped onto the WGS phylogenetic tree (Figure 2). In this collection, isolates with the same MLST type predictably clustered into the same branch. Although isolates sharing the same ST clustered together, the high resolution of WGS demonstrated that the distance between individual isolates within each cluster varied substantially (Figure S2). This may suggest that the close clusters represent successful lineages that have undergone recent clonal expansion, whereas the distantly related clades may represent rarer, less successful lineages.
Figure 2.

SNP-based phylogenetic analysis and MLST. Multilocus strain types were computationally mapped onto the phylogenetic tree. The coloured outer ring denotes the MLST type.
To identify whether particular strains clustered together by geographic region, the location of the practice site was mapped onto the WGS phylogenetic tree. The MLST and phylogenetic analysis revealed that specific clones were not necessarily specific to particular geographic locations (Figure S3), with the exception that both S. aureus strains from Bulverde, TX, clustered closely together. These two strains from unrelated patients were identified to be most closely related to an S. aureus strain (RF-122) associated with severe bovine mastitis (Figure S4).25
Antimicrobial resistance determinants among community-associated S. aureus
The antibiograms of 143 isolates (32 nasal isolates and 111 SSTI isolates) were analysed. Genetic determinants were specifically isolated within specific clades on the phylogenetic tree (Figure S5). Notably, the presence of resistance determinants for aminoglycosides, tetracycline, mupirocin and trimethoprim occurred primarily among ST8 isolates.
The predictive patterns of antimicrobial resistance determinants based on genetic mechanisms are detailed in Table 1. After discrepancy testing, the overall VMEs and MEs were 0% and 1.4%, respectively (Table S5). Upon subsequent discrepancy testing, all 9 VMEs were rectified and aligned with the genotype (Table 2). Furthermore, most MEs were also resolved after repeat phenotypic testing. The remaining MEs were from the detection of oxacillin resistance. The mecA gene was detected while phenotypically displaying susceptibility to oxacillin.
Table 1.
Comparison of whole-genome detection of antimicrobial resistance determinants and phenotype
| Phenotype: susceptible |
Phenotype: resistant |
Error rate (%) |
Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| genotype |
genotype |
|||||||||||
| Antibiotic | #S | S | R | #R | S | R | VMEs | MEs | ||||
| Ciprofloxacin | 87 | 78 | 7 | 56 | 3 | 53 | 2.1 | 4.9 | 95 | 90 | 85 | 96 |
| Clindamycin | 133 | 133 | 0 | 10 | 0 | 10 | 0 | 0 | 100 | 100 | 100 | 100 |
| Erythromycin | 70 | 62 | 8 | 73 | 3 | 70 | 2.1 | 5.6 | 96 | 88 | 90 | 94 |
| Gentamicin | 141 | 141 | 0 | 2 | 0 | 2 | 0 | 0 | 100 | 100 | 100 | 100 |
| Mupirocin | 140 | 140 | 0 | 3 | 1 | 2 | 0.7 | 0 | 67 | 100 | 100 | 99 |
| Oxacillin | 71 | 67 | 4 | 72 | 2 | 70 | 1.4 | 2.8 | 97 | 93 | 93 | 97 |
| Rifampicin | 143 | 143 | 0 | 0 | 0 | 0 | 0 | 0 | n/a | 100 | n/a | 100 |
| Tetracycline | 141 | 141 | 0 | 2 | 0 | 2 | 0 | 0 | 100 | 100 | 100 | 100 |
| Trimethoprim | 139 | 139 | 0 | 4 | 1 | 3 | 0.7 | 0 | 75 | 100 | 100 | 99 |
| Vancomycin | 143 | 143 | 0 | 0 | 0 | 0 | 0 | 0 | n/a | 100 | NA | 100 |
S, susceptible; R, resistant; #S, number of isolates susceptible; #R, number of isolates resistant; PPV, positive predictive value; NPV, negative predictive value.
Table 2.
Discrepancy testing of whole-genome detection of antimicrobial resistance determinants and phenotype
| Error type/ antimicrobial | Isolate | Initial phenotype | Repeat phenotype [MIC, mg/L (interpretation)] | Genotype | Resolved |
|---|---|---|---|---|---|
| VMEs | |||||
| ciprofloxacin | I5 | R | 0.125 S | no gyrA or grlA mutation detected | yes |
| I23 | R | 0.94 S | no gyrA or grlA mutation detected | yes | |
| I39 | R | 0.125 S | no gyrA or grlA mutation detected | yes | |
| erythromycin | B23 | R | 0.125 S | no erm or msrA gene detected | yes |
| I7 | R | 0.125 S | no erm or msrA gene detected | yes | |
| I17 | R | 0.125 S | no erm or msrA gene detected | yes | |
| oxacillin | B23 | OXA MIC >4 (R) FOX+ | 0.5 S | no mecA gene detected | yes |
| I7 | OXA MIC >4 (R) FOX+ | 0.25 S | no mecA gene detected | yes | |
| trimethoprim | B23 | R | 0.04 S | no dfrA, dfrG or dfrB mutation detected | yes |
| MEs | |||||
| ciprofloxacin | A8 | S | ≥32 R | gyrA mutation detected | yes |
| I7 | S | 8 R | gyrA mutation detected | yes | |
| I17 | S | 6 R | gyrA mutation detected | yes | |
| I42 | S | 16 R | gyrA mutation detected | yes | |
| erythromycin | B30 | S | 32 R | msr gene detected | yes |
| C11 | S | 32 R | msr gene detected | yes | |
| I5 | S | 48 R | msr gene detected | yes | |
| I11 | S | 24 R | msr gene detected | yes | |
| I21 | S | 64 R | msr gene detected | yes | |
| I23 | S | 64 R | msr gene detected | yes | |
| I24 | S | 48 R | msr gene detected | yes | |
| J17 | S | 32 R | msr gene detected | yes | |
| oxacillin | A8 | OXA MIC < 0.25 (S) FOX− | 64 R sub-colonies | mecA gene detected | yes |
| C11 | OXA MIC = 1 (S) FOX+ | 1 S | mecA gene detected | no | |
| I5 | OXA MIC < 0.25 (S) FOX− | 1 S | mecA gene detected | no | |
| I11 | OXA MIC = 0.5 (S) FOX− | 32 R sub-colonies | mecA gene detected | yes | |
FOX+/FOX−, positive or negative, respectively, on a cefoxitin screen; OXA, oxacillin.
Epidemiological features of MDR
Approximately 31% of the isolates were MDR. Most MDR isolates were ST8 (91%); one isolate was an ST30, one isolate was ST121 and two had novel ST designations. Approximately 42% of ST8 strains were MDR. A higher proportion of SSTI isolates compared with colonizing isolates were MDR (37% versus 11%; P < 0.05). Among SSTIs, features associated with MDR isolates included African American race (P < 0.01) and obesity (P = 0.04) (Table 3). Notably, there were no differences in reported prior antibiotic exposures between cases with MDR isolates compared with non-MDR isolates (17% versus 14%; P = 0.60).
Table 3.
Characteristics of patients with MDR compared with non-MDR community-associated S. aureus SSTI strains
| Characteristic | MDR (n = 38) | No MDR (n = 73) | P value |
|---|---|---|---|
| Mean age, years (± SD) | 43 ± 12 | 41 ± 13 | 0.56 |
| Gender, n (%) | |||
| male | 20 (53) | 36 (49) | 0.67 |
| Race/ethnicity, n (%) | |||
| African American | 6 (16) | 2 (3) | 0.01* |
| Hispanic | 29 (76) | 52 (72) | 0.64 |
| Diabetes, n (%) | 11 (30) | 18 (25) | 0.63 |
| Obese (BMI ≥30), n (%) | 21 (55) | 29 (40) | 0.04* |
| MRSA phenotype, n (%) | 31 (81) | 36 (49) | <0.01* |
| Prior SSTI in last 90 days, n (%) | 7 (18) | 9 (12) | 0.41 |
| Prior antibiotic in last 90 days, n (%) | 5 (13) | 10 (14) | 0.61 |
There were no cases of patients with the peripheral vascular disease, HIV infection, cancer or receipt of chemotherapy.
Statistical significance.
When evaluating geographic clustering, we found more than half of the MDR isolates clustered within the Inner West Side of San Antonio, a predominantly Hispanic and African American community with household incomes significantly below the state average (Figure S6a). However, the proportion of MDR strains (among areas with two or more isolates) was disproportionately higher (50% of isolates in this geographic region were MDR) in the north-west side of San Antonio, the location of the South Texas Medical Center (Figure S6b).
Plasmid characterization
On average, the isolates carried approximately three plasmids (range 0–6). The occurrence of pUSA300-like plasmids (e.g. pUSA02 and pUSA03) was rare and was only detected in two ST8 isolates. However, the distribution of plasmids was widespread. In the case of pSJH101 (rep16), its rep genes were detected in strains belonging to several different lineages (ST8, ST5, ST59, ST45 and ST30). This supports the notion of the potential transfer of MGEs and resistance genes among different S. aureus clonal lineages (Table S6).
Fluoroquinolone resistance among USA300
To explore the association of the role of antimicrobial resistance determinants in the evolution of S. aureus, the presence and absence of specific resistance mechanisms was mapped onto the WGS phylogenetic tree. The phylogeny of S. aureus strains revealed that strains containing the mutation that confers fluoroquinolone resistance within the gyrA (Leu84Ser) gene clustered among USA300 strains in the ST8 cluster (Figure 3a). When evaluating the population structure of USA300 strains, the strains broadly clustered within two major clades based on the presence of fluoroquinolone resistance (Figure 3b). Furthermore, three closely related MSSA USA300 strains were found to contain the arginine catabolic mobile element (ACME), suggesting some potential instability or transfer of the SCCmec. These findings indicate the mosaicism of composite islands in S. aureus, and reveal concerns for potential under-reporting of MRSA using genotypic methods.
Figure 3.
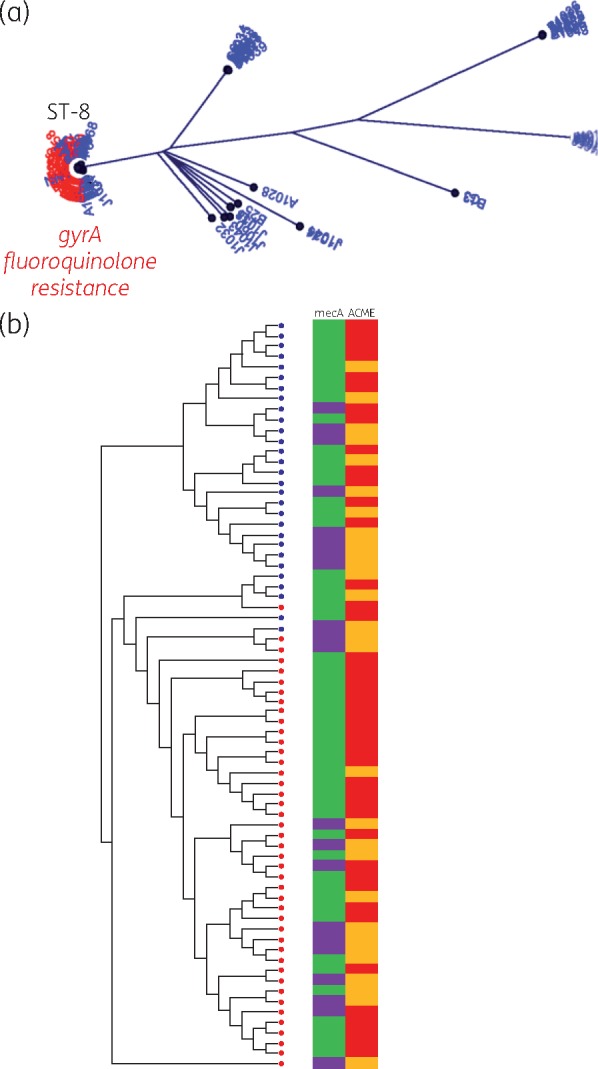
Fluoroquinolone resistance among S. aureus. (a) Radial tree of all isolates evaluated for the chromosomal mutation in gyrA conferring fluoroquinolone resistance (presence of chromosomal mutation is marked in red). (b) Maximum likelihood tree of USA300 S. aureus strains. Red indicates the presence and blue indicates the absence of chromosomal mutation in a strain. Adjacent to the tree is a panel showing the presence (green) and absence (purple) of the mecA gene and the presence (red) and absence (orange) of the arginine catabolic mobile element (ACME).
Discussion
The emergence of MDR S. aureus strains in outpatients complicates disease management and contributes to the development of persistent or recurrent community-associated MRSA infections. This study detected a high level of MDR among community-associated S. aureus strains in South Texas. Approximately 42% of USA300 S. aureus isolates were MDR. This is a sharp increase in comparison with a 2004 investigation of USA300 strains from 11 US cities, where only two MDR isolates were identfied.26 However, there have been sporadic reports of MDR in specific patient groups. This includes reports of USA300 MDR rates as high as 46% in MSM in San Francisco and Boston. In Taiwan, 61% of CA-MRSA (ST59) strains were resistant to four or more non-β-lactam agents among paediatric patients.3,27 To our knowledge, the present study is the first to describe high-level S. aureus MDR in Texas.
Resistance determinants were distributed among different clades and strain types. This suggests that the loss and acquisition of resistance genes varied not only between but also within the clades. Almost all MDR S. aureus were found in the ST8 and ST30 clades. Furthermore, while we found a higher proportion of MDR S. aureus among SSTI compared with carrier strains, the proportion of colonizing strains containing three or more resistance mechanisms was >10%. This further supports the notion that the nares are an important reservoir and site for transmission of antimicrobial resistance determinants.
This study identified a cluster of three isolates resistant to trimethoprim/sulfamethoxazole. Two of the trimethoprim-resistant isolates harboured dfrG, one of which also harboured dfrK. Previously, both dfrG and dfrK were found to play a role in trimethoprim resistance in S. aureus of animal origin. However, recent work evaluating trimethoprim resistance among S. aureus strains from Europe, Asia and Africa found dfrG to be the predominant genetic determinant.28,29 To the best of our knowledge, this is the first report of dfrG and dfrK in S. aureus from a human infection in this region. Consistent with prior studies, we found co-resistance to trimethoprim and tetracycline (tetK), suggesting there may be a similar link between these two resistance genes.29,30 These findings have major implications for outpatient treatment. In a prior study describing outpatient treatment patterns of purulent SSTIs, trimethoprim/sulfamethoxazole was the most commonly prescribed antibiotic, followed by doxycycline and clindamycin.31
This investigation identified African American race and obese individuals with a higher proportion of MDR S. aureus. While studies have reported African American race as a risk factor for MRSA infections,26,32,33 this is the first study to observe that this population may also be at increased risk of acquiring MDR S. aureus strains. Furthermore, the majority of MDR strains clustered in an area of lower socio-economic status composed predominantly of African Americans and Hispanics. It can be speculated that geographic areas with different socio-economic settings and networks, and uneven distribution of MRSA can contribute to the observed disparity; further investigations are required.33
The density of MDR S. aureus was geographically highest in the north-west side of San Antonio, the location of the South Texas Medical Center. Consistent with other studies, this indicates the possibility of direct acquisition of resistance determinants from MDR HA-MRSA strains.34,35 Further studies are needed to determine the role hospitals play, including antibiotic selection pressures, in the spread of MDR S. aureus strains back into the community.
The phylogeny of S. aureus strains revealed that strains containing the mutation that confers fluoroquinolone resistance within gyrA (Leu84Ser) clustered only among USA300 strains. When evaluating the population structure of USA300 strains, we identified the strains as clustered within two major clades based on the presence of fluoroquinolone resistance. This is consistent with prior studies suggesting that the use of fluoroquinolones might have further promoted the clonal expansion of USA300, and that dominant S. aureus strains might have been selected because of resistance to fluoroquinolones and not just methicillin.4,36 Fluoroquinolone resistance has been previously described as a marker of successful healthcare-associated MRSA clones, which may be what is driving the selection of fluoroquinolone resistance and MDR among isolates found in close proximity to the medical centre observed in this study.36
This study described the predictive patterns of antimicrobial resistance determinants based on genetic mechanisms most commonly observed among community-associated S. aureus isolates. The final results demonstrated a high level of concordance, comparable with the error rates for current phenotypic methodologies, including the Vitek 2 and the Phoenix automated microbiology systems.37
Interestingly, almost all errors were rectified with subsequent discrepancy testing that aligned to the genotype. The remaining errors were due to the genotypic detection of the mecA gene while phenotypically displaying susceptibility to oxacillin. Phenotypically oxacillin-susceptible and mecA-positive S. aureus strains have been increasingly reported; however, their clinical significance is unknown.38,39 These discrepancies underscore the impact of potential laboratory errors, mixed infections or within-host variation of populations on observed phenotypic variation. Stanczak-Mrozek et al.40 identified a range from one to eight S. aureus sub-isolates with genetic and antimicrobial resistance variation per patient.
This study has limitations. Firstly, the geographic associations were limited to the locations of the clinics that served the patients and not specific household addresses. The sequences were mapped onto a reference genome, FPR3757; therefore, DNA not found in the reference strain was not evaluated. This study is subject to sampling bias from the participating clinics. Lastly, other important epidemiological factors that may be associated with S. aureus transmission (e.g. previous healthcare exposures, contacts, social networks) were not analysed.
In summary, this study found that MDR of community-associated S. aureus strains has emerged and is an important cause of SSTIs in Texas. This underscores the need to develop outpatient surveillance strategies and technologies to detect antimicrobial resistance rapidly.
Supplementary Material
Acknowledgements
Data were generated in the Genome Sequencing Facility, which is supported by UT Health Science Center at San Antonio, NIH-NCI P30 CA054174 (Cancer Therapy and Research Center at UT Health Science Center at San Antonio) and NIH Shared Instrument grant 1S10OD021805–01 (S10 grant). The authors thank our clinical practice partners at UT Health Science Center for supplying the isolates.
Funding
This study was funded, in part, by the US National Institutes of Health in the form of a KL2 Career Development Award (5KL2 RR025766/RR/NCRR and KL2 TR000118/TR/NCATS) (C. R. F.).
Transparency declarations
None to declare.
Supplementary data
Tables S1 to S6 and Figures S1 to S6 appear as Supplementary data at JAC Online.
References
- 1. Chambers HF, Deleo FR.. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 2009; 7: 629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeLeo FR, Otto M, Kreiswirth BN. et al. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010; 375: 1557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diep BA, Chambers HF, Graber CJ. et al. Emergence of multidrug-resistant, community-associated, methicillin-resistant Staphylococcus aureus clone USA300 in men who have sex with men. Ann Intern Med 2008; 148: 249–57. [DOI] [PubMed] [Google Scholar]
- 4. Uhlemann AC, Dordel J, Knox JR. et al. Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York community. Proc Natl Acad Sci USA 2014; 111: 6738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chua K, Laurent F, Coombs G. et al. Antimicrobial resistance: not community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA)! A clinician's guide to community MRSA - its evolving antimicrobial resistance and implications for therapy. Clin Infect Dis 2011; 52: 99–114. [DOI] [PubMed] [Google Scholar]
- 6. Stevens DL, Bisno AL, Chambers HF. et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59: 147–59. [DOI] [PubMed] [Google Scholar]
- 7. Koser CU, Ellington MJ, Cartwright EJ. et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog 2012; 8: e1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koser CU, Ellington MJ, Peacock SJ.. Whole-genome sequencing to control antimicrobial resistance. Trends Genet 2014; 30: 401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koser CU, Fraser LJ, Ioannou A. et al. Rapid single-colony whole-genome sequencing of bacterial pathogens. J Antimicrob Chemother 2014; 69: 1275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strommenger B, Bartels MD. et al. Evolution of methicillin-resistant Staphylococcus aureus towards increasing resistance. J Antimicrob Chemother 2014; 69: 616–22. [DOI] [PubMed] [Google Scholar]
- 11. Alam MT, Petit RA 3rd, Crispell EK. et al. Dissecting vancomycin-intermediate resistance in Staphylococcus aureus using genome-wide association. Genome Biol Evol 2014; 6: 1174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Howden BP, McEvoy CR, Allen DL. et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog 2011; 7: e1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mwangi MM, Wu SW, Zhou Y. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci USA 2007; 104: 9451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forcade NA, Parchman ML, Jorgensen JH. et al. Prevalence, severity, and treatment of community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) skin and soft tissue infections in 10 medical clinics in Texas: a South Texas Ambulatory Research Network (STARNet) study. J Am Board Fam Med 2011; 24: 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee GC, Hall RG, Boyd NK. et al. Predictors of community-associated Staphylococcus aureus, methicillin-resistant and methicillin-susceptible Staphylococcus aureus skin and soft tissue infections in primary-care settings. Epidemiol Infect 2016; 144: 3198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Fourth Informational Supplement M100-S24. CLSI, Wayne, PA, USA, 2014. [Google Scholar]
- 17. Magiorakos AP, Srinivasan A, Carey RB. et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012; 18: 268–81. [DOI] [PubMed] [Google Scholar]
- 18. Diep BA, Gill SR, Chang RF. et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006; 367: 731–9. [DOI] [PubMed] [Google Scholar]
- 19. Homer N, Merriman B, Nelson SF.. BFAST: an alignment tool for large scale genome resequencing. PLoS One 2009; 4: e7767.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. David MZ, Glikman D, Crawford SE. et al. What is community-associated methicillin-resistant Staphylococcus aureus? J Infect Dis 2008; 197: 1235–43. [DOI] [PubMed] [Google Scholar]
- 21. David MZ, Siegel J, Lowy FD. et al. Asymptomatic carriage of sequence type 398, spa type t571 methicillin-susceptible Staphylococcus aureus in an urban jail: a newly emerging, transmissible pathogenic strain. J Clin Microbiol 2013; 51: 2443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carattoli A, Zankari E, Garcia-Fernandez A. et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 2014; 58: 3895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gupta SK, Padmanabhan BR, Diene SM. et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 2014; 58: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Long SW, Beres SB, Olsen RJ. et al. Absence of patient-to-patient intrahospital transmission of Staphylococcus aureus as determined by whole-genome sequencing. MBio 2014; 5: e01692–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herron-Olson L, Fitzgerald JR, Musser JM. et al. Molecular correlates of host specialization in Staphylococcus aureus. PLoS One 2007; 2: e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klevens RM, Morrison MA, Nadle J. et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298: 1763–71. [DOI] [PubMed] [Google Scholar]
- 27. Liu C, Graber CJ, Karr M. et al. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco, 2004-2005. Clin Infect Dis 2008; 46: 1637–46. [DOI] [PubMed] [Google Scholar]
- 28. Nurjadi D, Olalekan AO, Layer F. et al. Emergence of trimethoprim resistance gene dfrG in Staphylococcus aureus causing human infection and colonization in sub-Saharan Africa and its import to Europe. J Antimicrob Chemother 2014; 69: 2361–8. [DOI] [PubMed] [Google Scholar]
- 29. Nurjadi D, Friedrich-Janicke B, Schafer J. et al. Skin and soft tissue infections in intercontinental travellers and the import of multi-resistant Staphylococcus aureus to Europe. Clin Microbiol Infect 2015; 21: 567.e1–10. [DOI] [PubMed] [Google Scholar]
- 30. Kadlec K, Schwarz S.. Identification of a novel trimethoprim resistance gene, dfrK, in a methicillin-resistant Staphylococcus aureus ST398 strain and its physical linkage to the tetracycline resistance gene tet(L). Antimicrob Agents Chemother 2009; 53: 776–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Labreche MJ, Lee GC, Attridge RT. et al. Treatment failure and costs in patients with methicillin-resistant Staphylococcus aureus (MRSA) skin and soft tissue infections: a South Texas Ambulatory Research Network (STARNet) study. J Am Board Fam Med 2013; 26: 508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fridkin SK, Hageman JC, Morrison M. et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med 2005; 352: 1436–44. [DOI] [PubMed] [Google Scholar]
- 33. Popovich KJ, Snitkin E, Green SJ. et al. Genomic epidemiology of USA300 methicillin resistant Staphylococcus aureus in an urban community. Clin Infect Dis 2016; 62: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Otter JA, French GL.. Community-associated meticillin-resistant Staphylococcus aureus strains as a cause of healthcare-associated infection. J Hosp Infect 2011; 79: 189–93. [DOI] [PubMed] [Google Scholar]
- 35. Alam MT, Read TD, Petit RA. et al. Transmission and microevolution of USA300 MRSA in U.S. households: evidence from whole-genome sequencing. MBio 2015; 6: e00054.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knight GM, Budd EL, Whitney L. et al. Shift in dominant hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) clones over time. J Antimicrob Chemother 2012; 67: 2514–22. [DOI] [PubMed] [Google Scholar]
- 37. Nonhoff C, Rottiers S, Struelens MJ.. Evaluation of the Vitek 2 system for identification and antimicrobial susceptibility testing of Staphylococcus spp. Clin Microbiol Infect 2005; 11: 150–3. [DOI] [PubMed] [Google Scholar]
- 38. Hososaka Y, Hanaki H, Endo H. et al. Characterization of oxacillin-susceptible mecA-positive Staphylococcus aureus: a new type of MRSA. J Infect Chemother 2007; 13: 79–86. [DOI] [PubMed] [Google Scholar]
- 39. Sakoulas G, Gold HS, Venkataraman L. et al. Methicillin-resistant Staphylococcus aureus: comparison of susceptibility testing methods and analysis of mecA-positive susceptible strains. J Clin Microbiol 2001; 39: 3946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stanczak-Mrozek KI, Manne A, Knight GM. et al. Within-host diversity of MRSA antimicrobial resistances. J Antimicrob Chemother 2015; 70: 2191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.