

Abstract
Background
Hypertonic saline (HS) has been successfully used for treatment of various forms of brain edema. Decreased expression of aquaporin (AQP)4 and pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β have been linked to edema pathogenesis. This study examined the effect of 3% HS on brain edema in a rat model of traumatic brain injury (TBI).
Material/Methods
Sprague-Dawley rats were subjected to TBI induced by a controlled cortical impactor. The HS group was injected with 3% NaCl until the end of the study period. AQP4, TNF-α, IL-1β, and caspase-3 levels were measured by Western blotting, immunohistochemistry, enzyme-linked immunosorbent assay, and quantitative real-time PCR. Brain water content was also measured. Apoptotic cells in brain tissue were detected with terminal deoxynucleotidyl transferase dUTP nick-end labeling. Brain water content decreased following treatment with 3% HS relative to the TBI group.
Results
This was accompanied by decreases in AQP4, TNF-α, and IL-1β mRNA and protein levels. TBI resulted in increases in caspase-3 mRNA expression and the number of apoptotic cells; treatment with 3% HS suppressed apoptosis as compared to the TBI group.
Conclusions
Treatment with 3% HS ameliorated TBI-induced brain edema, possibly by suppressing brain edema, pro-inflammatory cytokine expression, and apoptosis.
MeSH Keywords: Apoptosis Inducing Factor; Aquaporin 4; Brain Edema; Saline Solution, Hypertonic
Background
Traumatic brain injury (TBI) can result in extensive physical, behavioral, and cognitive deficits [1–3]. Primary injury occurs simultaneously with TBI impact, leading to death or disability through disruption of the blood brain barrier and vasculature contributing to edema [4]. This is followed by secondary injury involving activation of microglia and astrocytes in the brain, which produce cytokines and chemokines, and recruitment of peripheral immune cells to the brain [5].
The aquaporin (AQP) family of water-channel proteins regulates water homeostasis [6]. To date, 13 conserved members have been identified in mammals; AQP3, AQP4, AQP5, AQP8, and AQP9 are expressed in cultured rat astrocytes [7]. AQP4 plays a key role in cerebral volume regulation following trauma, inflammation, and ischemia, as well as in tumors and metabolic disorders [8, 9]. Several studies have shown that AQP4 modulates memory and synaptic plasticity via regulation of glutamate transporter expression and contributes to the development of brain edema [10–13].
Hyperosmolar therapy with hypertonic saline (HS) solutions is a commonly used treatment for cerebral edema caused by cerebral ischemia, cerebral hemorrhage, or TBI [14,15]. HS was shown to be more effective in reducing brain edema as compared to equimolar doses of mannitol in a rat model of cerebral edema [16]. Interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and the complement cascade may play an important role in the development of cerebral swelling, and TNF-α may trigger caspase-3 activation, which leads to apoptosis [17]. We therefore speculated that HS exerts therapeutic effects on brain edema by modulating the expression of genes encoding these various factors.
To test this hypothesis, we investigated the effects of HS infusion on cerebral water content, inflammation, and apoptosis in a rat model of TBI. Our results indicate that HS treatment alleviates TBI-induced brain edema by inhibiting the expression of factors related to brain water regulation, inflammation, and apoptosis.
Material and Methods
Animals
Animal procedures were performed at the Animal Experiment Center of Zhejiang Chinese Medicine University. Experiments were performed on male Sprague-Dawley rats (250–300 g, 3–4 months old) that were group-housed in a temperature- and humidity-controlled environment (22±1°C and 55±%, respectively), had free access to food and water, and were maintained on a 12: 12-h light/dark cycle.
TBI model
TBI was induced in rats using a controlled cortical impactor (TBI-0310 Head Impactor; Precision Systems and Instrumentation, USA) with a 3-mm diameter metal tip [18]. Rats were anesthetized through intraperitoneal injection of 3.6% chloral hydrate (4 g/kg) and then fastened in a stereotactic fixing frame. Rectal temperature was monitored and body temperature was maintained at approximately 37°C by placing the animals on an electric heating blanket [19]. A 3-mm diameter hole was made in the cranium using an electric hand drill after exposing the skull. The right hemisphere was subjected to contusion injury at a speed of 3.0 m/s and depth of 2 mm [20–24]. The scalp was sterilized and sutured shut after the impact. The sham group rats (n=36) underwent craniotomy but were not subjected to impact injury. After 6 h, rats in the TBI and sham groups were injected with normal saline via the caudal vein; another group of rats (n=36) were injected with 3% NaCl (3% HS). Animals were sampled at 0, 6, 24, and 48 h.
Brain water content
Rats were sacrificed and brain tissue was removed and immediately placed in Petri dishes containing saline-moistened filter paper to prevent water evaporation. Blood was blotted with a filter paper and the cerebellum, brainstem, and pia were removed. The remaining brain tissue was weighed to determine wet weight within 5 min of brain removal. The brain tissue was weighted immediately after removal (wet weight) and again after drying in an oven at 100°C for 48 h. Water content was calculated according to the formula: ([wet weight]−[dry weight])/(wet weight)×100%.
Quantitative real-time (qRT)-PCR
Total RNA was extracted from the rat brain using an RNeasy Lipid Tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA was prepared from 1 μg total RNA using a Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics, Mannheim, Germany) and used for qRT-PCR, which was performed on a CFX96 system (Bio-Rad, Hercules, CA, USA) using primers and SYBR Green mix (Takara Bio, Otsu, Japan). Amplification was performed over 45 cycles of 95°C for 15 s and 60°C for 30 s after holding at 68°C for 15 s and 95°C for 1 min. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. Relative expression levels were determined with the cycle threshold method. Primer sequences are listed in Table 1.
Table 1.
Primer sequences.
| Primer | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| IL-1β | GCTTCAGGCAGGCAGTATCA | TGCAGTTGTCTAATGGGAACG |
| TNF-α | TACTGAACTTCGGGGTGATCG | CCACTTGGTGGTTTGCTACG |
| Caspase 3 | AGAGCTGGACTGCGGTATTGAG | GAACCATGACCCGTCCCTTG |
| GAPDH | GTGCCAGCCTCGTCTCATAG | CTTTGTCACAAGAGAAGGCAG |
Western blotting
AQP4 levels in brain tissue at 0, 6, 24, and 48 h were determined by Western blotting. Brain tissue was homogenized in lysis buffer (Pierce, Rockford, IL, USA) containing protease and phosphatase inhibitors on ice. Proteins were separated by polyacrylamide gel electrophoresis and transferred to an Immobilon-P polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA) that was probed with horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized with Super Signal chemiluminescent substrate (Pierce). GAPDH expression was detected with an antibody (1: 1500; Abcam, Cambridge, MA, USA) as a loading control.
Immunohistochemistry
Rats were perfused with saline solution followed by 4% paraformaldehyde after deep anesthesia with sodium pentobarbital. Their brains were removed and immediately post-fixed in 4% paraformaldehyde, embedded in paraffin, and then cut into 4-μm coronal sections that were incubated overnight at 4°C with primary antibody against AQP4 (1: 200; Abcam). After 3 washes with phosphate-buffered saline (PBS), sections were incubated for 60 min with Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA) at room temperature. Immunoreactivity was visualized with a BX51T-PHD-J11 fluorescence microscope (Olympus, Tokyo, Japan) and analyzed with Image-Pro Plus v.4.5 software (Media Cybernetics, Rockville, MD, USA) [25].
Analysis of TNF-α and IL-1β expression by enzyme-linked immunosorbent assay (ELISA)
The right hemisphere of the brain was dissected from rats at 0, 6, 24, and 48 h and immediately weighed. The tissue was frozen in 1 ml PBS (pH 7.4) and homogenized, centrifuged at 12 000×g at 4°C for 20 min. The protein levels of TNF-α and IL-1β were determined using ELISA kits for TNF-α (Immuno-Biological Laboratories, Hamburg, Germany; cat. no. JP27194) and IL-1β (Immuno-Biological Laboratories, Minneapolis, MN, USA; cat. no. 27193) according to the manufacturers’ protocols. Absorbance at 450 nm was measured within 30 min using an ELISA microplate reader.
Detection of apoptotic cells with the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
Brain tissue was harvested and apoptotic cells were detected with a TUNEL assay kit (DeadEnd fluorometric TUNEL System; Promega, Madison, WI, USA). The number of TUNEL-positive cells was counted at 400× magnification and results from 3 sections per brain were averaged [26].
Statistical analysis
Data are expressed as mean ± standard error. Differences between groups were analyzed with SPSS v.19.0 (IBM, Armonk, NY, USA). Semi-quantitative analysis of histological staining was performed by a single investigator blinded to treatment groups; differences between groups were evaluated by the Student’s t test. For Western blotting results, differences in protein expression levels were evaluated by two-way analysis of variance (ANOVA) with repeated measures, followed by a post hoc test. Differences in brain water content and qRT-PCR results were assessed with one-way ANOVA followed by a post hoc test. P values <0.05 were considered statistically significant.
Results
HS treatment reduces brain water content in a TBI model
Brain water content was measured to determine the extent of edema in the brain tissue of TBI rats. The brain water content was higher in TBI than in sham rats (Figure 1). Treatment with 3% HS decreased brain water content after 6, 24, and 48 h as compared to the TBI group (Figure 1).
Figure 1.
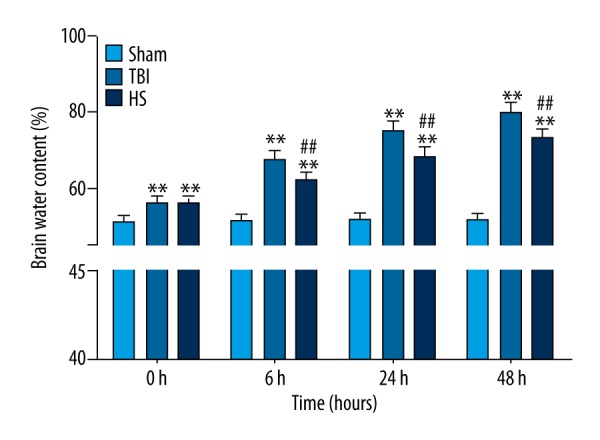
Brain water content following TBI and HS treatment. Brain water content was increased at 0, 6, 24, and 48 h in the TBI group relative to sham animals, which was reversed by HS treatment. * P<0.05, ** P<0.01 vs. sham group; # P<0.05, ## P<0.01 vs. TBI group.
To investigate the mechanism by which TBI increases brain water content, AQP4 protein expression in the brain was detected by Western blotting and immunohistochemistry. AQP4 level was increased in the TBI group as compared to sham animals at each time point (Figure 2A, 2B). Treatment with 3% HS inhibited AQP4 expression at 6, 24, and 48 h as compared to the TBI group (Figure 2A, 2B). Immunohistochemical detection of AQP4 distribution in brain tissue showed similar findings as in the Western blot analysis. AQP4 expression in the TBI group showed an increasing trend (Figure 2C), but this was abrogated by administration of 3% HS at each time point examined (Figure 2D). These results indicate that TBI induces the expression of AQP4, leading to an increase in brain water content, which was reversed by HS treatment.
Figure 2.

AQP4 expression and distribution in brain tissue following TBI and HS treatment. (A) AQP4 levels at 0, 6, 24, and 48 h in the TBI and HS groups. (B) Quantitative analysis of protein expression. * P<0.05, ** P<0.01 vs. sham group; # P<0.05, ## P<0.01 vs. TBI group. (C, D) AQP4 distribution in the brain tissues of the TBI, HS, and sham groups as detected by immunohistochemistry at 0, 6, 24, and 48 h. Scale bar: 50 μm.
Inflammation in the TBI brain is reduced by HS treatment
To investigate the effects of HS on TBI-induced inflammation in the brain, TNF-α and IL-1β mRNA were measured by qRT-PCR and ELISA, respectively. The levels of both factors were upregulated in the TBI group at 0, 6, 24, and 48 h relative to sham animals, but this effect was abolished following 3% HS administration (Figure 3A, 3C). The same trend was observed for TNF-α and IL-1β protein expression (Figure 3B, 3D). Thus, the increase in pro-inflammatory cytokine levels resulting from TBI is countered by HS treatment.
Figure 3.

TNF-α and IL-1β expression following TBI and HS treatment. (A, C) mRNA expression levels of TNF-α (A) and IL-1β (C), as determined by qRT-PCR at 0, 6, 24, and 48 h in the TBI, HS, and sham groups; GAPDH served as an internal reference. (B, D) Protein expression levels of TNF-α (B) and IL-1β (D) were measured at 0, 6, 24, and 48 h the TBI, HS, and sham groups. * P<0.05, ** P<0.01 vs. sham group; # P<0.05, ## P<0.01 vs. TBI group.
HS treatment inhibits apoptosis following TBI
We also investigated whether the beneficial effects of HS treatment include inhibition of apoptosis. Caspase-3 mRNA expression was rapidly upregulated following TBI as compared to the sham group, but slowly increased at 24 and 48 h (Figure 4). Treatment with 3% HS reduced caspase-3 mRNA levels at 6, 24, and 48 h as compared to the TBI group (Figure 4). A quantitative analysis of apoptotic cells detected with the TUNEL assay indicated that the apoptosis rate was increased at 0, 6, 24, and 48 h in the TBI group as compared to sham animals (Figure 5A, 5B), although there were no significant differences between the latter 2 time points. As compared to the TBI group, administration of 3% HS reduced the rate of apoptosis at each time point.
Figure 4.
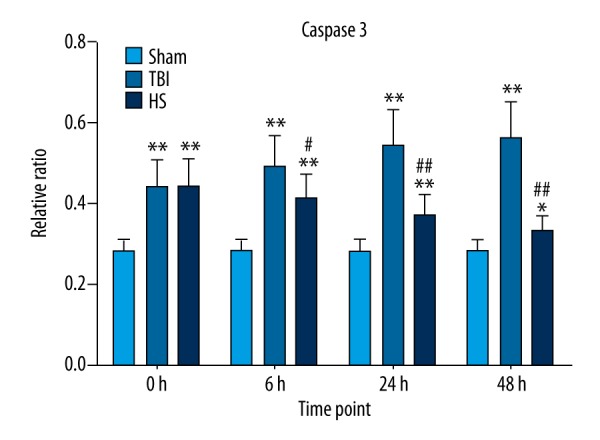
Caspase-3 mRNA expression following TBI and HS treatment. Caspase-3 mRNA level was detected by qRT-PCR at 0, 6, 24, and 48 h in the TBI, HS, and sham groups; GAPDH served as an internal reference. * P<0.05, ** P<0.01 vs. sham group; # P<0.05, ## P<0.01 vs. TBI group.
Figure 5.

Detection of apoptotic cells in brain tissue following TBI and HS treatment. (A) Apoptosis rate was determined at indicated time points the TBI, HS, and sham groups. * P<0.05, ** P<0.01 vs. sham group; # P<0.05, ## P<0.01 vs. TBI group. (B) Detection of apoptotic cells with the TUNEL assay.
Discussion
In this study, we demonstrated that TBI resulted in brain edema in rats and that this was reversed by treatment with 3% HS. TBI caused general pathological cellular and biochemical dysfunction that ultimately led to neuronal and glial cell death [27]. Osmotherapeutic agents such as HS and mannitol are currently used to treat cerebral edema and reduce intracranial pressure in ischemic stroke [28]. It was recently suggested that HS is more effective in improving cerebral edema than an equal volume of 20% mannitol [29]. We demonstrated here that brain water content continuously increased for 48 h following TBI, but that this effect was abrogated by treatment with 3% HS.
The mechanistic basis for the brain edema induced by TBI is not fully understood; however, the expression levels of the pro-inflammatory cytokines such as TNF-α and IL-1β and the complement cascade may be important factors [30]. We confirmed that TBI caused an upregulation of IL-1β and TNF-α. In this study, we found that treatment with 3% HS treatment for 6 h decreased TNF-α and IL-1β levels at 24 and 48 h. Moreover, the expression of caspase-3 – which may be activated by TNF-α via caspase-8 [31] – increased over time in the TBI group relative to sham animals, which was confirmed by the results of the TUNEL assay. However, these effects were countered by administration of 3% HS.
AQP4, the primary water-channel protein expressed in the brain, is abundant in astrocyte foot processes at the borders between the brain parenchyma and major fluid compartments, and in ependymal cells lining the ventricles containing cerebrospinal fluid [32]. AQP4 is upregulated in the ischemic brain and is thought to play an important role in the cerebrospinal fluid resorption and regulation of brain edema [33]. Indeed, mortality is reduced in AQP4-null mice, which are less prone to cytotoxic brain edema than their wild-type counterparts, whereas transgenic mice overexpressing AQP4 show increased brain swelling [34]. Our results show that TBI increased AQP4 protein level, which was reversed by 3% HS; this can lead to other non-osmotic effects in the treatment of brain edema. Many studies have shown that HS inhibited the expression of AQP4 in brain swelling induced by ischemic stroke and meningitis [35,36]. IL-1β induced the expression of AQP4 in microglia and astrocytes [34], and TNF-α is a key mediator of brain edema [37]. Thus, TBI induces the upregulation of AQP4 by increasing inflammation.
Conclusions
In the present study, we showed that administration of 3% HS plays a key role in the inhibition of AQP4, IL-1β, and TNF-α expression induced by TBI. Taken together, our results indicate that 3% HS is an effective treatment for suppressing inflammation and preventing neuronal damage following TBI.
Footnotes
Source of support: This research was supported in part by the Science and Technology Commission-funded projects of Hangzhou [grant number 20150633B28]
Conflict of interest
None.
References
- 1.McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014;10:242–53. doi: 10.1016/j.jalz.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–23. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 3.Molgaard CA, Stanford EP, Morton DJ, et al. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 1990;9(5):233–42. doi: 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- 4.Pop V, Badaut JA. Neurovascular perspective for long-term changes after brain trauma. Transl Stroke Res. 2011;2(4):533–45. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simon-O’Brien E, Gauthier D, Riban V, Verleye M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J Neuroinflammation. 2016;13(1):203. doi: 10.1186/s12974-016-0687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verkman AS, Anderson MO, Papadopoulos MC. Aquaporins: Important but elusive drug targets. Nat Rev Drug Discov. 2014;13:259–77. doi: 10.1038/nrd4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto N, Yoneda K, Asai K, et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Brain Res Mol Brain Res. 2001;20(1):26–38. doi: 10.1016/s0169-328x(01)00064-x. [DOI] [PubMed] [Google Scholar]
- 8.Badaut J, Fukuda AM, Jullienne A, Petry KG. Aquaporin and brain diseases. Biochim Biophys Acta. 2014;1840(5):1554–65. doi: 10.1016/j.bbagen.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14(4):265–77. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li YK, Wang F, Wang W, et al. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: Involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology. 2012;37(8):1867–78. doi: 10.1038/npp.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szu JI, Binder DK. The role of astrocytic Aquaporin-4 in synaptic plasticity and learning and memory. Front Integr Neurosci. 2016;10:8. doi: 10.3389/fnint.2016.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Y, Wu P, Su J, et al. Effects of Aquaporin-4 on edema formation following intracerebral hemorrhage. Exp Neurol. 2010;223(2):485–95. doi: 10.1016/j.expneurol.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Jin BJ, Zhang H, Binder DK, Verkman AS. Aquaporin-4-dependent K(+) and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol. 2013;141(1):119–32. doi: 10.1085/jgp.201210883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziai WC, Toung TJ, Bhardwaj A. Hypertonic saline: First-line therapy for cerebral edema. J Neurol Sci. 2007;261(1):157–66. doi: 10.1016/j.jns.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 15.Peterson B, Khanna S, Fisher B. Prolonged hypernatremia controls elevated intracranial pressure in head-injured pediatric patients. Crit Care Med. 2000;28(4):1136–43. doi: 10.1097/00003246-200004000-00037. [DOI] [PubMed] [Google Scholar]
- 16.Zeng HK, Wang QS, Deng YY. A comparative study on the efficacy of 10% hypertonic saline and equal volume of 20% mannitol in the treatment of experimentally induced cerebral edema in adult rats. BMC Neurosci. 2010;11(1):153. doi: 10.1186/1471-2202-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu QY, Dian CF, Rong QW, Shi MY. Effect of NF-κB, survivin, Bcl-2 and Caspase3 on apoptosis of gastric cancer cells induced by tumor necrosis factor related apoptosis inducing ligand. World J Gastroenterol. 2004;10(1):22–25. doi: 10.3748/wjg.v10.i1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao ZZ, Li P, Ye SY, et al. Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A2A receptor inactivation. Sci Rep. 2017;7:2254. doi: 10.1038/s41598-017-02505-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson CS, Garcia R, Gaddam SS, et al. Treatment of mild traumatic brain injury with an erythropoietin-mimetic peptide. J Neurotrauma. 2013;30:765–74. doi: 10.1089/neu.2012.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon CE, Farin FM, Lighthall JW, et al. A controlled cortical impact model of traumatic brain injury in the rat. J neurosci Methods. 1991;39:253–62. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 21.Anderson GD, Farin FM, Bammler TK, et al. The effect of progesterone dose on gene expression after traumatic brain injury. J Neurotrauma. 2011;28:1827–43. doi: 10.1089/neu.2011.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim DH, Ko IG, Kim BK, et al. Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol Behav. 2010;101:660–65. doi: 10.1016/j.physbeh.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 23.Haber M, Abdel Baki SG, Grin NM, et al. Minocycline plus N-acetylcysteine synergize to modulate of mild traumatic brain injury. Exp Neurol. 2013;249:167–77. doi: 10.1016/j.expneurol.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Briones TL, Woods J, Rogozinska M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol Commun. 2013;1:57. doi: 10.1186/2051-5960-1-57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Ren Z, lliff JJ, Yang L, et al. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab. 2013;33:834–45. doi: 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai RK, Chang CH, Wang HZ. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp Eye Res. 2008;87(3):242–50. doi: 10.1016/j.exer.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Morganti-Kossmann MC, Satgunaseelan L, Bye N, et al. Modulation of immune response by head injury. Injury. 2007;38(12):1392–400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Simard JM, Kent TA, Chen M, et al. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6(3):258–68. doi: 10.1016/S1474-4422(07)70055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng HK, Wang QS, Deng YY, et al. A comparative study on the efficacy of 10% hypertonic saline and equal volume of 20% mannitol in the treatment of experimentally induced cerebral edema in adult rats. BMC Neurosci. 2010;11(1):153. doi: 10.1186/1471-2202-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander JJ, Jacob A, Cunningham P, et al. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52(3):447–56. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croker BA, O’Donnell JA, Nowell CJ, et al. Fas-mediated neutrophil apoptosis is accelerated by Bid, Bak, and Bax and inhibited by Bcl-2 and Mcl-1. Proc Natl Acad Sci. 2011;108(32):13135–40. doi: 10.1073/pnas.1110358108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tait MJ, Saadoun S, Bell BA, et al. Water movements in the brain: Role of aquaporins. Trends Neurosci. 2008;31(1):37–43. doi: 10.1016/j.tins.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 33.King LS, Agre P. Pathophysiology of the aquaporin water channels. Annu Rev Physiol. 1996;58(1):619–48. doi: 10.1146/annurev.ph.58.030196.003155. [DOI] [PubMed] [Google Scholar]
- 34.Cao C, Yu X, Liao Z, et al. Hypertonic saline reduces lipopolysaccharide-induced mouse brain edema through inhibiting aquaporin 4 expression. Crit Care. 2012;16(5):R186. doi: 10.1186/cc11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, Li L, Luo ZQ, et al. Superior effect of hypertonic saline over mannitol to attenuate cerebral edema in a rabbit bacterial meningitis model. Crit Care Med. 2011;39(6):1467–73. doi: 10.1097/CCM.0b013e3182120d13. [DOI] [PubMed] [Google Scholar]
- 36.Zeng HK, Wang QS, Deng YY, et al. A comparative study on the efficacy of 10% hypertonic saline and equal volume of 20% mannitol in the treatment of experimentally induced cerebral edema in adult rats. BMC Neurosci. 2010;11(1):153. doi: 10.1186/1471-2202-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexander JJ, Jacob A, Cunningham P, et al. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–56. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]