

Abstract
Introduction
Gastrointestinal stromal tumours (GIST) are mesenchymal neoplasms that usually carry an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRA) genes with predictive and prognostic significance. We investigated the extended mutational status of GIST in a patient population of north-western Greece in order to look at geopraphic/genotypic distinctive traits.
Patient and methods
Clinicopathological and molecular data of 38 patients diagnosed from 1996 to 2016 with GIST in the region of Epirus in Greece were retrospectively assessed. Formalin-fixed paraffin-embedded tumours were successfully analysed for mutations in 54 genes with oncogenic potential. Next generation sequencing was conducted by using the Ion AmpliSeqCancer Hotspot Panel V.2 for DNA analysis (Thermofisher Scientific).
Results
Among 38 tumours, 24 (63.16%) and seven (18.42%) of the tumours harboured mutations in the KIT and PDGFRA genes, respectively, while seven (18.42%) tumours were negative for either KIT or PDGFRA mutation. No mutations were detected in five (13.16%) cases. Concomitant mutations of BRAF and fibroblast growth factor receptor 3 (FGFR3) genes were observed in two patients with KIT gene mutation. Two patients with KIT/PDGFRA wild-type GIST had mutations in either KRAS or phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) genes. There was no significant survival difference regarding the exonic site of mutation in either KIT or PDGFRA gene. The presence of a mutation in pathway effectors downstream of KIT or PDGFRA, such as BRAF, KRAS or PIK3CA, was associated with poor prognosis. Adverse prognosticators were also high mitotic index and the advanced disease status at diagnosis.
Conclusions
We report comparable incidence of KIT and PDGFRA mutation in patients with GIST from north-western Greece as compared with cohorts from other regions. Interestingly, we identified rare mutations on RAS, BRAF and PIK3CA genes in patients with poor prognosis.
Keywords: gastrointestinal stromal tumors, mutational status, next generation sequencing
Key questions.
What is already known about this subject?
Prognostic factors such as the tumour site and size, the mitotic count and the type of certain KIT or PDGFRA mutations on gastrointestinal stromal tumours (GIST) have been already described.
Data beyond KIT and PDGFRA mutation are scarce and lacks a population-based study from a region of south-east Europe.
What does this study add?
This is a population-based study of patients with GIST from north-western Greece.
We examined the extended mutational profile of patients with GIST by using next generation sequencing.
We report concomitant mutations of BRAF and FGFR3 genes in two patients with KIT mutation. We also detected rare mutations on PIK3CA and KRAS genes in two patients with KIT /PDGFRA wild-type GIST.
We show that the patients with a mutation in a downstream effector of KIT and PDGFRA signalling such as BRAF, KRAS, PIK3CA genes had poor prognosis.
How might this impact on clinical practice?
In the near future, methods to investigate the comprehensive molecular profile of patients with GIST may be implemented in the clinical practice to uncover mutations with prognostic and predictive significance.
Introduction
Gastrointestinal stromal tumours (GISTs) are mesenchymal neoplasms originating at any segment of the gastrointestinal tract.1 The term was coined by Mazur et al in 1983 to describe a morphologically broad spectrum of tumours of the gastric wall.2 GISTs are rare tumours and comprise about 1% of all gastrointestinal malignancies. They most frequently arise in the stomach (60%) and less frequently in the small intestine (30%). They can also occur in other parts of the gut (10%) and rarely in the mesentery and omentum. Thirty per cent of GISTs have a malignant clinical phenotype with increased frequency of intra-abdominal and liver metastasis.‘’
The annual incidence of GIST is estimated at 10–15 per million per year.3 The median age at diagnosis is 65 and the prevalence is equal between men and women. Most of the cases are sporadic (95%), but they can also be associated with genetic syndromes such as familial GIST, neurofibromatosis type 1, Carney’s triad and Carney-Stratakis triad.
The majority of GISTs carry an activating mutation of KIT proto-oncogene or the plateled-derived growth factor receptor alpha (PDGFRA) gene. Both genes encode proteins that belong to the receptor tyrosine kinase class III family. KIT and PDGFRA orchestrate signalling transduction pathways that promote proliferation and inhibit apoptosis when activated by their respective ligand stem cell factor or platelet-derived growth factor. Gain-of-function mutations lead to constitutively active signalling and result in neoplasia.4 5 GISTs are suggested to originate from the stem cell precursors of the interstitial Cajal cell (ICC).6 Both GISTs and ICC express the receptor tyrosine kinase KIT and their development relies on KIT receptor signalling.4
Macroscopically, GISTs are well circumscribed and vary in size ranging from less than 1 cm to more than 40 cm.7 Tumour sections reveal grey to pink colour and areas of cystic degeneration, haemorrhage and infarction. Microscopically, GISTs have either spindle cell or epithelioid cell morphology and contain eosinophilic collagen structures stained with periodic acid-Schiff stain. Positive staining for CD117 (KIT) (>95% GISTs) and/or discovered on GIST 1 markers is the hallmark of diagnosis.8 However, negative staining does not rule out the diagnosis, and mutational analysis of KIT and PDGFRA can be helpful in these cases.
The mutational profile of GISTs is of great biological significance.9 Approximately, 85% of GISTs have an active mutation in the KIT and 5% in the PDGFRA gene. The most common mutation of KIT is located in exon 11 (70%) which is the region that encodes the juxtamembrane domain of the receptor. The second most common mutation is in exon 9 (10%) encoding the extracellular domain of KIT. Exon 9 mutants show less sensitivity to imatinib.10 Mutations of exons 13 and 17 encoding the ATP-binding pocket and the activation loop, respectively, are rare (1%–2%). Mutations in exon 18 account for the majority (>80%) of the PDGFRA mutations and may be associated with imatinib resistance. Rare mutations in exons 12 and 14 of the PDGFRA gene have also been reported. Finally, 5%–15% of GIST are characterised as wild type. The latter do not harbour mutations of either KIT or PDGFRA gene and are potentially resistant to imatinib.
Prognostic factors such as the tumour site and size, the mitotic count and the type of certain KIT mutations have been well evaluated.11 Although GIST is considered to be responsive to tyrosine kinase inhibitors, primary and acquired resistance to such treatment is frequently reported and it is often attributed to secondary mutations.12 Moreover, ethnic and geographical variations in the mutational profile and genetic determinants of resistance have not been extensively studied. Nevertheless, data from comprehensive analysis of genomic alterations are scarce. In this study, we present data from a retrospective analysis of patients diagnosed with GIST within a 20-year period in the region of north-western Greece. We examined clinicopathological features of this rare tumour and we investigated the prognostic significance of the mutational status of KIT and PDGFRA as well as of 52 additional oncogenes and tumour suppressor genes by the use of next generation sequencing (NGS) technology.
Patients and methods
Patient characteristics and tumour samples
Clinical and molecular data of 38 patients diagnosed with GIST in north-western Greece within a period of 20 years (1996–2016) were retrieved. GIST diagnosis was made by distinctive histopathology and the presence of KIT expression in biopsies obtained from the primary tumour in treatment-naive patients. Demographic and clinical factors were also evaluated. Informed consent was obtained from all patients before testing.
Tissue selection and DNA extraction
H&E stained tissue sections of formalin-fixed paraffin-embedded (FFPE) tumour samples were used as a guide for the localisation of neoplastic areas. Only specimens with a minimum of 75% tumour cell content were selected for this study. The selected neoplastic areas were manually microdissected and DNA was extracted from unstained 10 µm thick FFPE sections using the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. The Qubit DNA HS assay kit (Life Technologies) was used to quantify purified DNA.
Ion Ampliseq NGS
NGS was conducted by using the Ion AmpliSeq Cancer Hotspot Panel v2 for DNA analysis (Thermofisher Scientific). The Ion AmpliSeq Cancer Hotspot Panel v2 (Ampliseq, Thermofisher Scientific) is designed to amplify 207 amplicons covering approximately 2800 COSMIC mutations from 50 oncogenes and tumour suppressor genes and four fusion-gene transcripts in FFPE tumour samples (ComPlit DX assay: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, NTRK1, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, ROS1, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL genes).
Library preparation
DNA concentrations were measured using the Qubit 2 fluorometer in combination with the Qubit2 dsDNA HS assay kit. For DNA library construction, 10 ng of DNA from each of the 38 FFPE samples were used. An amplicon library was thus generated from total DNA using the Ion AmpliSeq Library Kit V.2.0 according to the manufacturer’s instructions. Briefly, amplicon amplification was performed using the Ion Ampliseq HiFi Master Mix. The amplicons were then digested with FUPA reagent and barcoded with the IonCode Barcode Adapters. Subsequently, the amplified products were purified from the other reaction components using AgencourtAMPure XP PCR purification system (Beckman Coulter, Brea, California, USA).
For libraries originated from genomic DNA, the Ion Library Equaliser Kit method was used for normalising library concentration at ~100 pM without the need of extra instrumentation. Finally, equal volumes of each normalised DNA library were combined and clonally amplified on Ion Sphere particles (ISP) by emulsion PCR performed on the Ion One Touch 2 instrument with the Ion PI HiQ OT2 200 Kit (Thermofisher Scientific) according to the manufacturer’s instructions.
Quality control was performed using the Ion Sphere Quality Control kit (Thermofisher Scientific) to ensure that 10%–30% of template positive ISP were generated in the emulsion PCR. Finally, the template-positive Ion PI ISP were enriched in the Ion OneTouch ES instrument, loaded on an Ion PI Chip v3 and sequenced on an Ion Proton Sequencer with the Ion PI HiQ Sequencing 200 Kit according to the manufacturer’s instructions.
Data analysis
NGS data analysis was performed with the Ion Reporter Software within Torrent Suite Software (Thermofisher Scientific). Statistical analysis was made with SPSS V.17.
Results
Patient demographics and tumour characteristics
Thirty-eight patients (26 males, 12 females) originating from north-western Greece were diagnosed with GIST and managed in the Department of Medical Oncology at Ioannina University Hospital, between 1996 and 2016. Twenty tumours were larger than 5 cm and 15 harboured necrosis. More than one and more than five mitoses per 10 high-power fields were observed in 17 and 15 tumours, respectively. Ki67 immunostaining was as follows: <2% of malignant cells in nine cases, 2%–20% in 22 and >20% in seven. The tumour relapsed in eight patients later during the course of the disease, while nine patients died at a median follow-up of 78 months (online Supplementary table 1). The median overall survival of the entire GIST cohort was 171 months (95% CI 135 to 199).
esmoopen-2018-000335supp001.pdf (308.9KB, pdf)
Mutation analysis
Mutation analysis revealed the presence of mutations in 33 of 38 GISTS (86.84%), while no mutations were detected in five cases (13.16%). Of the 54 oncogenes and tumour suppressor genes sequenced, alterations were detected in BRAF, PIK3CA, KRAS, FGFR3, PDGFRA and KIT genes. The most common site of mutation was in the KIT gene (24 cases, 63.16%) followed by the PDGFRA gene (seven cases, 18.42%) (figure 1). Notably, seven patients (18.42%) had tumours negative for KIT/PDGFRA gene mutation. All mutations are summarised in table 1.
Figure 1.
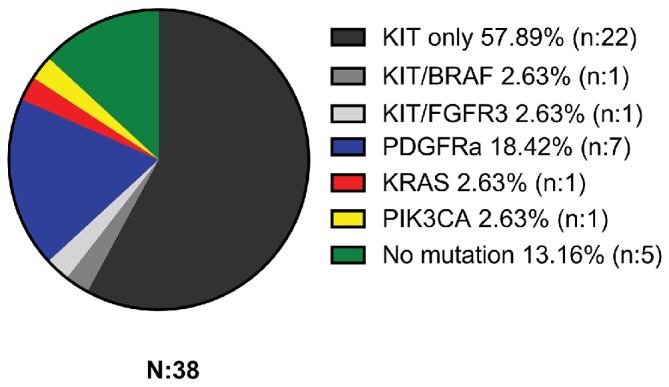
Percentage of mutations detected in 38 gastrointestinal stromal tumours analysed.
Table 1.
The mutational profile and the allelic frequencies of 38 gastrointestinal stromal tumour cases
| Gene | Case no. | Exon | Nucleotide | Codon | Allelic frequency (%)/case no. |
| KIT | 2, 24, 38 | 9 | c.1509_1510insGCCTAT | p.Y503_F504insAY | 32%, 43%, 35% |
| KIT | 26, 36 | 11 | c.1651_1662del12 | p.P551_E554del | 52%, 65% |
| KIT | 9, 23 | 11 | c.1652_1654delCCA | p.P551_M552>L | 51%, 49% |
| KIT | 13 | 11 | c.1665_1679del15 | p.V555_V560>V | 47% |
| KIT | 20 | 11 | c.1663_1716del53 | p.V555_D572del | 94% |
| KIT | 8 | 11 | c.1666_1671delCAGTGG | p.Q556_W557del | 47% |
| KIT | 22 | 11 | c.1669T>G | p.W557G | 41% |
| KIT | 25, 34 | 11 | c.1669_1674delTGGAAG | p.W557_K558del | 52%, 43% |
| KIT | 16* | 11 | c.1669_1680del12 | p.W557_V560del | 67% |
| KIT | 3, 35 | 11 | c.1669_1683del15 | p.W557_E561del | 86%, 90% |
| KIT | 6 | 11 | c.1676T>C | p.V559A | 49% |
| KIT | 10 | 11 | c.1679T>A | p.V560D | 60% |
| KIT | 31 | 11 | c.1676T>A | p.V559D | 49% |
| KIT | 14 | 11 | c.1682T>A | p.V561D | 9% |
| KIT | 7, 29 | 11 | c.1727T>C | p.L576P | 66%, 50% |
| KIT | 12, 15† | 13 | c.1924A>G | p.K642E | 60%, 48% |
| PDGFRA | 5, 18, 27, 30 | 18 | c.2525A>T | p.D842V | 39%, 55%, 40%, 51% |
| PDGFRA | 37 | 18 | c.2525_2534delinsC | p.D842_H845delinsA | 57% |
| PDGFRA | 19, 28 | 18 | c.2527_2538del12 | p.I843_D846delIMHD | 39%, 54% |
| BRAF | 16* | 11 | c.1405G>A | p.G469R | 6% |
| FGFR3 | 15† | 9 | c.1150T>C | p.F384L | 49% |
| KRAS | 21 | 2 | c.35G>A | p.G12D | 70% |
| PIK3CA | 17 | 9 | c.1633G>A | p.E545K | 37% |
*Concomitant KIT/BRAF mutation (case 16).
†Concomitant KIT/FGFR3 mutation (case 15).
KIT mutations
Of the 24 KIT mutated GIST, 19 (79.17%) harboured mutations in exon 11, three (12.5%) in exon 9 and two (8.33%) in exon 13. No mutations were detected in the remaining exons (2, 10, 14, 15 and 17) of the KIT gene. More specifically, most of the activating exon 11 mutations were clustered in the proximal part between codons 550 and 560 and consisted of small in-frame deletions and point mutations. In the distal part of exon 11, one point mutation (p.L576P) was found in two samples. In the remaining two mutated exons of the KIT gene, exons 9 and 13, a 6-nucleotide insertion GCCTAT between 1509 and 1510 nucleotides causing an insertion of two amino acids (p.Y503_F504insAY) and an amino acid substitution at position 642 (p.K642E) were detected, respectively. The median overall survival of patients with KIT-mutated tumours was 171 months (95% CI 125 to 199), with no significant survival differences observed between various exonic mutations.
PDGFRA mutations
Regarding PDGFRA, all mutations were localised in exon 18. In three samples (42.86%), two deletions and one deletion/insertion mutation were detected, whereas the drug resistance-associated p.D842V mutation was detected in four (57.14%) samples. The median overall survival of patients with PDGFRA-mutated tumours was 159 months (95% CI 115 to 180), with no significant survival differences observed between various exonic PDGFRA mutations or patients with PDGFRA D842V mutation (median overall survival of 125 months).
Concomitant mutations
Considering the scarcity of comprehensive genomic data in GIST literature, we investigated the incidence of mutations in additional oncogenes that may be implicated in GIST development and progression. As shown in table 1, two cases with concomitant KIT and BRAF (case 16) and KIT and FGFR3 (case 15) mutations were detected. The patient with concurrent KIT/BRAF mutations was placed on adjuvant treatment with imatinib but he developed progressive disease with liver metastases 12 months later. He was then switched to sunitinib and he had an overall survival of 55 months. The patient with simultaneous KIT/FGFR3 mutations received adjuvant treatment with imatinib with no tumour relapse so far (17 months). In addition to the concomitant mutations above, we found two rare mutations in KIT/PDGFRA wild-type GIST. In particular, we report a patient with a KRAS c.35G>A (G12D) and a patient with a PIK3CA c.1633G>A (E545K) mutation, respectively. The patient with the KRAS mutation had a gastric GIST, he was managed with subtotal gastrectomy but he relapsed quickly locoregionally and despite imatinib therapy, he succumbed at an overall survival of 15 months. The patient with the PIK3CA mutation had locally advanced GIST at diagnosis which relapsed quickly and she was then placed on imatinib but with a limited survival of 4 months.
Correlation of histopathological and molecular features
In our study, 25 of the 38 cases were composed of spindle cells (65.79%), five were composed of epithelioid cells (13.16%), while eight were of mixed subtype (21.05%). From the 24 KIT mutated cases, seven showed epithelioid or mixed histology (two epithelioid, five mixed), whereas the rest showed spindle histology. From the seven PDGFRA mutated cases, five were of epithelioid or mixed cell morphology (two epithelioid, three mixed; 71.43%) and two were of spindle cell morphology (28.57%). From the seven GIST that were negative for KIT/PDGFRA mutation, one was of epithelioid histology subtype and the rest were of spindle histology subtype. Overall, PDGFRA-mutated GISTs were more often epithelioid than the KIT mutated and the KIT/PDGFRA wild-type GIST. Interestingly, the KIT-mutated case with concomitant BRAF mutation was also of epithelioid histology (figure 2). Regarding KIT immunohistochemistry, all KIT-mutated GIST expressed KIT protein in tissue sections. KIT immunohistochemical staining, even focally, was also detected in five out of seven PDGFRa-mutated GISTs and in three out of seven KIT/PDGFRA wild-type GIST.
Figure 2.

Histological–molecular correlation in gastrointestinal stromal tumour (GIST). (A and B) Spindle cell GIST with KIT mutation ((A) H&E ×400 and (B) KIT immunohistochemistry, DAB ×400). (C and D) Epithelioid GIST with concominant KIT and BRAF mutation ((C) H&E ×400 and (D) KIT immunohistochemistry, DAB ×400). (E) Epithelioid GIST with PDGFRA mutation (H&E ×200). (F) Spindle cell GIST with undetected mutation (H&E ×200).
Prognostic parameters
Among the clinical and molecular variables analysed, those that retained prognostic significance were the Ki67 immunostaining, the disease extent and the presence of an activating mutation downstream of KIT and PDGFRa genes such as RAS, BRAF or PIK3CA.
The median overall survival of patients with low Ki67 staining (<2%) was not reached versus 170 months (95% CI 67 to 215) in patients with intermediate Ki67 expression (2%–20%) versus only 42 months (95% CI 25 to 58) in patients with high Ki67 staining (>20%) (log-rank, two-sided p=0.01). The median overall survival of patients with localised tumours at diagnosis was 171 months (95% CI 110 to 197) versus 55 months (95% CI 13 to 79) in patients with metastatic dissemination at diagnosis (log-rank, two-sided p=0.011). The presence of an activating mutation in the RAS/RAF axis (KRAS or BRAF genes) or the PI3K/AKT axis (PIK3CA gene), downstream of KIT/PDGFRA signalling pathway, was associated with a median overall survival of 15 months (95% CI 15 to 68) versus a median overall survival of 171 months (95% CI 125 to 197) in the absence of KRAS, BRAF and PIK3CA mutations (log-rank, two-sided p=0.003) (figure 3).
Figure 3.

Patient survival by KRAS, BRAF, PIK3CA mutation status. Comparison between patients with a mutation in a downstream effector of KIT (KRAS, BRAF, PIK3CA) and all other cases.
Discussion
In the present study, we investigated the mutational profile in 38 Greek patients with GIST managed in an academic department in north-western Greece. Although other Greek groups have published clinicopathological and molecular data,13 14 this is the first population-based study on Greek patients with GIST. To the best of our knowledge, it also consists the first report from a region of south-east Europe.
The overall extended mutational rate (54 gene panel) in our registry was 86.84%, approximating the frequencies observed in previous studies. However, the mutation rate for KIT was 63.16% and for PDGFRA 18.42%, whereas the estimated percentages from clinical trials were 80%–85% and 5%–10%, respectively.10 15 This disparity could be attributed to different patient characteristics, as most of these case series were phase III studies that recruited patients with advanced GIST, whereas our report is a population-based study of patients with GIST of various stages. Similarly, a prospective epidemiological study of patients with GIST in the Rhone-Alpes region in France demonstrated comparable incidence of KIT (67%) and PDGFRA mutations (16%).16 Presumably, the favourable prognosis of patients with PDGFRA mutation accounts for its decreased incidence in patients with advanced diseased in phase III clinical trials.17
Population studies have shown variable rates of KIT and PDGFRA mutation as well. Steigen et al demonstrated mutation rates of 75% for KIT and 10% for PDGFRA in northern Norway,18 while Braconi et al found KIT and PDGFRA mutations in 79% and 12% of tumour samples, respectively, in Ancona, Italy. Additionally, Mazzola et al showed mutation rates of 65% for KIT and 10% for PDGFRA in the south of Switzerland,19 while Wozniak et al demonstrated mutation rates of 69.3% and 12.9% for KIT and PDGFRA, respectively.20 Regarding data beyond Europe, Braggio et al reported mutation frequencies of 74.5% for KIT and 7.3% for PDGFRA mutation in Brazilian patients,21 while Du et al found KIT mutation rates of 76.6% and PDGFRA rates of 2.8% in China.22 These findings indicate ethnic/genetic variations. Environmental as well as genetic risk factors related to polymorphisms in DNA repair mechanisms or xenobiotic metabolism may contribute to the generation of a distinctive population.23 Interestingly, the north-west Hellenic region is a mountainous territory, prone to isolation and development of distinct genetic ‘niches’ with conserved, proprietary genetic traits.
Regarding the exonic site of mutation, exon 11 of the KIT gene was the most common site (79.17%) and exon 9 the second most frequent location (12.5%) followed by mutations in exon 13 (8.33%). Notwithstanding some fluctuations in the frequencies, our results are in line with other European cohorts.16 20 Several lines of evidence indicate that exon 9 mutations are associated with an increased risk for tumour progression.24 Furthermore, certain types of mutations such as exon 11 duplications may be correlated with favourable prognosis.17 Conversely, deletions of the codon 557–558 may be associated with poor clinical outcome.25 26 We did not observe significant differences in the overall survival and progression-free survival regarding the type of KIT mutation. With respect to PDGFRA, all mutations were located in exon 18 and the patients had benign clinical course. PDGFRA mutation is associated with a favourable disease outcome based on the reports of Wozniak et al from a multicenter analysis of a European registry24 and Joensuu et al from a pooled analysis of population-based studies and individual data.17 Moreover, we did not find significant difference in the overall survival between patients with PDGFRA D842V mutation and other PDGFRA mutations. However, the lack of difference in the overall survival between groups with different types of mutations in our cohort may be attributed to the small sample size, different tumour stage at diagnosis, varying follow-up times (76.31% of patients being alive at the time of the analysis) and distinct ‘all comers’ clinicopathological characteristics.
In addition to KIT and PDGFRA, we investigated the mutational status of several other oncogenes that may drive GIST development.27 BRAF mutation affects a small subset of patients with KIT/PDGFRA wild-type GIST, and the V600E mutation in exon 15 is the most commonly found one.28–31 BRAF mutation is of great therapeutic relevance given the fact that BRAF kinase is a downstream effector of KIT/PDGFRA signalling, while imatinib targets exclusively the upstream tyrosine kinases.32 33 In the current study, we found a case with concomitant mutations of KIT and BRAF. Rosi et al showed that concomitant mutations of BRAF and KIT are an extremely rare event.34 However, such cases may be underrepresented because investigations for genomic alterations of BRAF are usually reserved for GIST negative for KIT and PDGFRA mutations. Furthermore, we demonstrated that the patient with this dual mutation had poor prognosis. This is in accordance with the study of Miranda et al who showed that BRAF mutation conferred resistance to imatinib.35
Fibroblast growth factor receptor (FGFR) axis is another tyrosine kinase receptor signalling pathway that is implicated in oncogenesis by inducing proliferation and inhibiting apoptosis through the RAS/RAF/MEK/ERK and PI3K/AKT pathway. Mutations, amplifications and fusions that lead to constitutively active conformation of the FGFR have been reported in diverse tumours.36 Shi et al reported actionable alterations of FGFR1 gene that consisted of kinase fusions in KIT/PDGFRA wild-type GIST.37 Here, we report a case of GIST with concomitant mutation of FGFR3 in exon 9 and KIT in exon 13. FGFR may drive oncogenesis in KIT/PDGFRA wild-type GIST, but its role as an auxiliary mutation is unknown. Preclinical studies have shown that activation of the FGFR pathway restored c-KIT phosphorylation during imatinib treatment possibly through crosstalk with the MAPK pathway.38 However, the patient of our cohort has been placed on imatinib treatment without having tumour relapse so far (17 months).
KRAS mutations are frequently detected in various tumours but little is known in GIST where it seems to be an extremely rare event. Lasota et al failed to detect such mutations by sanger sequencing and pyrosequencing in a large cohort of 514 GIST.39 Other studies failed to detect KRAS mutation as well.29–31 40 Nevertheless, Miranda et al demonstrated the rare occurrence of KRAS mutation concurrently with exon 11 mutations in KIT gene (two cases) and exon 18 mutation in PDGFRA gene (one case) in GIST samples retrieved from the Ticino Registry.35 In the same study, it was also experimentally shown that the expression of KRAS mutants mediated resistance to imatinib treatment in a cell culture line cotransfected with a constitutively active KIT gene. Here, we found the presence of KRAS mutation in a patient with an overtly malignant GIST by next generation sequencing. Likewise, Hechtman et al found a KRAS G12V (c.35G>T) mutation in a patient with a clinically aggressive GIST by next generation sequencing.41 Similarly, Gao et al also reported the presence of KRAS mutation in a small subset of patients that have been previously characterised as wild-type GIST by sanger sequencing.42 We speculate that the adoption of NGS technology in population-based studies on GIST would unveil uncommon genomic alterations, providing us the opportunity to study their prognostic relevance.
PI3K pathway is another major cell signalling pathway that drives carcinogenesis by inducing cell survival and proliferation.43 Daniels et al and Lasota et al reported concurrent PIK3CA mutations with exon 11 KIT mutations in imatinib naive GIST that may represent secondary oncogenic events related to GIST progression. Such mutations are rare and associated with clinically aggressive phenotype.30 44 Furthermore, Falchook et al demonstrated the presence of PI3KC mutation in BRAF mutated GIST that was detected after treatment with BRAF inhibitor, thus indicating acquired resistance to this investigational compound.33 Here, we demonstrate the presence of a PIK3CA mutation in a patient with KIT/PDGFRA wild-type GIST who did not respond to imatinib and had short survival. This is the first report of a PIK3CA mutation in wild-type GIST. We believe that the lack of comprehensive genomic screening in clinical studies may underestimate such rare mutations.
In conclusion, we report comparable KIT and PDGFRA mutation rate in patients with GIST from north-western Greece compared with registries from other European regions. Although the retrospective, small nature of our cohort precluded us from studying the prognostic impact of KIT and PDGFRA mutation status, we report the presence of rare mutations in downstream effectors of KIT such as BRAF, KRAS or PIK3CA in patients with GIST with poor prognosis. Our data highlight the oddities between different populations with GIST and underscore the significance of the comprehensive molecular profiling in population-based studies. In the era of personalised medicine, the latter is of particular importance in view of the emergence of primary and acquired resistance to tyrosine kinase inhibitors.
Footnotes
Contributors: All the authors were involved in the acquisition and/or analysis of data and approved the final version for submission. LM and GP drafted and revised the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Obtained.
Ethics approval: Ethics Committee/Institutional Review Board of the university Hospital of Ioannina.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006;130:1466–78.doi:10.1043/1543-2165(2006)130[1466:GSTROM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 2. Mazur MT, Clark HB, tumors Gstromal. Reappraisal of histogenesis. Am J Surg Pathol 1983;7:507–19. [DOI] [PubMed] [Google Scholar]
- 3. Søreide K, Sandvik OM, Søreide JA, et al. . Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol 2016;40:39–46. 10.1016/j.canep.2015.10.031 [DOI] [PubMed] [Google Scholar]
- 4. Hirota S, Isozaki K, Moriyama Y, et al. . Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–80. 10.1126/science.279.5350.577 [DOI] [PubMed] [Google Scholar]
- 5. Heinrich MC, Corless CL, Duensing A, et al. . PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–10. 10.1126/science.1079666 [DOI] [PubMed] [Google Scholar]
- 6. Kindblom LG, Remotti HE, Aldenborg F, et al. . Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;152:1259–69. [PMC free article] [PubMed] [Google Scholar]
- 7. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 2008;3:557–86. 10.1146/annurev.pathmechdis.3.121806.151538 [DOI] [PubMed] [Google Scholar]
- 8. Novelli M, Rossi S, Rodriguez-Justo M, et al. . DOG1 and CD117 are the antibodies of choice in the diagnosis of gastrointestinal stromal tumours. Histopathology 2010;57:259–70. 10.1111/j.1365-2559.2010.03624.x [DOI] [PubMed] [Google Scholar]
- 9. Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol 2006;23:91–102. 10.1053/j.semdp.2006.08.006 [DOI] [PubMed] [Google Scholar]
- 10. Heinrich MC, Corless CL, Demetri GD, et al. . Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9. 10.1200/JCO.2003.04.190 [DOI] [PubMed] [Google Scholar]
- 11. ESMO/European Sarcoma Network Working Group. Gastrointestinal stromal tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3::iii21-6 10.1093/annonc/mdu255 [DOI] [PubMed] [Google Scholar]
- 12. Kee D, Zalcberg JR. Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther Adv Med Oncol 2012;4:255–70. 10.1177/1758834012450935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koumarianou A, Economopoulou P, Katsaounis P, et al. . Gastrointestinal stromal tumors (GIST): a prospective analysis and an update on biomarkers and current treatment concepts. Biomark Cancer 2015;7(Suppl 1):BIC.S25045–7. 10.4137/BIC.S25045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kontogianni-Katsarou K, Dimitriadis E, Lariou C, et al. . KIT exon 11 codon 557/558 deletion/insertion mutations define a subset of gastrointestinal stromal tumors with malignant potential. World J Gastroenterol 2008;14:1891–7. 10.3748/wjg.14.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Debiec-Rychter M, Sciot R, Le Cesne A, et al. . KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–103. 10.1016/j.ejca.2006.01.030 [DOI] [PubMed] [Google Scholar]
- 16. Cassier PA, Ducimetière F, Lurkin A, et al. . A prospective epidemiological study of new incident GISTs during two consecutive years in Rhône Alpes region: incidence and molecular distribution of GIST in a European region. Br J Cancer 2010;103:165–70. 10.1038/sj.bjc.6605743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joensuu H, Rutkowski P, Nishida T, et al. . KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol 2015;33:634–42. 10.1200/JCO.2014.57.4970 [DOI] [PubMed] [Google Scholar]
- 18. Steigen SE, Eide TJ, Wasag B, et al. . Mutations in gastrointestinal stromal tumors – a population-based study from Northern Norway. APMIS 2007;115:289–98. 10.1111/j.1600-0463.2007.apm_587.x [DOI] [PubMed] [Google Scholar]
- 19. Mazzola P, Spitale A, Banfi S, et al. . Epidemiology and molecular biology of gastrointestinal stromal tumors (GISTs): a population-based study in the South of Switzerland, 1999-2005. Histol Histopathol 2008;23:1379–86. 10.14670/HH-23.1379 [DOI] [PubMed] [Google Scholar]
- 20. Wozniak A, Rutkowski P, Piskorz A, et al. . Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol 2012;23:353–60. 10.1093/annonc/mdr127 [DOI] [PubMed] [Google Scholar]
- 21. Braggio E, Braggio DA, Small IA, et al. . Prognostic relevance of KIT and PDGFRA mutations in gastrointestinal stromal tumors. Anticancer Res 2010;30:2407–14. [PubMed] [Google Scholar]
- 22. Du CY, Shi YQ, Zhou Y, et al. . The analysis of status and clinical implication of KIT and PDGFRA mutations in gastrointestinal stromal tumor (GIST). J Surg Oncol 2008;98:175–8. 10.1002/jso.21104 [DOI] [PubMed] [Google Scholar]
- 23. O’Brien KM, Orlow I, Antonescu CR, et al. . Gastrointestinal stromal tumors, somatic mutations and candidate genetic risk variants. PLoS One 2013;8:e62119 10.1371/journal.pone.0062119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wozniak A, Rutkowski P, Schöffski P, et al. . Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: a european multicenter analysis based on ConticaGIST. Clin Cancer Res 2014;20:6105–16. 10.1158/1078-0432.CCR-14-1677 [DOI] [PubMed] [Google Scholar]
- 25. Wardelmann E, Losen I, Hans V, et al. . Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887–95. 10.1002/ijc.11323 [DOI] [PubMed] [Google Scholar]
- 26. Yan L, Zou L, Zhao W, et al. . Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: a systematic review and meta-analysis. Sci Rep 2015;5:13718 10.1038/srep13718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol 2014;27 Suppl 1(Suppl 1):S1–S16. 10.1038/modpathol.2013.173 [DOI] [PubMed] [Google Scholar]
- 28. Agaram NP, Wong GC, Guo T, et al. . Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes: chromosomes & cancer, 2008:47:853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agaimy A, Terracciano LM, Dirnhofer S, et al. . V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol 2009;62:613–6. 10.1136/jcp.2009.064550 [DOI] [PubMed] [Google Scholar]
- 30. Daniels M, Lurkin I, Pauli R, et al. . Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 2011;312:43–54. 10.1016/j.canlet.2011.07.029 [DOI] [PubMed] [Google Scholar]
- 31. Martinho O, Gouveia A, Viana-Pereira M, et al. . Low frequency of MAP kinase pathway alterations in KIT and PDGFRA wild-type GISTs. Histopathology 2009;55:53–62. 10.1111/j.1365-2559.2009.03323.x [DOI] [PubMed] [Google Scholar]
- 32. Hostein I, Faur N, Primois C, et al. . BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010;133:141–8. 10.1309/AJCPPCKGA2QGBJ1R [DOI] [PubMed] [Google Scholar]
- 33. Falchook GS, Trent JC, Heinrich MC, et al. . BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013;4:310–5. 10.18632/oncotarget.864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rossi S, Sbaraglia M, Dell’Orto MC, et al. . Concomitant KIT/BRAF and PDGFRA/BRAF mutations are rare events in gastrointestinal stromal tumors. Oncotarget 2016;7:30109–18. 10.18632/oncotarget.8768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miranda C, Nucifora M, Molinari F, et al. . KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res 2012;18:1769–76. 10.1158/1078-0432.CCR-11-2230 [DOI] [PubMed] [Google Scholar]
- 36. Chae YK, Ranganath K, Hammerman PS, et al. . Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget 2017;8:16052–74. 10.18632/oncotarget.14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi E, Chmielecki J, Tang CM, et al. . FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med 2016;14:339 10.1186/s12967-016-1075-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Javidi-Sharifi N, Traer E, Martinez J, et al. . Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res 2015;75:880–91. 10.1158/0008-5472.CAN-14-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lasota J, Xi L, Coates T, et al. . No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol 2013;26:1488–91. 10.1038/modpathol.2013.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Origone P, Gargiulo S, Mastracci L, et al. . Molecular characterization of an Italian series of sporadic GISTs. Gastric Cancer 2013;16:596–601. 10.1007/s10120-012-0213-y [DOI] [PubMed] [Google Scholar]
- 41. Hechtman JF, Zehir A, Mitchell T, et al. . Novel oncogene and tumor suppressor mutations in KIT and PDGFRA wild type gastrointestinal stromal tumors revealed by next generation sequencing. Genes: chromosomes & cancer, 2015:54:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao J, Li J, Li Y, et al. . Intratumoral KIT mutational heterogeneity and recurrent KIT/ PDGFRA mutations in KIT/PDGFRA wild-type gastrointestinal stromal tumors. Oncotarget 2016;7:30241–9. 10.18632/oncotarget.7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Millis SZ, Ikeda S, Reddy S, et al. . Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol 2016;2:1565–73. 10.1001/jamaoncol.2016.0891 [DOI] [PubMed] [Google Scholar]
- 44. Lasota J, Felisiak-Golabek A, Wasag B, et al. . Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod Pathol 2016;29:275–82. 10.1038/modpathol.2015.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2018-000335supp001.pdf (308.9KB, pdf)