

Retroviral TET1 or hypoxia stabilize Foxp3 in iTregs generated in vitro
Keywords: DNA methylation, hypoxia, regulatory T cell, reprogram
Abstract
Since induced regulatory T cells (iTregs) can be produced in a large quantity in vitro, these cells are expected to be clinically useful to induce immunological tolerance in various immunological diseases. Foxp3 (Forkhead box P3) expression in iTregs is, however, unstable due to the lack of demethylation of the CpG island in the conserved non-coding sequence 2 (CNS2) of the Foxp3 locus. To facilitate the demethylation of CNS2, we over-expressed the catalytic domain (CD) of the ten-eleven translocation (TET) protein, which catalyzes the steps of the iterative demethylation of 5-methylcytosine. TET-CD over-expression in iTregs resulted in partial demethylation of CNS2 and stable Foxp3 expression. We also discovered that TET expression was enhanced under low oxygen (5%) culture conditions, which facilitated CNS2 DNA demethylation and stabilization of Foxp3 expression in a TET2- and TET3-dependent manner. In combination with vitamin C treatment, which has been reported to enhance TET catalytic activity, iTregs generated under low oxygen conditions retained more stable Foxp3 expression in vitro and in vivo and exhibited stronger suppression activity in a colitis model compared with untreated iTregs. Our data indicate that the induction and activation of TET enzymes in iTregs would be an effective method for Treg-mediated adoptive immunotherapy.
Introduction
Regulatory T cells (Tregs) suppress excess immunity against a diverse range of antigens, including self-antigens, commensal bacteria-derived antigens and environmental allergens (1). Tregs are specified by an expression of the transcription factor Foxp3 (Forkhead box P3), which plays crucial roles in the differentiation, maintenance and function of Tregs (2–5). One population of Tregs, which develop in the thymus, is called thymus-derived Tregs (tTregs). On the other hand, another population of Tregs is differentiated from naive CD4+ T cells in the periphery upon reception of antigen stimulation with an appropriate combination of cytokines, including IL-2 and TGF-β. Foxp3+ Tregs generated outside the thymus are called induced Tregs (iTregs) when generated in vitro, or peripherally induced Tregs (pTregs) when generated in vivo. The ultimate goal of the adoptive transfer of antigen-specific iTregs is to control inflammation with minimum adverse effects in cases such as systemic immunosuppression and opportunistic infection. Given the low frequency of Tregs in human peripheral blood, a feasible approach is the generation of stable Tregs in vitro from non-Tregs. Unlike tTregs, however, iTregs are unstable, which is a fundamental difference between the two types of Tregs. This is a significant obstacle to the use of ex vivo-expanded iTregs for adoptive immunotherapy (6, 7).
Several intronic enhancers, designated ‘conserved non-coding sequences (CNSs)’, in addition to a promoter, have been identified at the Foxp3 gene locus and were determined to play important roles in stable Foxp3 expression (8, 9). Foxp3 expression in tTregs is stabilized by demethylation of the CpG island of the CNS2 region of the Foxp3 locus (10, 11), because demethylation of CNS2 leads to the recruitment of various transcription factors, including Stat5, NFAT, Runx1/Cbfβ, CREB and Foxp3 itself (12, 13). The unstable expression of Foxp3 in iTregs is believed to be associated with the strong methylation of CNS2 (14, 15). This idea is supported by the fact that iTregs induced in the presence of azacytidine, an inhibitor of DNA methylation, or iTregs with sustained IL-2 signals by an IL-2-anti-IL-2 antibody complex induced CNS2 demethylation in iTregs and stabilized Foxp3 expression (16, 17).
The CpG methylation patterns between tTregs and other CD4+ T-cell subsets were surprisingly similar globally, yet the Treg-specific demethylated regions (TSDRs) were distributed in genes that are known to be important for Treg differentiation and functions. Major TSDRs were observed in the CNS2 enhancer of Foxp3, Ctla4, Il2ra, Ikzf4 and Tnfrsf18 (14). TSDR demethylation is probably achieved through an active mechanism that includes recently discovered intermediate steps involving active DNA demethylation pathways, 5-hydroxymethylcytosine (5hmC) and enzymes of the ten-eleven translocation (TET) family (18–20). CNS2 was shown to be demethylated in a TET-dependent manner in Foxp3+ iTregs remained after transfer to lymphopenic mice (18). Thus, induction of TET enzymatic activity in iTregs provides a new strategy to generate stable iTregs (21). Indeed, vitamin C, a potent activator of TET enzymes (22, 23), has been shown to promote the demethylation of the Foxp3 CNS2 region and increase the stability of Foxp3 expression in iTregs (18, 24). The targeting of the pathways outlined here should allow the generation of stable iTregs, which may be used to achieve long-term tolerance in clinical settings.
Here, we established a new strategy to induce TET protein expression to generate stable iTregs. We showed that retroviral induction of the TET catalytic domain (TET-CD) in iTregs was sufficient for demethylation of the CNS2. Furthermore, we found that TET expression was drastically increased under low oxygen culture conditions. Low oxygen conditions also enhanced 5hmC levels in iTregs and induced demethylation of CNS2 of the Foxp3 gene. The combination of vitamin C treatment and low oxygen conditions generated extremely stable iTregs in vitro and showed the strongest potential for treatment in the inflammatory bowel disease model in vivo. Therefore, the induction of TET protein content and the enhancement of TET activities in iTregs may be used as an innovative strategy for the adoptive immunotherapy of transplant rejection and autoimmune diseases.
Methods
Mice
All research involving animals was carried out in accordance with the Guidelines for Animal Care approved by Keio University. Animals were maintained in specific pathogen-free conditions. The TET2flox/flox mice were obtained from the Jackson laboratory (25), and TET3flox/flox mice were as described previously (26). Foxp3Cre-YFP mice were kindly provided by Dr A. Rudensky (27). Foxp3hCD2-hCD52 mice were kindly provided by Dr S. Hori (28). All mouse strains were maintained on a C57BL/6 genetic background.
Plasmid construction
Murine TET1 catalytic domain (TET1-CD) (aa 1367–2036) cDNA was cloned by PCR from several murine cDNA libraries as described (21). cDNA was sub-cloned into eMIGR retrovirus vector containing N-terminal 2xFlag tag and 3′ IRES-EGFP (21, 29). The mutant TET1-CD with defective catalytic activity was generated by introducing H1620Y/D1622A substitutions (21). The retrovirus was prepared as described (30).
Retrovirus transduction to primary T cells and iTreg induction
Naive CD4+ T cells were stimulated for 72 h with plate-coated anti-CD3ε (2C11, 4 µg ml−1), anti-CD28 (PV1.17.10, 1 µg ml−1), anti-IFN-γ (R4-6A2, 5 µg ml−1) and anti-IL-4 (11B11, 5 µg ml−1) antibodies, supplemented with 10 ng ml−1 IL-2 and 2 ng ml−1 TGF-β1 (iTreg conditions). Then retrovirus solutions were added to the cells on day 3 in the presence of polybrene (4 µg ml−1), followed by centrifugation at 2500 rpm for 2 h at 35°C. Virus supernatants were removed 6 h after infection and cultured for an additional 48 h. Then, retrovirus-transduced cells were sorted into GFP-positive cells and, subsequently, used for DNA methylation assay as well as in vitro and in vivo stability and suppression assays. For in vitro stability assay, transduced iTregs were cultured for an additional 72 h with plate-coated anti-CD3ε in the presence or absence of 10 ng ml−1 IL-12 (Th1 conditions).
Treatment of T cells with vitamin C and low oxygen conditions
Hypoxic iTregs were induced by culturing naive CD4+ T cells under iTreg conditions for 4–5 days under 5% O2 and 5% CO2 in modular incubator chambers (Billups-Rothenberg) (31). Normoxic cells were incubated at 5% CO2 and 20% O2. Over 95% of T cells were Foxp3 positive; therefore, iTregs were used directly for subsequent DNA methylation assays as well as in vitro and in vivo stability and suppression assays. Vitamin C (10 µg ml−1 was used unless specified otherwise; Sigma-Aldrich) treatment was performed as described (24).
Assay for 5-methylcytosine and 5hmC content
Genomic DNA (2.5 µg) was incubated with 5 units of DNase I (Sigma, St Louis, MO, USA) and 4 mM MgCl2 at 37°C for 18 h. The sample was further treated with 3 units of nuclease P1 in 10 mM NaOAc (pH 5.2) and 50 µg ml−1 ZnSO4 at 37°C for 7 h, and then with 2.5 units of Escherichia coli alkaline phosphatase in 0.1 M NH4HCO3 at 37°C for 16 h. After purification, the samples underwent liquid chromatography on a Shimadzu high-performance liquid chromatography (HPLC) system (Shimadzu Corporation, Kyoto, Japan). Next, the mass spectrometer was operated under positive ionization mode with ion spray voltage of 5000 V and a source temperature of 500°C. The curtain and collision gas flows were 40.0 and 4.0 l min−1, respectively. Different forms of cytosines were identified by running mass spectrometry (MS)/MS multiple reaction monitoring in positive ion mode (MRM+) and by monitoring transition pairs of m/z 228 (precursor ion)/112 (product ion) for 2′-deoxycytidine (dC), m/z 242/126 for 5-methyl-2′-deoxycytidine (5mdC) and m/z 258/142 for 5-hmdC.
CpG methylation analysis by bisulfite sequencing
Bisulfite sequencing of TSDR was performed as described (32). Briefly, genomic DNA isolated from ~5 × 105 cells by phenol–chloroform extraction and isopropanol precipitation was digested with BamHI. The digested DNA concentration was adjusted to the range of 200 ng to 1 μg in 19 μl of dH2O, and then 1.2 µl of 5 N NaOH was added; the mixture was incubated at 37°C for 15 min. Next, 120 µl of the bisulfite mixture consisting of 3.6 N sodium bisulfite, 0.57 mM hydroquinone and 0.3 N NaOH was added. Then, the sample was treated with 15 cycles of 95°C for 30 s to 50°C for 15 min. After purification by the Wizard DNA Clean-up System (Promega) and elution with 50 µl of TE buffer, 3 µl of 5 N NaOH was added and incubated for 5 min at room temperature. The bisulfite product was isopropanol-precipitated and dissolved with 20 µl of TE buffer. Modified DNA was amplified by PCR with the primer sets described by Ohkura et al. (14), and T/A-cloned into pGEM-T Easy Vector (Promega). Eight inserted plasmids from each colony were purified and sequenced for each amplified PCR product.
SureSelect-PostBisulfite Adaptor tagging library preparation and data analysis
One hundred and nine megabases of mouse genome, which contain CpG island, tissue-specific differentially methylated regions and open regulatory regions, were captured using the SureSelect Mouse Methyl-Seq kit and the SureSelect Target Enrichment kit (Agilent Technologies). In brief, 500 ng of dsDNA was fragmented into 500–600 bp using Covaris S2 (Covaris), and shared DNA was purified with AMPure XP beads (Beckman Coulter). Purified DNA was hybridized with a biotinylated RNA probe and enriched using Dynabeads MyOne Streptavidin T1 (Life Technologies). Unmethylated lambda DNA was added to check bisulfite conversion efficiency. Bisulfite conversion was performed using an EZ DNA Methylation-Direct kit (Zymo Research), and bisulfite sequencing libraries were prepared according to the PostBisulfite Adaptor tagging (PBAT) protocol (http://www.chem-agilent.com/pdf/PBAT_SureSelect_Methyl_DraftB_19AUG15.pdf#search=‘SureSelectPBAT’). After first and second strand synthesis with Klenow fragment (3′→5′ exo-) (NEB), the concentration of library DNA was measured using a Library Quantification kit for Illumina (Kapa Biosystems). Two cycles of PCR amplification were performed to obtain sufficient DNA for sequencing. PhiX Control v3 was spiked at a final concentration of 3.3 pM and 71 bp single-end sequencing was performed using a Hiseq with a rapid run reagent kit v2 (Illumina). Sequenced single-end raw reads were trimmed based on read length and read quality using Trimmomatic (v. 2.0) and mapped to the mouse mm9 reference genome using Bmap software (http://itolab.med.kyushu-u.ac.jp/BMap/index.html) with default parameter settings. We extracted CpG sites that were covered at least five times in all samples and used them for analysis. Box plots and violin plots were made using R software package ggplot2 (v. 2.1.0) and EasyGgplot2 (v. 1.0.09000).
In vivo suppression assay
In vivo Treg suppression assays were performed as described (33). For in vivo suppression assay, 2 × 105 naive T cells from wild-type (WT) (Ly5.2+) mice purified with FACS were injected intravenously into Rag2−/− mice in combination with 2 × 105 iTregs (Ly5.1+) as described previously (34). Mice were observed and weighed daily. T cells were analyzed with flow cytometry.
Statistical analyses
Statistical analyses of all end-points were performed using the two-sided Student’s t-test or one-way analysis of variance (ANOVA). The variance among the groups was estimated using the F test or the Bartlett test. All data were presented as the mean ± SEM. The P values were represented as follows: ***P < 0.001, **P < 0.01 and *P < 0.05. P <0.05 was considered statistically significant.
Results
Over-expression of the TET catalytic domain in T cells promotes 5hmC synthesis
Previous reports demonstrated that the induction of CNS2 demethylation of iTregs in vivo is dependent on TET enzymes (18). Thus, we attempted to increase the expression of TET enzymes to induce CNS2 demethylation in TGF-β-induced iTregs. First, we attempted to introduce TET cDNA into naive T cells by retrovirus. However, we could not infect vectors carrying full-length cDNAs with high efficiency, because TET enzymes are extremely large (5–6 kb of DNA). Therefore, we constructed a retrovirus vector carrying the TET1-CD (35). Schematic structures of the constructs are shown in Fig. 1(A). We introduced these constructs into the 68-41 T-cell line and measured 5hmC levels by HPLC. As shown in Fig. 1(B), 5hmC levels in the parental 68-41 cells or 68-41 cells expressing mutant TET-CD (H1620Y/D1622A) were below the detection levels, whereas 5hmC was detected in cells expressing TET-CD. These data indicate that TET-CD can access DNA even though TET-CD lacked N-terminal DNA and protein interaction domains.
Fig. 1.

Expression of the TET catalytic domain enhances the stability of Foxp3 expression in iTregs. (A) Schematic representation of constructs carrying the TET1-CD and its inactive mutant (H1620Y/D1622A). (B) Detection of 5hmC by LC-MS/MS. 68-41 T-cell lines carrying empty (eMIGR), TET-CD and TET-mutated CD (H1620Y/D1622A). (C) The stability of Foxp3 expression in iTregs under Th1 conditions. iTregs were induced for 3 days, and control eMIGR or TET-CD retroviruses were infected and cultured a further 2 days. After sorting using GFP, iTregs were further incubated for 3 days under iTreg (upper panels) or Th1 (lower panels) conditions. Quantification of percentages of Foxp3+ cells after Th1 switch is shown in the right graph. (D) The stability of Foxp3 in iTregs after transfer to Rag2−/− mice. iTregs (1 × 106) infected with empty eMIGR or TET-CD retroviruses were transferred into Rag2−/− mice. After 2 weeks, Foxp3 positivity in lymph nodes (cLN, cervical lymph nodes; mLN, mesenteric lymph node; pLN, popliteal lymph node) and spleen is shown in the right graphs. Representative FACS profile of pLN CD4+ T cells is shown in the left panels. n = 3; ***P < 0.001, *P < 0.05.
Over-expression of the TET catalytic domain in iTregs leads to more stable Foxp3 expression and CNS2 demethylation
To introduce TET-CD constructs in primary T cells, we first stimulated naive CD4+ T cells under iTreg conditions (anti-CD3ε and anti-CD28 antibodies in the presence of TGF-β1 and IL-2) for 3 days; retrovirus was then infected under iTreg conditions for a further 2 days. At this point, the Foxp3+ fraction was >95% regardless of the virus infection. After sorting of infected cells with GFP, infected iTregs were incubated for a further 3 days under Th1 conditions (anti-CD3ε and anti-CD28 antibodies in the presence of IL-12). Empty vector-infected T cells lost Foxp3 expression, whereas some of the T cells over-expressing TET-CD retained Foxp3 expression under Th1 conditions (Fig. 1C). To examine the stability of iTregs over-expressing TET-CD in vivo, we transferred infected iTregs into Rag2−/− mice. Fourteen days after transfer, Foxp3 levels were greatly reduced in empty vector-infected iTregs, whereas 60–80% of Foxp3 was retained in iTregs infected with TET-CD retrovirus (Fig. 1D). These data indicate that TET-CD stabilized Foxp3 expression in iTreg under Th1 and lymphopenic conditions. Next, we examined the DNA methylation status of TSDRs (14). The CpG motifs of Foxp3 CNS2, Tnfrsf18 (GITR), Ctla4, Ikzf4 (Eos) and Il2ra (CD25) in tTregs were highly demethylated, whereas those in naive T cells were almost completely methylated (Fig. 2). Methylation of the CpGs of CNS2 in iTregs infected with empty vector was maintained during iTreg differentiation, while CpGs of CNS2 in iTregs infected with TET-CD vector were highly demethylated (Fig. 2). In contrast, none of the other TSDRs except for Il2ra were demethylated by TET-CD over-expression. These data indicate that TET-CD over-expression can introduce CNS2 site-specific demethylation in iTregs.
Fig. 2.

DNA methylation status of major TSDRs in iTregs carrying TET-CD. Methylation status determined by bisulfite conversion of the CpG sites of Foxp3 CNS2, Tnfrsf18 (GITR), Ctla4, Ikzf4 (Eos) and Il2ra (CD25) in iTregs carrying empty eMIGR or TET-CD. The methylation status of tTregs and, naive T cells is shown as positive and negative controls.
Lastly, we compared the suppression activity of iTregs over-expressing TET-CD in vitro and in vivo. The in vitro suppression activity of iTregs expressing TET-CD was comparable to that of iTregs infected with empty vector (data not shown). For in vivo colitis suppression assay, naive T cells (Ly5.1+) were co-transferred with iTregs (Ly5.2+) into Rag2−/− mice (Fig. 3). Body weight loss resulting from colitis was observed in Rag2−/− mice that received naive CD4+ T cells only (Fig. 3A). Co-transfer with iTregs infected with empty vector resulted in only partial suppression of colitis and body weight loss (Fig. 3A, eMIGR). In contrast, when naive T cells were co-transferred with iTregs expressing TET-CD, colitis was almost completely suppressed (Fig. 3A, TET-CD). Histological examination confirmed the infiltration of mononuclear cells in the colon tissues of empty vector-infected iTreg-transferred mice but not in those of TET-CD-introduced iTreg-transferred mice (Fig. 3B). Foxp3+ cells remained at much higher levels in lymphatic tissues of iTreg-TET-CD-transferred mice compared with control iTreg-transferred mice (Fig. 3C). These data indicate that the stability of iTregs expressing TET-CD was much higher in vivo inflammatory conditions than control iTregs and suggest that iTregs expressing TET-CD possess a high therapeutic potential for colitis.
Fig. 3.

Over-expression of TET-CD in iTreg results in higher suppression activity in vivo. Naive T cells (2 × 105) from WT mice purified with cell sorting were injected intravenously into Rag2−/− mice with or without 2 × 105 Ly5.1+ iTregs infected with eMIGR or TET-CD vectors. Body weight changes are shown in (A), and hematoxylin and eosin (HE) staining of the colon tissues is shown in (B). Foxp3 positivity in Ly5.1+CD4+ T cells in lamina propria lymphocyte (LPL) or mesenteric lymph node (mLN) was analyzed by flow cytometry (C). n = 3; **P < 0.01, *P < 0.05.
Low oxygen conditions increase the expression of TETs and promote 5-hydroxylation of methylated cytosine
We showed that TET-CD over-expression stabilized iTregs; however, gene manipulation remains a significant obstacle to the therapeutic application of iTregs. Therefore, we attempted to establish a method to up-regulate TET expression in iTregs. A previous paper demonstrated that the hypoxic condition increases TET1 expression, thereby inducing global 5hmC levels and facilitating the DNA demethylation of response elements (31). We confirmed that the 5% O2 condition up-regulated all TET expression in iTregs (Fig. 4A). The expression levels of DNA methyltransferase (DNMT) 1, DNMT3A and DNMT3B mRNA were not affected by the low oxygen condition. Next, we compared global 5hmC levels by HPLC in iTregs generated under low oxygen conditions (low-O2 iTregs). Since vitamin C has been shown to promote TET enzymatic activity in T cells (18, 24), we examined effects of the combination of the low oxygen culture condition and vitamin C treatment (10 µg ml−1). As shown in Fig. 4(B), although addition of vitamin C slightly increased global 5hmC levels, the low oxygen condition increased 5hmC more strongly than vitamin C. The combined treatment with the low oxygen and vitamin C did not show a further increase in global 5hmC levels. Low-O2 iTregs expressed similar levels of Foxp3 and GITR as normoxic iTregs after 5 days induction, whereas the levels of CTLA4 and CD25 were rather higher in low-O2 iTregs than in normoxic iTregs (data not shown).
Fig. 4.

Low oxygen conditions induce TET enzyme expression and Foxp3 CNS2 demethylation in iTregs. (A) iTregs were induced in the presence of 10 µg ml−1 vitamin C (Vit.C) or under the 5% O2 condition. The expressions of indicated genes were analyzed by quantitative real-time PCR. (B) 5hmC content in iTregs induced with indicated conditions was determined by LC-MS/MS analysis. (C, D) Methylation status determined by bisulfite conversion of the CpG sites of Foxp3 CNS2 in iTregs generated under the 5% O2 condition and/or 10 µg ml−1 vitamin C (C), Ctla4, Ikzf4, Tnfrsf18 and Il2ra in iTregs generated under the 5% O2 condition and 10 µg ml−1 vitamin C (D). The methylation status of tTregs is shown as a positive control in (C). (E) Violin plot showing the genome-wide methylation levels of CpG sites under 5% oxygen and normoxic conditions as detected by SureSelect-PBAT. *P < 0.05.
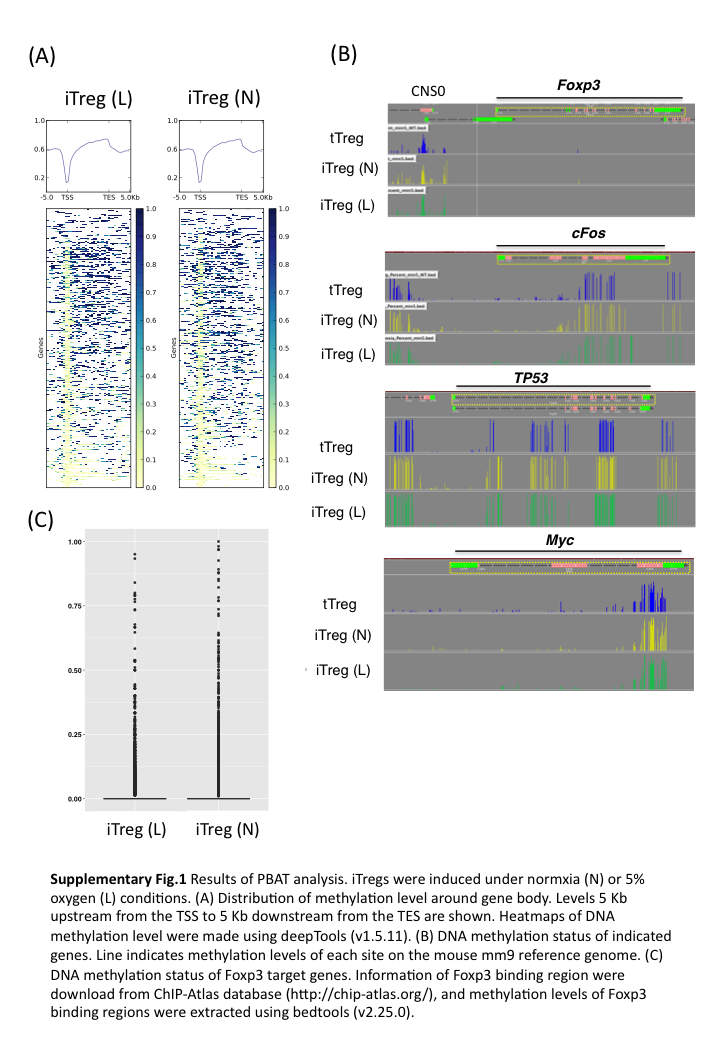
Next, we examined methylation status in TSDRs. The demethylation of Foxp3 CNS2 in iTregs was facilitated by both the treatment with vitamin C and the low oxygen condition (Fig. 4C). The combination of the low oxygen and vitamin C treatment further promoted the demethylation of Foxp3 CNS2 (Fig. 4C), while Tnfrsf18, Ctla4 and Ikzf4 were not effectively demethylated (Fig. 4D). PBAT analysis demonstrated that the genome-wide 5-methylcytosine (5mC) levels in major CpG islands did not differ between iTregs induced under the normoxia condition and the low oxygen condition (Fig. 4E). There may be a concern about unfavorable effects of TET activation and DNA demethylation in iTreg cells, such as oncogene activation. We have compared the DNA methylation status at whole genome levels by using PBAT (Supplementary Figure 1, available at International Immunology Online). Overall, there were no significant differences in whole genome DNA methylation levels and DNA methylation patterns in promoter and enhancer regions between iTregs induced under normoxia and low oxygen conditions (Fig. 4E; Supplementary Figure 1A, available at International Immunology Online). Although we observed demethylation in CNS2, there was no significant difference of DNA methylation in the promoter regions in the Foxp3 locus between these two types of iTregs (Supplementary Figure 1B, available at International Immunology Online). We compared the DNA methylation status of oncogenes and anti-oncogenes such as c-myc, c-fos and p53, but we did not find significant differences (Supplementary Figure 1B, available at International Immunology Online). There are slight increases of demethylation within the Foxp3 target genes, but these are not statistically significant (Supplementary Figure 1C, available at International Immunology Online). These data indicate that the low oxygen condition induced a very limited amount of site-specific demethylation in iTregs, which can be further promoted in combination with vitamin C treatment.
We further examined the stability of iTregs generated with the low oxygen concentration and vitamin C. The low oxygen and vitamin C treatment did not affect Foxp3 expression during the iTreg induction phase. After switching to the Th1 condition, Foxp3 expression was severely decreased in conventional iTregs, and the low oxygen and vitamin C only slightly prevented the loss of Foxp3 (Fig. 5A). The combination of the low oxygen and vitamin C treatment drastically improved the retention of Foxp3 expression (Fig. 5A). To confirm the effect of low oxygen and vitamin C on iTreg stability in vivo, iTregs were transferred into Rag2−/− mice. Combination of low oxygen and vitamin C stabilized the Foxp3 expression in iTregs in vivo (Fig. 5B).
Fig. 5.

Low oxygen conditions with vitamin C treatment confer Foxp3 stability and higher suppression activity on iTregs. (A) iTregs induced under the 5% O2 condition and/or 10 µg ml−1 vitamin C for 5 days were further cultured in Th1 conditions for 3 days. Foxp3 expression was analyzed by flow cytometry. Representative data of three independent experiments are shown. (B) The stability of Foxp3 in iTregs generated under the 5% O2 condition and/or 10 µg ml−1 vitamin C treatment after transfer to Rag2−/− mice. iTregs (1 × 106) were transferred into Rag2−/− mice. After 2 weeks, Foxp3 positivity in lamina propria lymphocyte (LPL), mesenteric lymph node (mLN) or spleen is measured. Data from duplicate experiments. (C) Naive T cells (2 × 105) from WT mice were injected intravenously into Rag2−/− mice together with or without 2 × 105 iTregs induced under the 5% O2 condition and/or 10 µg ml−1 vitamin C. Body weight changes were measured after transfer. n = 3 **P < 0.01, *P < 0.05.
Then, we compared the suppression activity of low-O2 iTregs and normoxic iTregs. iTregs induced by the low oxygen concentration and vitamin C more effectively suppressed colitis in Rag2−/− mice induced by the transfer of naive T cells compared with normal iTregs or iTregs induced in the presence of vitamin C alone (Fig. 5C). These data indicate that iTregs induced under the low oxygen condition are more stable and possess stronger suppression activity in vivo.
Finally, we investigated whether the effect of the low oxygen condition on Foxp3 stability and CNS2 demethylation is mediated through TET proteins. We isolated naive T cells from Tet2f/fTet3f/fFoxp3Cre-YFP (DKO) mice, in which TET2 and TET3 are deleted using Foxp3Cre, and then induced iTregs under low oxygen conditions. As shown in Fig. 6(A), Foxp3 expression in low-O2 WT iTregs were stabilized under Th1 conditions but not in low-O2 DKO iTregs. Foxp3 CNS2 demethylation was reduced in DKO iTregs (Fig. 6B). These data indicate that TET enzymes are involved in the Foxp3 CNS2 demethylation and iTreg stabilization induced by low oxygen concentrations and vitamin C.
Fig. 6.

Demethylation of CNS2 by low oxygen conditions is TET2/3 dependent. (A) Foxp3 stability in TET2/3-deficient (DKO) iTregs. iTregs were induced from naive T cells isolated from WT or DKO mice for 5 days under the 5% O2 condition, then cultured for a further 3 days under the Th1 condition. Foxp3 positivity of T cells was determined by flow cytometry. (B) Methylation status determined by the bisulfite conversion of the CpG sites of Foxp3 CNS2.
Discussion
Understanding the molecular basis for the development and generation of iTregs and pTregs in addition to tTregs may promote the development of novel methods for the application of iTregs in immunotherapy. Various reagents such as retinoic acid, vitamin D, rapamycin and aryl hydrocarbon receptor ligands have been proposed to up-regulate and stabilize Foxp3 expression (36). Vitamin C is, however, the only reagent that has been reported to induce the demethylation of CNS2 of the Foxp3 locus to date. Here, we demonstrated that the low oxygen condition increases the expression of TET enzymes and promotes the demethylation of CNS2.
We found that catalytic domain of TET is enough to induce demethylation of CNS2 in iTregs. This is surprising because N-terminal regions of TET1 and TET3 contain the CXXC domain which is shown to interact with methylated DNA. This may be because the expression levels of our TET-CD may be higher than endogenous TET expression levels, and TET-CD can access DNA in the open chromatin regions. This idea is supported by the co-crystal structure of TET2-CD and DNA, which shows a direct interaction between CD and DNA (37). Enhanced TET expression resulted in demethylation of CNS2 but not Tnfrsf18, Ctla4 and Ikzf4, which is consistent with previous reports using vitamin C (18, 24). The mechanism of this locus-specific demethylation has not yet been clarified. Previously, Thillainadesan et al. reported that TGF-β triggers the active demethylation of the p15ink4b gene by the co-recruitment of Smad2/3 and the thymine DNA glycosylase (TDG), which is the last step enzyme that removes the methyl group from 5mC in combination with activation-induced deaminase (AID) (38, 39). Thus, Smads may be involved in CNS2-specific DNA demethylation.
Low oxygen conditions have been shown to improve the efficiency of iPS cell generation, suggesting that they can induce the site-specific epigenetic modification of important genes for reprogramming (40). Several reports indicate that the low oxygen condition promotes DNA demethylation through the up-regulation of TETs (31, 41). Another report shows that the hypoxic condition suppresses TET enzymatic activity and enhances DNA methylation in tumor cells (42). In that report, the authors found that the low oxygen condition reduced 5hmC levels without affecting TET mRNA expression levels in many tumor cell lines and concluded that the hypoxic condition inhibits TET enzymatic activity. In SK-N-Be2c and SHSY5Y neuroblastoma cell lines, however, the hypoxic condition increased 5hmC and TET mRNA levels. Therefore, hypoxia-mediated TET induction may be cell type-dependent.
Low oxygen conditions may also be important for CNS2 demethylation in vivo. It has been shown that most pTregs in the gut express RORγt (43, 44), and CNS2 of the Foxp3 locus in pTregs has been shown to be demethylated (16). TET enzymes were demonstrated to be required for the demethylation of Foxp3 CNS2 in pTregs (18, 24). Hypoxic conditions in the gut may facilitate TET expression, resulting in the promotion of CNS2 demethylation.
We showed that the low oxygen condition increased all TET mRNA levels in murine CD4+ T cells. However, importantly, TET activation induced DNA demethylation only in limited genes. Oxygen concentration may also be important for total TET enzymatic activity, as the hydroxylation of 5mC requires oxygen. It has been reported that modest hypoxia (2–5% O2) does not affect TET activity (42, 45). We used 5% O2, which had little effect on T-cell proliferation and viability.
Antigen-specific iTregs can be expanded in vitro by co-culturing naive T cells with dendritic cells in the presence of known antigens and TGF-β, making this procedure applicable for antigen-specific immunotherapy. Huter et al. demonstrated that antigen-specific iTregs revealed a stronger suppressive capacity than polyclonal tTregs did (46). Similarly, antigen-specific iTregs generated in vitro have been shown to be more effective than antigen-non-specific polyclonally expanded iTregs in a heart transplantation model (47, 48). For many autoimmune diseases, however, the target antigens are unknown or the antigens vary among patients. To overcome these difficulties, it would be ideal if iTregs could be generated from effector T cells that are accumulated in the inflamed regions or from peripheral memory T cells. Our method using the low oxygen and vitamin C treatment may be used as the epigenetic reprogramming of antigen-specific memory T cells into stable and long-lived iTregs. We did not observe any unfavorable effects of TET activation and the overall DNA demethylation status was not changed in iTreg cells by the low oxygen conditions (Supplementary Figure 1, available at International Immunology Online). When iTregs stably expressing TET-CD or induced under the low oxygen and vitamin C condition were transferred into Rag2-deficient mice, iTregs did not abnormally expand, and mice did not show any proliferative diseases within 50 days after transfer. These data suggest that the iTregs with active TETs are normal and at little risk of transformation.
Supplementary data
Supplementary data are available at International Immunology Online.
Funding
This work was supported by a JSPS KAKENHI Grant-in-Aid for (S) No. 17H06175, and Advanced Research & Development Programs for Medical Innovation (AMED-CREST) No. 17gm0510019h0005, the Takeda Science Foundation, the Uehara Memorial Foundation, the Kanae Foundation, the SENSHIN Medical Research Foundation and Keio Gijuku Academic Developmental Funds.
Conflicts of interest statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
{kind=link}
Acknowledgements
K.S. and H.N. conceived of the study. Significant contributions to the measurement of DNA methylation and data processing were made by K.-i.T., T.K., I.K., T.S., J.K. and T.T.-E. TET3-flox mice were generated by Y.T. The manuscript was reviewed and improved by all authors, including M.I., T.K. and Y.A.
References
- 1. Sakaguchi S. Yamaguchi T. Nomura T. and Ono M. 2008. Regulatory T cells and immune tolerance. Cell 133:775. [DOI] [PubMed] [Google Scholar]
- 2. Fontenot J. D. Gavin M. A. and Rudensky A. Y. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330. [DOI] [PubMed] [Google Scholar]
- 3. Khattri R. Cox T. Yasayko S. A. and Ramsdell F. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337. [DOI] [PubMed] [Google Scholar]
- 4. Hori S. Nomura T. and Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057. [DOI] [PubMed] [Google Scholar]
- 5. Kim J. M. Rasmussen J. P. and Rudensky A. Y. 2007. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8:191. [DOI] [PubMed] [Google Scholar]
- 6. Koenecke C., Czeloth N., Bubke A., et al. 2009. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur. J. Immunol. 39:3091. [DOI] [PubMed] [Google Scholar]
- 7. Schmidt A. Eriksson M. Shang M. M. Weyd H. and Tegnér J. 2016. Comparative analysis of protocols to induce human CD4+Foxp3+ regulatory T cells by combinations of IL-2, TGF-beta, retinoic acid, rapamycin and butyrate. PLoS ONE 11:e0148474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schlenner S. M. Weigmann B. Ruan Q. Chen Y. and von Boehmer H. 2012. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 209:1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zheng Y. Josefowicz S. Chaudhry A. Peng X. P. Forbush K. and Rudensky A. Y. 2010. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng Y. Arvey A. Chinen T. van der Veeken J. Gasteiger G. and Rudensky A. Y. 2014. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 158:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X. Liang Y. LeBlanc M. Benner C. and Zheng Y. 2014. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 158:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitagawa Y. Ohkura N. and Sakaguchi S. 2013. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front. Immunol. 4:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sekiya T. Nakatsukasa H. Lu Q. and Yoshimura A. 2016. Roles of transcription factors and epigenetic modifications in differentiation and maintenance of regulatory T cells. Microbes Infect. 18:378. [DOI] [PubMed] [Google Scholar]
- 14. Ohkura N., Hamaguchi M., Morikawa H., et al. 2012. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37:785. [DOI] [PubMed] [Google Scholar]
- 15. Hilbrands R., Chen Y., Kendal A. R., et al. 2016. Induced Foxp3(+) T cells colonizing tolerated allografts exhibit the hypomethylation pattern typical of mature regulatory T cells. Front. Immunol. 7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Polansky J. K., Kretschmer K., Freyer J., et al. 2008. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 38:1654. [DOI] [PubMed] [Google Scholar]
- 17. Chen Q. Kim Y. C. Laurence A. Punkosdy G. A. and Shevach E. M. 2011. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J. Immunol. 186:6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yue X., Trifari S., Äijö T., et al. 2016. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 213:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toker A., Engelbert D., Garg G., et al. 2013. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J. Immunol. 190:3180. [DOI] [PubMed] [Google Scholar]
- 20. Yang R., Qu C., Zhou Y., et al. 2015. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 43:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okada M. Kanamori M. Someya K. Nakatsukasa H. and Yoshimura A. 2017. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin 10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blaschke K., Ebata K. T., Karimi M. M., et al. 2013. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minor E. A. Court B. L. Young J. I. and Wang G. 2013. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 288:13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sasidharan Nair V. Song M. H. and Oh K. I. 2016. Vitamin C facilitates demethylation of the Foxp3 enhancer in a Tet-dependent manner. J. Immunol. 196:2119. [DOI] [PubMed] [Google Scholar]
- 25. Moran-Crusio K., Reavie L., Shih A., et al. 2011. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsukada Y. Akiyama T. and Nakayama K. I. 2015. Maternal TET3 is dispensable for embryonic development but is required for neonatal growth. Sci. Rep. 5:15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rubtsov Y. P., Rasmussen J. P., Chi E. Y., et al. 2008. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28:546. [DOI] [PubMed] [Google Scholar]
- 28. Komatsu N. Mariotti-Ferrandiz M. E. Wang Y. Malissen B. Waldmann H. and Hori S. 2009. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl Acad. Sci. USA 106:1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamiya T., Ichiyama K., Kotani H., et al. 2013. Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J. Immunol. 191:2360. [DOI] [PubMed] [Google Scholar]
- 30. Ichiyama K., Sekiya T., Inoue N., et al. 2011. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity 34:741. [DOI] [PubMed] [Google Scholar]
- 31. Mariani C. J., Vasanthakumar A., Madzo J., et al. 2014. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 7:1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sekiya T., Kashiwagi I., Yoshida R., et al. 2013. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol. 14:230. [DOI] [PubMed] [Google Scholar]
- 33. Muto G., Kotani H., Kondo T., et al. 2013. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS ONE 8:e74639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takimoto T., Wakabayashi Y., Sekiya T., et al. 2010. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 185:842. [DOI] [PubMed] [Google Scholar]
- 35. Weber A. R., Krawczyk C., Robertson A. B., et al. 2016. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 7:10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanamori M. Nakatsukasa H. Okada M. Lu Q. and Yoshimura A. 2016. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 37:803. [DOI] [PubMed] [Google Scholar]
- 37. Hu L., Li Z., Cheng J., et al. 2013. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155:1545. [DOI] [PubMed] [Google Scholar]
- 38. Thillainadesan G., Chitilian J. M., Isovic M., et al. 2012. TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol. Cell 46:636. [DOI] [PubMed] [Google Scholar]
- 39. Wotton D. 2012. TGF-β drives DNA demethylation. Mol. Cell 46:556. [DOI] [PubMed] [Google Scholar]
- 40. Yoshida Y. Takahashi K. Okita K. Ichisaka T. and Yamanaka S. 2009. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 5:237. [DOI] [PubMed] [Google Scholar]
- 41. Wu M. Z., Chen S. F., Nieh S., et al. 2015. Hypoxia drives breast tumor malignancy through a TET-TNFα-p38-MAPK signaling axis. Cancer Res. 75:3912. [DOI] [PubMed] [Google Scholar]
- 42. Thienpont B., Steinbacher J., Zhao H., et al. 2016. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohnmacht C., Park J. H., Cording S., et al. 2015. Mucosal immunology. The microbiota regulates type 2 immunity through RORγt⁺ T cells. Science 349:989. [DOI] [PubMed] [Google Scholar]
- 44. Yang B. H., Hagemann S., Mamareli P., et al. 2016. Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 9:444. [DOI] [PubMed] [Google Scholar]
- 45. Laukka T., Mariani C. J., Ihantola T., et al. 2016. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 291:4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huter E. N. Stummvoll G. H. DiPaolo R. J. Glass D. D. and Shevach E. M. 2008. Cutting edge: antigen-specific TGF beta-induced regulatory T cells suppress Th17-mediated autoimmune disease. J. Immunol. 181:8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takasato F., Morita R., Schichita T., et al. 2014. Prevention of allogeneic cardiac graft rejection by transfer of ex vivo expanded antigen-specific regulatory T-cells. PLoS ONE 9:e87722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Joffre O., Santolaria T., Calise D., et al. 2008. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 14:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.