

Abstract
Histone deacetylase (HDAC) enzymes govern the post-translational acetylation state of lysine residues on protein substrates, leading to regulatory changes in cell function. Due to their role in cancers, HDAC proteins have emerged as promising targets for cancer treatment. Four HDAC inhibitors have been approved as anti-cancer therapeutics, including SAHA (Suberoylanilide hydroxamic acid, Vorinostat, Zolinza). SAHA is a nonselective HDAC inhibitor that targets most of the eleven HDAC isoforms. The nonselectivity of SAHA might account for its clinical side effects, but certainly limits its use as a chemical tool to study cancer-related HDAC cell biology. Herein, the nonselective HDAC inhibitor SAHA was modified at the C4 position of the linker to explore activity and selectivity. Several C4-modified SAHA analogs exhibited dual HDAC6/8 selectivity. Interestingly, (R)-C4-benzyl SAHA displayed 520- to 1300-fold selectivity for HDAC6 and HDAC8 over HDAC1, 2, and 3, with IC50 values of 48 and 27 nM with HDAC6 and 8, respectively. In cellulo testing of the inhibitors was consistent with the observed in vitro selectivity. Docking studies provided a structural rationale for selectivity. The C4-SAHA analogs represent useful chemical tools to understand the role of HDAC6 and HDAC8 in cancer biology and exciting lead compounds for targeting of both HDAC6 and HDAC8 in various cancers.
Keywords: Histone Deacetylase, HDAC inhibitor, dual HDAC6/HDAC8 selective inhibitor, SAHA (Vorinostat), HDAC isoform selectivity
Graphical abstract

1. Introduction
Histone deacetylase (HDAC) proteins are key enzymes involved in epigenetic regulation of gene expression. Specifically, HDAC-mediated deacetylation of acetyllysine residues on nucleosomal histones leads to tight binding to genomic DNA, which affects accessibility and transcription [1-2]. In addition, HDAC proteins influence protein-protein interaction, protein-DNA interaction, protein localization, and protein stability through deacetylation of non-histone substrates [3-4]. The eighteen human HDAC proteins are divided into four classes according to their homology with yeast proteins, size, cellular localization, and number of catalytic active sites [5]. Class III (SIRT1-7) HDAC proteins are NAD+-dependent. Classes I (HDAC1, 2, 3 and 8), II (HDAC4, 5, 6, 7, 9, and 10), and IV (HDAC11) HDAC proteins are metal-dependent, and are the focus of this work [5].
HDAC proteins regulate the expression of several cancer-related proteins involved in cell signaling, transcription, and tumor suppression through the deacetylation of nucleosomal histone proteins [6-7]. Overexpression of HDAC proteins results in unregulated transcription and aberrant protein activity and function, which is linked to several diseases, including cancer [7]. For example, HDAC1 was overexpressed in lung [8], breast [9], and colon cancers [10]. HDAC2 was overexpressed in colorectal cancer[11]. HDAC8 was highly expressed in neuroblastoma patients, leading to cancer progression and poor survival rates [12]. In addition, selective inhibition of HDAC8 induced apoptosis in leukemia and T-cell lymphoma cell lines [13]. Class II HDAC6 was overexpressed in oral squamous cell carcinoma and ovarian cancer [14-15]. Overexpression of both HDAC6 and HDAC8 was linked to breast cancer metastasis and invasion [16].
Due to their key role in cancer, several anti-cancer agents targeting HDAC proteins have been developed [17]. HDAC inhibitors promoted apoptosis and reduced proliferation and migration through their effect on both histone and non-histone substrates.[17-19]. Several HDAC inhibitors have been approved by the FDA for treatment of cancer, and several others are in clinical trials [20]. SAHA (suberoylamide hydroxamic acid, Vorinostat, Zolinza™), and Belinostat (PXD101, Belodaq™, Figure 1) are FDA-approved for treatment of T-cell lymphoma [20-22], while Panobinostat (LBH-589, Farydak™, Figure 1) was approved for treatment of multiple myeloma [23]. SAHA is a nonselective inhibitor that targets most of the eleven metal-dependent HDAC isoforms [24]. The nonselectivity of the FDA-approved drugs, including SAHA, might explain the side effects observed in the clinic, such as cardiac arrhythmia and thrombocytopenia [25-26]. Moreover, the use of SAHA as a chemical tool to study the role of specific HDAC isoforms in cancer cell biology is limited due to its nonselectivity.
Figure 1.
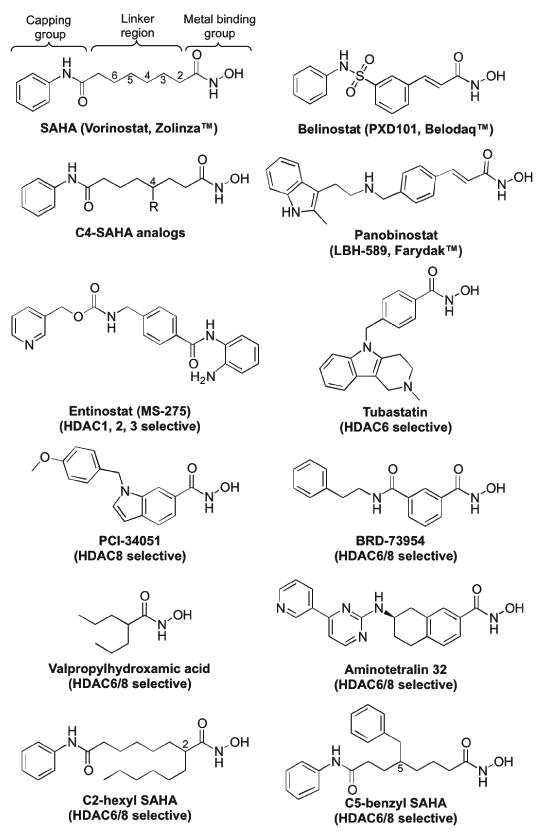
Chemical structures of the FDA-approved drugs SAHA, Belinostat, and Panobinostat, the C4-modified SAHA analogs reported here, and several isoform selective HDAC inhibitors discussed in the text (MS-275, Tubastatin, PCI-34051, BRD-73954, Valpropylhydroxamic acid, Aminotetralin 32, C2-hexyl SAHA, and C5-benzyl SAHA).
To overcome the limitations of nonselective drugs, several isoform selective HDAC inhibitors have been developed, with some in clinical trials. As illustrative examples, entinostat (MS-275, Figure 1) is selective for HDAC1, 2, and 3 [24, 27], whereas tubastatin (Figure 1) is HDAC6-selective [28-29]. Recently, several dual HDAC6/8 selective inhibitors have been reported, including BRD-73954 and valpropylhydroxamic acid (Figure 1) [30-31]. HDAC inhibitors that target one or two HDAC isoforms will be valuable for development of new drugs with minimal side effects [32-35]. In addition, recent reports suggested that inhibition of two HDAC isoforms is desirable by maintaining synergistic therapeutic effects in various cancers [30, 36-37]. Related to this work, dual inhibition of HDAC6 and HDAC8 might have potential application in breast cancer angiogenesis and metastasis [13, 30]. Moreover, selective HDAC inhibitors will be useful as chemical tools to study cancer-related HDAC cell biology.
To understand the structural requirements of HDAC inhibitors, we previously synthesized SAHA analogs substituted in the linker region at carbon 2 (C2), carbon 3 (C3), carbon 5 (C5), or carbon 6 (C6) (Figure 1) [38-41]. C2-hexyl SAHA (Figure 1) showed 49- to 300-fold HDAC6/8 dual selectivity over HDAC1, 2, and 3, with 0.6 and 2.0 μM potency against HDAC6 and HDAC8, respectively [42]. Among the C5-SAHA analogs, C5-benzyl SAHA (Figure 1) displayed 8- to 21-fold HDAC6/8 selectivity with IC50 values of 270 and 380 nM with HDAC6 and HDAC8, respectively [41]. Some of the C3-modified SAHA analogs displayed preference for HDAC6 over HDAC1 and 3 [39], while some of the C6-modified SAHA analogs inhibited HDAC1 and 6 over HDAC3 [40]. In addition, SAHA analogs modified at the hydroxamic acid moiety had a preference for HDAC1 [43]. In this work, SAHA analogs modified at the C4 position were synthesized and screened in vitro and in cellulo for their activity and selectivity. The C4-modified SAHA analogs showed high selectivity towards HDAC6 and 8 over HDAC1, 2, and 3, with nanomolar potency against HDAC6 and HDAC8. Docking studies provided a structural rationale for the observed selectivity. These studies emphasize that modification of the SAHA linker can enhance isoform selectivity. In addition, the HDAC6/8 dual selective C4-SAHA analogs reported here have the potential to be useful pharmacological tools for biomedical research and lead compounds for anti-cancer drug development.
2. Results and discussion
2.1. Synthesis of C4-modified SAHA analogs
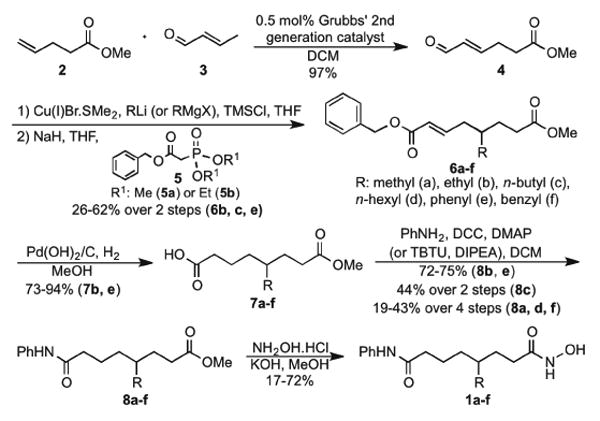
Synthesis of the C4-SAHA analogs started with a cross metathesis reaction of methyl-4-pentenoate (2) with crotonaldehyde (3) using second generation Grubbs' catalyst to afford the α,β-unsaturated aldehyde (4) (Scheme 1). Different substituents were appended to 4 via 1,4-addition using organolithium cuprates, followed by Horner–Wadsworth–Emmons reaction with benzyl phosphonoacetate (5) to give the unsaturated benzyl esters (6a-f). Reduction and hydrogenolysis of 6a-f gave free acids (7a-f), which were coupled with aniline to afford 8a-f. Finally, esters (8a-f) were reacted with hydroxylamine to afford the C4-substituted SAHA derivatives (1a-f) as racemic mixtures.
Scheme 1. Synthesis of C4-SAHA analogs (1a-f).
2.2. In vitro screening of C4-modified SAHA analogs
SAHA analogs 1a-f were tested for global HDAC inhibition with HeLa cell lysates as the source of all HDAC proteins (Table 1). SAHA also included as a broad spectrum inhibitor, while Tubastatin and BRD-73954 were tested as isoform selective inhibitors. HDAC activity was measured using the commercially available HDAC-Glo™ I/II substrate (Promega). The results of the screening showed that all of the synthesized derivatives were less potent than SAHA (Tables 1 and S1, and Figure S141). The most potent derivative was C4-methyl SAHA (1a), which showed an IC50 value of 3.3 μM. Compared to the parent molecule SAHA, C4-methyl SAHA is 18-fold less potent, while the rest of the analogs showed 78- to 344-fold reduction in potency. Both tubastatin and BRD-73954 also showed 36- to 60-fold less potency compared to SAHA (9.9 and 6.7 μM IC 50 values). Because HeLa cell lysates contain all HDAC isoforms, the poor potency of the C4-SAHA analogs suggests that they might be selective for specific isoforms, similar to tubastatin and BRD-73954.
Table 1. IC50 values for SAHA, Tubastatin, BRD-73954, and C4-SAHA analogs (1a-1f) with HeLa cell lysates.a.
| Compound | IC50 (μM) |
|---|---|
| SAHA | 0.20 ± 0.02 |
| Tubastatin | 9.9 ± 0.4 |
| BRD-73954 | 6.7 ± 0.2 |
| 1a (methyl) | 3.3 ± 0.1 |
| 1b (ethyl) | 14 ± 1 |
| 1c (n-butyl) | 53 ± 2 |
| 1d (n-hexyl) | 60 ± 1 |
| 1e (phenyl) | 65 ± 6 |
| 1f (benzyl) | 62 ± 1 |
Mean IC50 value and standard error of at least three independent trials are shown (Figure S141 and Table S1).
To assess selectivity, an initial screen was performed with analogs 1a-f and the parent molecule SAHA at a single concentration against HDAC1, 2, 3, and 6 using an ELISA-based HDAC activity assay [28]. These select isoforms were chosen due to their high deacetylation activity among the family members and their representation of both class I (HDAC1, 2, and 3) and class II (HDAC6). SAHA, as expected, showed no selectivity among HDAC1, 2, 3, and 6, inhibiting their activity to a similar extent (Figure 2). Interestingly, all C4-SAHA analogs (1a-f) displayed more potent inhibition against HDAC6 compared to HDAC1, HDAC2, and HDAC3 (Figure 2 and Table S2). The analogs that showed the greatest difference in potency with HDAC6 versus the other isoforms were C4-n-butyl (1c) and C4-benzyl (1f). The C4-methyl SAHA analog (1a) showed the smallest difference in potency comparing HDAC6 to HDAC1 and HDAC3 (Figure 2 and Table S2). This single concentration screen suggested that C4-modification of the SAHA structure results in selectivity for HDAC6.
Figure 2.
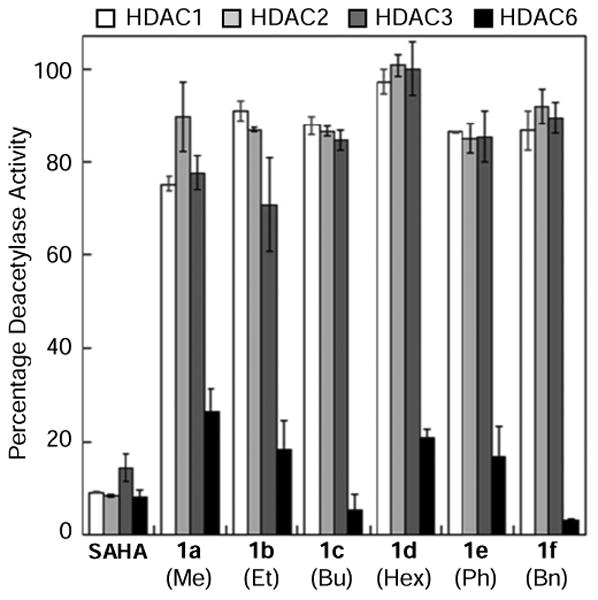
In vitro isoform selectivity screening of C4-modified SAHA analogs (1a-f) against HDAC1, HDAC2, HDAC3, and HDAC6 using an ELISA-based HDAC activity assay [28]. Analogs 1a-f were tested at 0.75, 0.75, 2.5, 1.25, 2.5, and 5 μM final concentration, respectively. SAHA was tested at 1 μM concentration [28]. Mean percent deacetylase activities from a minimum of two independent trials with standard errors were plotted (Table S2).
To further assess selectivity, IC50 values for derivatives 1b-f were determined with HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8 isoforms (Table 2). HDAC8 was included due to its similar active site structure compared to HDAC6 [31]. For comparison, the non-selective parent molecule SAHA and the HDAC6-selective inhibitor tubastatin (Figure 1) were also tested as control compounds (Table 2) [28]. As expected, the non-selective inhibitor SAHA showed similar low nanomolar IC50 values with HDAC1, 2, 3, 6, but a 6- to 27-fold reduction in potency against HDAC8 [28]. In contrast, the HDAC6-selective inhibitor tubastatin displayed 87- to 130-fold selectivity for HDAC6 over HDAC1, 2, and 3, and 11-fold selectivity for HDAC6 over HDAC8, which is consistent with prior studies [28, 42]. As expected based on the single concentration screen, analogs 1b-f displayed preference for HDAC6 and HDAC8, with 28- to 740-fold selectivity compared to HDAC1, 2, and 3 (Tables 2 and S10). Importantly, analogs 1b-f maintained low nanomolar IC50 values in the 57 to 290 nM range with HDAC6 and HDAC8 (Tables 2), similar to SAHA. Among the analogs, C4-benzyl SAHA (1f) displayed the highest selectivity, with 210- to 740-fold selectivity for HDAC6 and 8 over HDAC1, 2, and 3 (Tables 2 and S10), and potent inhibition with low nanomolar IC50 values (140 and 57 nM with HDAC6 and HDAC8, respectively, Table 2). Similarly, C4-n-butyl (1c) and C4-n-hexyl (1d) SAHA demonstrated 170- to 480-fold HDAC6/8-selectivity compared to HDAC1, 2, and 3 isoforms, with low nanomolar IC50 values (Tables 2 and S10). The C4-ethyl (1b) SAHA analog displayed the weakest selectivity among the analogs, with 28- and 46-fold selectivities for HDAC6 and 8 over HDAC1, 2, and 3 (Tables 2 and S10). Comparing the IC50 values of the analogs to the parent SAHA (Table 2), the higher HDAC6 selectivities are due to comparable potency against HDAC6 (2.5 to 3-fold reduction), but dramatic loss of potency against HDAC1, 2 and 3 (51- to 2,100- fold reduction, Table 2). Remarkably, the analogs displayed enhanced potency with HDAC8 compared to the parent SAHA compound (2- to 9-fold enhancement, Table 2), which lead to the observed HDAC8 selectivities, along with HDAC6.
Table 2. IC50 values for SAHA, Tubastatin, SAHA analogs 1b-1f, (R)-1f, and (S)-1f against HDAC1, 2, 3, 6 and 8, and EC50 values with U937 cells.
| Compound | IC50 values (nM)a | EC50 (μM)b,c | ||||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 | U937 cells | |
| SAHAd | 33 ± 1 | 96 ± 10 | 20 ± 1 | 33 ± 3 | 540 ± 10 | 0.88 ± 0.13 |
| Tubastatind | 2,700 ± 200 | 3,900 ± 400 | 2,900 ± 500 | 31 ± 4 | 330 ± 10 | 83 ± 8 |
| PCI-34051 | – | – | – | – | – | 210 ± 30 |
| 1b (ethyl) | 4,400 ± 300 | 4,900 ± 400 | 6,000 ± 1200 | 160 ± 10 | 130 ± 3 | – |
| 1c (n- butyl) | 15,000 ± 1000 | 18,000 ± 2000 | 23,000 ± 3000 | 88 ± 7 | 74 ± 2 | 34 ± 2 |
| 1d (n-hexyl) | 35,000 ± 1000 | 38,000 ± 3000 | 30,000 ± 3000 | 140 ± 10 | 79 ± 3 | 16 ± 3 |
| 1e (phenyl) | 11,000 ± 1000 | 12,000 ± 1000 | 23,000 ± 2000 | 110 ± 10 | 290 ± 20 | – |
| 1f (benzyl) | 29,000 ± 1000 | 32,000 ± 2000 | 42,000 ± 4000 | 140 ± 10 | 57 ± 2 | 28 ± 7 |
| (R)-1f | 25,000 ± 2000 | 36,000 ± 3000 | 27,000 ± 2000 | 48 ± 8 | 27 ± 2 | – |
| (S)-1f | 40,000 ± 1000 | 51,000 ± 1000 | 37,000 ± 2000 | 95 ± 9 | 150 ± 10 | – |
Mean IC50 value and standard error of at least three independent trials are shown (Figures S142-S148 and Tables S3-S9).
Mean EC50 value and standard error of at least three independent trials are shown. Data in table is associated with Figures S150 and Table S11.
“–” represents not determined.
In terms of a structure-activity relationship (SAR) analysis, modifying SAHA at the C4 position with long alkyl substituents led to enhanced selectivity; the C4-hexyl 1d analog with the longest alkyl chain demonstrated elevated selectivity (210- to 480-fold) compared to the C4-butyl 1c analog (170- to 310-fold), which were both more selective than the C4-ethyl analog 1b with the smallest alkyl chain (28- and 46-fold, Tables 2 and S10). For these alkyl analogs, selectivity was due to discrimination against HDAC1, 2, and 3, with the least selective C4-ethyl analog 1b demonstrating greater potency to HDAC1, 2, and 3 (4.4 - 6.0 μM, Table 2) compared to the most selective C4-hexyl analog 1d (30 – 38 μM, Table 2). The length of the substituent also influenced the selectivities of the analogs with aryl groups at the C4 position; the lack of a methylene in C4-phenyl analog 1e led to decreased selectivity (38- to 350-fold) compared to the C4-benzyl analog 1f (210- to 740-fold). Similar to the alkyl series, the reduced selectivity of C4-phenyl analog 1e was due to greater potency with HDAC1 and 2 (11 and 12 μM, Table 2) compared to C4-benzyl analog 1f (29 and 32 μM, Table 2), in addition to decreased potency with HDAC8 (290 nM, Table 2) compared to C4-benzyl analog 1f (57 nM, Table 2). In total, the SAR analysis indicated that longer substituents at the C4 position led to greater HDAC6/8 selectivity primarily due to discrimination against HDAC1, 2, and 3.
Compared to previously reported inhibitors, the observed HDAC6 selectivities of the analogs 1c, 1d, and 1f (at least 170-, and 210-fold) were higher than the selectivity observed with the HDAC6-selective inhibitor tubastatin (at least 87-fold, Tables 2 and S10). Moreover, analogs 1c, 1d, and 1f showed comparable HDAC8 selectivities (at least 200-, 380-, and 510-fold) relative to the HDAC8-selective inhibitor PCI-34051 (Figure 1) (at least 400-fold) [44]. Finally, C4-benzyl analog 1f showed higher dual HDAC6/8 selectivity (at least 210- and 510-fold) compared to the known HDAC6/8-selective inhibitor BRD-73954 (75- and 250-fold selectivity to HDAC6, and 8 over HDAC1, 2, and 3) [30].
2.3. In cellulo selectivity testing
To further assess the observed HDAC6 selectivity in cellulo, the C4-benzyl (1f) SAHA analog was tested for inhibition of HDAC activities in cells. The inhibition of HDAC6 was monitored by detecting the levels of its known substrate acetyl-α-tubulin (AcTub), whereas Class I HDAC proteins (HDAC1, 2, and 3) inhibition was monitored through the levels of their known substrate acetyl-histone H3 (AcH3). SAHA or the analog 1f were incubated with U937 leukemia cells before lysis and western blot analysis of protein acetylation. As expected, SAHA showed increased levels of both acetyl-α-tubulin and acetyl-histone H3 (Figure 3A, lane 2), which is consistent with its non-selective inhibition of HDAC1, 2, 3, and 6 isoforms. In contrast, C4-benzyl (1f) showed a dose-dependent increase in levels of acetyl-α-tubulin (Figure 3A, lanes 3-5, AcTub) compared to the DMSO control (Figure 3A, lane 1), which was elevated compared to the levels of acetyl histone H3 (Figure 3A, lanes 3-5, AcH3). Greater acetylation of the HDAC6 substrate tubulin compared to the class I HDAC substrate histone 3 in cells (Figure 3A) is consistent with the HDAC6 selectivity of 1f observed in the in vitro screening (Table 2 and Figure 1).
Figure 3.

A) Cell based selectivity testing of the C4-benzyl SAHA analog 1f. Western blot analysis of acetyl-histone H3 (AcH3) and acetyl-α-tubulin (AcTub) is shown after treatment with SAHA or the C4-benzyl SAHA analog 1f. U937 cells were treated with DMSO (1%), SAHA (5 μM), or increasing concentrations of C4-benzyl SAHA (1f) analog (20-60 μM), before lysis, SDS-PAGE separation, transfer to a PVDF membrane, and western analysis with AcH3 or AcTub antibodies. GAPDH levels in the samples were also probed as a gel load control. A DMSO control sample was included for comparison to inhibitor treated samples. Repetitive trials are shown in Figure S149. B) Cytotoxicity of tubastatin (Tub) and PCI-34051 (PCI) alone or in combination using an MTT assay with the U937 cell line. Concentrations close to the EC50 values of each inhibitor were used (80 μM for tubastatin and 200 μM for PCI-34051). Mean percentage cell viability from three independent trials with standard errors was plotted (Table S12).
2.4. In vitro cancer cell growth inhibition
To assess the cytotoxic effect of the HDAC6/8 selective inhibitors in cancer cells, select SAHA derivatives were tested with the leukemia cell lines, U937 and Jurkat, due to the prominent role of HDAC6 in leukemia [45]. Initially, as controls, the nonselective parent SAHA, along with the HDAC6-selective inhibitor tubastatin and the HDAC8-selective inhibitor PCI-34051, were tested. SAHA showed an EC50 value of 0.88 μM (Table 2), which is consistent with the cytotoxicity previously reported [46]. The non-selective inhibition of all the HDAC proteins by SAHA likely contributes to its high cytotoxicity. In contrast, the isoform selective inhibitors tubastatin and PCI-34051 displayed lower cytotoxicity compared to SAHA, with 83 and 210 μM EC50 values, respectively (Table 2). To provide evidence that dual inhibition of both HDAC6 and HDAC8 enhances anti-proliferative activity, U937 cells were incubated alone or with both tubstatin and PCI-34051 at concentrations near their EC50 values (Figure 3B). The combination of both tubastatin and PCI-34051 reduced cell viability compared to each inhibitor alone, which suggests that dual HDAC6/8 inhibition provides beneficial anti-viability effects.
Next, the EC50 values of analogs, C4-n-butyl (1c), C4-n-hexyl (1d), and C4-benzyl (1f), were determined with the U937 cell line [45]. SAHA analogs 1c, 1d, and 1f displayed 34, 16, and 28 μM EC50 values, respectively, with the U937 cell line (Table 2). The analogs were 18- to 39-fold less cytotoxic compared to SAHA. To confirm the cytotoxicity study in U937 cells, SAHA and the analogs were also tested for cytotoxicity with the Jurkat cell line. The analogs showed reduced cytotoxicity compared to SAHA (Figure S151, Table S13), consistent with the study in U937 cells. The reduced cytotoxicity of tubastatin, PCI-34051, and the C4-modified SAHA analogs in U937 and Jurkat cells compared to SAHA might be due to their selectivity toward HDAC6 and/or 8. Consistent with this hypothesis, similar micromolar cytotoxicities were also observed with the several HDAC6-selective inhibitors in previous reports [37, 47-48].
2.5. Enantioselective synthesis and screening of (R)- and (S)-C4-benzyl SAHA analog (1f)
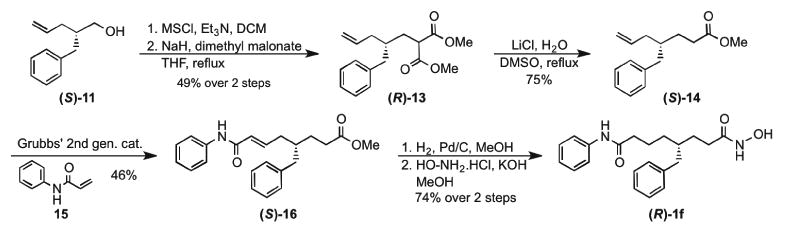
Since all analogs were synthesized and screened as racemic mixtures, single enantiomers of the most selective analog C4-benzyl SAHA 1f were synthesized to test selectivities. The asymmetric syntheses of both enantiomers were carried out utilizing Evan's oxazolidinone chiral auxiliary (R)-8 (Scheme 2) [49]. To ultimately prepare (R)-1f, 4-pentenoyl chloride was reacted with chiral auxiliary (R)-8 using n-butyllithium, which yielded pentenoyl oxazolidinone intermediate (R)-9 (Scheme 2A). Enantioselective reaction of benzyl bromide with (R)-9 gave the diastereomeric intermediate (RS)-10. To ultimately prepare (S)-1f, a similar procedure was employed (Scheme 2B), but reacting 3-phenylpropanoyl chloride with chiral auxiliary (R)-8, followed by enantioselective reaction with allyl bromide to give diastereomeric intermediate (RR)-10. Both diastereomers were obtained with high diastereomeric ratios (dr), which were assessed by 1HNMR analysis (99:1 dr for (RS)-10, and 97:3 dr for (RR)-10, Figures S79 and S86).
Scheme 2. Enantioselective synthesis of intermediate alcohols (S)-11 (a) and (R)-11 (b).

To remove the chiral auxiliary from (RS)-10 and (RR)-10, each diastereomer was reacted with lithium aluminum hydride, which produced alcohols (S)-11 and (R)-11 (Scheme 2). To assess the optical purity and enantiomeric excess (ee) of the alcohol intermediates (S)-11 and (R)-11, Mosher esters were synthesized by coupling each alcohol with (R)-(+)-α-Methoxy-α-trifluoromethylphenylacetic acid ((R)-MTPA) using EDCI and DMAP (Scheme S1) [50]. Analysis of the 1HNMR spectra showed that both Mosher esters were observed in high diastereomeric ratios (99:1 dr for (S)-11-(R)-MTPA, and 98:2 dr for (R)-11-(R)-MTPA, Figures S94, S95, S100, and S101), which implies that the alcohol intermediates (S)-11 and (R)-11 were obtained with high enantiomeric ratios (98% ee for (S)-11 and 96% ee (R)-11).
To synthesize enantiopure C4-benzyl SAHA analogs (R)-1f and (S)-1f, both alcohols (S)-11 and (R)-11 were then converted to methanesulfonate esters, followed by substitution with dimethyl malonate to give diesters (R)-13 and (S)-13, respectively (Schemes 3 and S2). Krapcho decarboxylation afforded methyl ester intermediates (S)-14 and (R)-14. Cross metathesis of the methyl esters with N-phenyl acrylamide (15) using Grubbs' second generation catalyst afforded ester amides (S)-16 and (R)-16 [51]. Finally, reduction followed by substitution with hydroxyl amine gave both enantiomers of C4-benzyl SAHA analog, (R)-1f and (S)-1f (Schemes 3 and S2).
Scheme 3. Synthesis of (R)-C4-benzyl SAHA analog.
To assess the selectivities of enantiomers (R)-C4-benzyl SAHA (R)-1f and the (S)-C4-benzyl SAHA (S)-1f, IC50 values of both were determined with HDAC1, 2, 3, 6, and 8 (Table 2). Similar to the racemic mixture, both enantiomers showed dual HDAC6/HDAC8 selectivity. (R)-C4-benzy SAHA (R)-1f displayed more potent inhibition against HDAC6 and HDAC8 compared to the (S)-C4-benzyl SAHA (S)-1f, with IC50 values of 48 and 27 nM for (R)-1f and 95 and 150 nM for (S)-1f (Table 2). In addition, (R)-1f showed greater fold preference for HDAC6 and 8 over HDAC1, 2, and 3 (520- to 1300-fold) compared to the racemic mixture (210- to 740-fold). In contrast, (S)-1f showed lower fold selectivities (240- to 540-fold) compared to both (R)-1f and the racemic mixture (Tables 2 and S10). In summary, in vitro screening revealed that both (R)-1f and (S)-1f are highly selective for HDAC6 and 8 over HDAC1, 2, and 3 with nanomolar potency. However, (R)-1f was more potent and selective than (S)-1f (Tables 2 and S10).
2.6. Docking studies
To rationalize the experimental results, docking studies with both enantiomers of C4-benzyl SAHA (1f) were performed using the AutoDock 4.2 and Autodock tools programs [52-53]. The recently reported crystal structure of HDAC6 (PDB: 5G0H) [54] along with HDAC3 crystal structure (PDB: 4A69) [55] were used in these studies. SAHA, the parent molecule, was also examined with both crystal structures for comparison and to validate the docking procedure. With HDAC6, both enantiomers of C4-benzyl SAHA (1f) were positioned similarly in the active site compared to SAHA, with the hydroxamic acid moiety near the catalytic metal, the linker region in the 11 Å channel, and the anilide group in the solvent exposed region. In addition, the hydroxamic acid groups bound within 1.9-2.7 Å of the active site residues (H573, H574, and Y745) and the active site catalytic zinc atom (Figures 4A and S153A), similar to that of the parent molecule SAHA (1.7-2.7 Å) (Figure S152A). Superimposition of each enantiomer with SAHA in the HDAC6 active site showed similar binding of the hydroxamic acid moiety (Figures 4C and S153C). The docking studies with the HDAC6 crystal structure are consistent with the high binding affinity of C4-benzyl SAHA and SAHA in vitro (Table 2).
Figure 4.
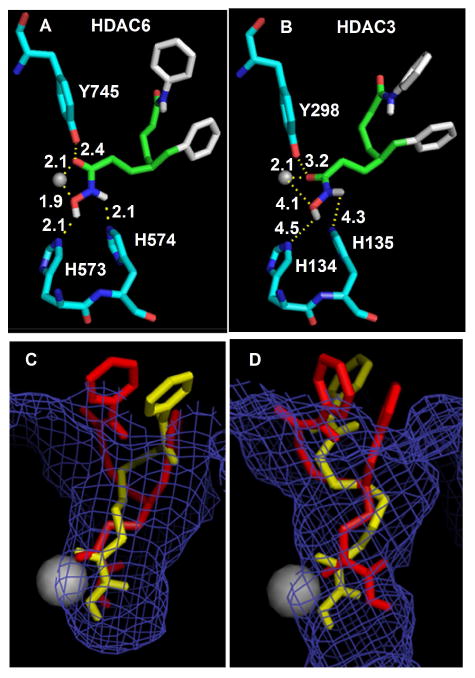
Docked poses of (R)-C4-benzyl SAHA (R)-1f in the crystal structures of HDAC6 (A,C) and HDAC3 (B, D) using Autodock 4.2. A and B) Binding distances between the hydroxamic acid atoms and active site residues (numbered in figure) or the metal are displayed in Angstroms. The atomic radius of the metal (Zn2+) in was set at 0.5 Å for clarity. Atom color-coding: (R)-C4-benzyl SAHA (C=green/white; O=red; N=blue; H=White); amino acids (C=deep teal; O=red, N=blue); Zn2+ metal ion (grey sphere). C and D) Shown is the superimposition of SAHA (yellow) and (R)-C4-benzyl SAHA (red) in the crystal structures of HDAC6 (C) or HDAC3 (D), with the metal ion (Zn2+) represented as a grey sphere (1.39 Å radius).
To explain the observed selectivities, (R)-1f and (S)-1f were also docked into the HDAC3 crystal structure. Longer and weaker binding interactions were observed with (R)-1f and (S)-1f in the HDAC3 structure (2.1-5.0 Å, Figures 4B and S153B) compared with the HDAC6 structure (1.9-2.7 Å). Superimposition of each enantiomer with SAHA in the HDAC3 structure showed a shift in the position of the hydroxamic acid moieties away from the catalytic metal compared to SAHA (Figure 4D and S153D). The docking analysis suggests that the presence of a bulky substituent on the C4 position weakens binding to HDAC3 due to steric clash with the relatively narrow and long 11Å channel of HDAC3 (Figures S154B, see arrows). In particular, the steric clash of the bulky substituent with active site residues positions the hydroxamic acid moieties away from the metal binding region (Figures 4B, S153B and S154B). In contrast, HDAC6 maintains a wider and shorter V-shaped 11Å channel (Figure S154A, see arrow), which can accommodate relatively large C4 substituents without affecting hydroxamic acid-metal binding (Figures 4A, S153A and S154A).
Structural analysis of prior HDAC6/8 selective inhibitors are consistent with our docking analysis suggesting that HDAC6 maintains a wider and shorter 11Å channel compared to HDAC1, 2, and 3 [29, 54]. Valpropylhydroxamic acid (Figure 1) with a propyl substituent in the linker region showed selectivity for HDAC6 and 8, although with micromolar potency and low fold selectivity (9-17-fold selectivity against with HDAC1, 2, and 3) [56]. Inhibitors bearing a bicyclic or phenyl ring in the linker displayed dual HDAC6 and HDAC8 selectivity (BRD-73953 and Aminotetralin 32, Figure 1) [30, 36-37]. Finally, SAHA analogs with a hexyl or benzyl substituent at the C2 or C5 positions in the linker also demonstrated dual HDAC6/8 selectivity (Figure 1) [42]. Based on these prior reports and our docking studies, we hypothesize that substitutuents at the C4 position of SAHA can be accommodated in the relatively wider and shorter V-shaped 11Å cavity of HDAC6 active site (Figures 4A, S153A, 4C, S153C, and S154A), but not the narrower and relatively longer 11Å cavity of HDAC1, HDAC2, and HDAC3 (Figures 4B, S153B, 4D, S153D, and S154B). In addition, the size of the substituent plays a critical role in the selectivity. For example, analogs bearing smaller substituents (as methyl or ethyl in 1a or 1b) demonstrated lower selectivity (Table S1) compared to analogs comprising bulkier substituents (as butyl, hexyl, phenyl, and benzyl 1c-1f) (Figure 2 and Table 2). In total, the docking results are consistent with previously reported structural studies suggesting that the wider HDAC6 active site entrance is the source of selectivity.
3. Conclusions
In conclusion, SAHA analogs modified at the C4 position were synthesized and screened for potency and selectivity. C4-SAHA analogs showed up to 1300-fold dual selectivity for HDAC6 and HDAC8 over HDAC1, HDAC2, and HDAC3. The best analogs were C4-n-butyl SAHA (1c) and C4-benzyl SAHA (1f). C4-benzyl SAHA (1f) showed the highest fold selectivity with 210- to 740-fold selectivity for HDAC6 and 8 compared to HDAC1, 2, and 3, and 140 and 57 nM IC50 with HDAC6 and HDAC8. Interestingly, the fold selectivity of the C4-benzyl SAHA analog was higher than the recently reported dual HDAC6/8 selective inhibitors (at least 23-, 75-, and 79-fold) [30, 36-37]. Furthermore, in cellulo testing of the C4-benzyl analog showed consistency with the in vitro screening. Enantioselective synthesis and screening of both enantiomers of the C4-benzyl SAHA revealed that (R)-C4-benzyl SAHA is more potent and selective than the (S) enantiomer, with 48 and 27 nM IC50 with HDAC6 and HDAC8, and 520- to 1300-fold selectivity for HDAC6 and 8 over HDAC1, 2, and 3. The dual HDAC6/HDAC8 selective C4-SAHA analogs reported in this work can be useful as biological tools to understand the role of HDAC6 and HDAC8 in cancer, and as well as lead compounds towards development of more effective anti-cancer drugs. More generally, these studies with SAHA analogs suggest that modifying current drugs can significantly improve their properties.
4. Experimental
4.1. Materials and Instrumentation
Unless otherwise noted, chemicals were purchased from Sigma-Aldrich, Acros Organics, or Fisher Scientific. “Iron-free” glassware was prepared by rinsing glass vessels with a 5M aqueous HCl solution, followed by washing with distilled de-ionized water. “Iron-free” silica gel was prepared by washing with 5M aqueous HCl, followed by washing with distilled de-ionized water until colorless, and subsequently drying under air or in the oven at 80° C. NMR spectra were taken on a Varian or Agilent 400 or 600 MHz instruments. 1HNMR spectra showed NMR solvents peaks at 3.3 ppm (for CD3OD) and at 4.9 ppm (for trace amounts of water in NMR solvent), while 13CNMR spectra showed NMR solvent peaks at 77 (for CDCl3) or at 47 (for CD3OD) [57]. Infrared (IR) spectra were taken on Perkin Elmer Spectrum Two ATR-FTIR. Low resolution mass spectra (LRMS) were taken on Waters ZQ LCSQMS, while high resolution mass spectra (HRMS) spectra were taken on a Waters LCT-MS premier TOF. HPLC analysis to assess the purity of final compounds was performed with a Waters 1525 binary HPLC pump and Waters 2998 Photodiode Array detector. The separation was performed on a reverse phase HPLC analytical column (YMC America, 250 × 4.6 mmI.D, 4μm, 8 nm) using a gradient of 90% to 10% of buffer A over 30 minutes (buffer A = 0.1% HPLC grade TFA in water; buffer B = HPLC grade acetonitrile) at a flow rate of 1.0 mL/min at room temperature. The peaks were detected at wavelength 254 nm. The synthesized final compounds were found to be ≥97% pure (Figures S48, S53, S58, S63, S68, and S73). Flash chromatography was performed with 40-60 micron LC60A silica gel (Davisil).
4.2. Synthesis procedures
4.2.1. Synthesis procedures for 1a-1f
4.2.1.1 Synthesis of methyl (E)-6-oxohex-4-enoate (4)
The compound was synthesized according to the reported procedure, with the use of a different catalystm [58]. Briefly, in a flame dried 2-neck 100 mL flask, purged with argon, Grubb's catalyst second generation (26.65 mg, 0.0314 mmol, 0.5 mol%) was dissolved in dry dichloromethane (20 mL). Crotonaldehyde 3 (2.6 mL, 31.38 mmol) and methyl pent-4-enoate 2 (0.78 mL, 6.28 mmol) were added, and the reaction was stirred with reflux for 3.5 hours under argon. The reaction was then cooled to room temperature, concentrated, and purified by silica gel flash chromatography (ethyl acetate:hexanes 1:4) to afford aldehyde 4 as an orange oily product (97%). The spectral data for the synthesized compound was consistent with the reported data in literature [58].
4.2.1.2 Synthesis of 1-benzyl 8-methyl (E)-5-ethyloct-2-enedioate (6b)
In a 200 mL, 2-neck flame dried flask, copper(I)bromide dimethyl sulfide (2.17 g, 10.56 mmol) was dissolved in dry THF (20 mL). Air was purged with argon, and then the temperature was reduced to -15°C. Ethyllithium (12.35 mL of a 1.7M solution in dibutyl ether, 756 mg, 21.0 mmol) was added drop wise with stirring and the mixture was allowed to stir for additional 20 minutes at -15°C. The temperature was then reduced to -78°, followed by drop wise addition of chlorotrimethylsilane (3.44 g, 31.66 mmol) and methyl pent-4-enoate 4 (500 mg, 3.52 mmol). The reaction was stirred for 5 hours at -78°C. The reaction was then quenched by addition of a saturated ammonium chloride:ammonia solution (1:1) portion wise until the reaction color turned blue. The organic layer was collected, and the aqueous layer was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and rotavaped to an oily crude product, which was used in the next reaction without purification.
In a 200 mL 2-neck flame dried flask, air was purged with argon, and NaH (169 mg of 60% NaH in mineral oil, 7.04 mmol) dissolved in dry THF (20 mL) was added. The reaction was then cooled to 0°C and benzyl diethyl phosphonoacetate 5b (1.80 mL, 7.04 mmol) was added drop wise with stirring. The reaction was allowed to stir for 15 minutes at 0°C, then the crude product from the previous reaction was added. The reaction was stirred for another 30 minutes at 0°C, followed by stirring for 90 minutes at room temperature. The reaction was quenched with a saturated ammonium chloride solution (20 mL). The organic layer was collected, and the aqueous layer was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The product was purified by flash silica gel chromatography (ethyl acetate:hexanes 1:9) to afford 6b (375 mg, 35% over two steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.86-0.90 (t, J = 7.2 Hz, 3H), 1.28-1.34 (m, 2H), 1.47 (m, 1H), 1.55-1.61 (m, 2H), 2.18-2.22 (m, 2H), 2.30-2.34 (t, J = 7.2 Hz, 2H), 3.63 (s, 3H), 5.15 (s, 2H), 5.89-5.93 (d, J = 16 Hz, 1H), 6.92-6.98 (m, 1H), 7.29-7.35 (m, 4H). 13CNMR (100 MHz, CD3OD) δ (ppm): 9.69, 25.31, 27.86, 30.79, 35.24, 35.33, 38.07, 50.66, 65.69, 122.02, 127.80 (2C), 128.15, 136.28, 148.47, 166.31, 174.4. IR: 2957, 2931, 2875, 1723, 1655, 1437 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C18H24O4H+, 305.2, found 305.2; calculated for [M+Na]+ C18H24O4Na+, 327.2, found 327.1.
4.2.1.3. Synthesis of 1-benzyl 8-methyl (E)-5-ethyloct-2-enedioate (6c)
The procedure was similar to that of 6b except the following reagents were used: copper(I)bromide dimethyl sulfide (4.34 g, 21.10 mmol), n-butyllithium (16.86 ml of a 2.5 M solution in hexanes, 2.7 g, 42.2 mmol), chlorotrimethylsilane (6.87 g, 63.3 mmol), methyl pent-4-enoate 4 (1 g, 7.03 mmol), NaH (478 mg of 60% NaH in mineral oil, 11.95 mmol) and benzyl diethyl phosphonoacetate 5b (3.42 g, 11.95 mmol). The reaction was left to stir at room temperature for 10 hours. The product was purified by flash silica gel chromatography (5% ethyl acetate in hexanes) to afford 6c (602 mg, 26% over two steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.91 (t, J = 6.8 Hz, 3H), 1.29 (m 7H), 1.58 (m, 3H), 2.22 (t, J = 6.0 Hz, 2H), 2.33 (t, J = 7.2 Hz, 2H), 3.63 (s, 3H), 5.16 (s, 2H), 5.91 (d, J = 15.2 Hz, 1H), 6.97 (dt, 1H), 7.34 (m, 4H). 13CNMR (100 MHz, CD3OD) δ (ppm): 12.96, 22.05, 28.35, 28.38, 30.78, 32.60, 35.77, 36.51, 50.64, 65.68, 122.01, 127.76 (2C), 128.12, 136.29, 148.47, 166.33, 174.44. IR: 3057, 3033, 2954, 2928, 2860, 1720, 1654, 1456, 1436 cm−1. LRMS (ESI, m/z): calculated for [M+Na]+ C20H28O4Na+, 355.19, found 355.16.
4.2.1.4. Synthesis of 1-benzyl 8-methyl (E)-5-phenyloct-2-enedioate (6e)
The procedure was similar to that of 6b except the following reagents were used: copper(I)bromide dimethyl sulfide (4.61 g, 22.40 mmol), phenyllithium (22.4 ml of a 2M in dibutyl ether, 44.8 mmol), chlorotrimethylsilane (8.53 mL, 76.24 mol), methyl pent-4-enoate 4 (1.06 g, 7.49 mmol), NaH (0.51 g of 60% NaH in mineral oil, 12.70 mmol) and benzyl dimethyl phosphonoacetate 5a (2.67 mL, 12.70 mmol). The reaction was heated to reflux for 1 hour 45 minutes. The product was purified by column chromatography (diethyl ether:petroleum ether 1:6 to 1:4) to afford 6e as an orange yellow oil (1.64 g, 62% over two steps). 1HNMR (400 MHz, CDCl3): 1.87 (m, 1H), 2.05 (m, 1H), 2.15 (m, 2H), 2.53 (t, J = 7.6 Hz, 2H), 2.67 (m, 1H), 3.61 (s, 3H), 5.14 (s, 2H), 5.82 (d, J = 15.2 Hz, 1H), 6.88 (dt, J = 15.6 Hz, and 7.2 Hz, 1H), 7.12 (d, J = 7.6 Hz, 2H), 7.22 (m, 1H), 7.37 (m. 7H). 13CNMR (100 MHz, CDCl3): 30.94, 31.96, 39.61, 44.50, 51.56, 66.05, 115.33, 120.33, 126.81, 127.53, 128.15, 128.53, 128.71, 136.04, 142.81, 147.45, 166.19, 173.82. IR: 3063, 3030, 2951, 1718, 1654, 1495, 1454, 1437 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C22H24O4H+, 353.2; found 353.4; calculated for [M+Na]+ C22H24O4Na+, 375.2; found 375.3.
4.2.1.5. Synthesis of 8-methoxy-5-ethyl-8-oxooctanoic acid (7b)
In a 50 mL flask, 6b (375 mg, 1.2 mmol) was dissolved in MeOH (20 mL), then Pd(OH)2 (87 mg of 20 wt. % Pd(OH)2 on carbon, 0.12 mmol) was added. The air inside the flask was purged with argon (three times), then with hydrogen gas (three times). The reaction was stirred under hydrogen for 4 hours. The reaction was filtered through a celite plug, and the solvent was evaporated. The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:1.5) to afford 7b (299 mg, 94%); 1HNMR (400 MHz, CD3OD) δ (ppm): 0.84 (t, J = 7.2 Hz, 3H), 1.28 (m, 5H), 1.60 (m, 4H), 2.30 (m, 4H), 3.66 (s, 3H) 11.40 (bs, 1H). 13CNMR (100 MHz, CD3OD) δ (ppm): 10.58, 21.69, 25.28, 28.00, 31.47, 32.11, 34.27, 38.12, 51.55, 174.52, 179.89. IR: 2956, 2930, 2873, 1738, 1709, 1614, 1459, 1439 cm−1. LRMS (ESI, m/z): calculated for [M+Na]+ C11H20O4Na+, 239.1, found 239.2.
4.2.1.6. Synthesis of 8-methoxy-8-oxo-5-phenyloctanoic acid (7e)
The procedure was similar to that of 7b except the following reagents were used: 6e (1.40 g, 3.98 mmol), Pd(OH)2 (1.12 g of 20 wt. % Pd(OH)2 on carbon, 1.59 mmol). The product was purified by column chromatography (ethyl acetate:petroleum ether 1:6 then 1:3) to afford 7e (764 mg, 73%); 1HNMR (400 MHz, CD3OD): 1.40 (m, 2H), 1.63 (m, 2H), 1.80 (m, 1H), 1.98 (m, 1H), 2.10 (m, 2H), 2.20 (m, 2H), 2.54 (m, 1H), 3.60 (s, 3H), 7.17 (m, 3H), 7.28 (m, 2H); 13CNMR (100 MHz, CDCl3): 22.30, 31.80, 32.20, 33.95, 35.90, 45.15, 51.80, 126.40, 127.50, 128.30, 143.95, 174.30, 179.90. IR: 3028, 2948, 1734, 1705, 1603, 1494, 1453, 1437 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C15H20O4H+, 265.1; found 265.4; calculated for [M+Na]+ C15H20O4Na+, 287.1; found 287.4.
4.2.1.7. Synthesis of methyl 4-methyl-8-oxo-8-(phenylamino)octanoate (8a)
The procedure was similar to 6b except the following: copper(I)bromide dimethyl sulfide complex (2.26 g, 10.98 mmol), dry THF (40 mL), methyllithium (13.7 ml of a 1.6M solution in diethyl ether, 482 mg, 21.9 mmol), chlorotrimethylsilane (3.58 g, 32.94 mmol), methyl pent-4-enoate 4 (520 mg, 3.66 mmol), NaH (293 mg of 60% NaH in mineral oil, 7.32 mmol), dry THF (20 mL), and benzyl diethyl phosphonoacetate 5b (2.10 g, 7.32 mmol). The reaction was stirred for 3.5 hours. The product was used in the next step without purification.
The procedure was similar to 7b except the following: crude 6a from prior step, MeOH (20 mL) and Pd(OH)2 (413 mg of 20 wt. % Pd(OH)2 on carbon, 0.59 mmol). The reaction was stirred under hydrogen for 3.5 hours. The reaction was filtered through a celite plug, and the solvent was evaporated. The crude product was used in the following reaction.
The crude product 7a was dissolved in acetonitrile (10 mL), followed by addition of DIPEA (946 mg, 7.32 mmol) and TBTU (1.76 g, 5.49 mmol), and the reaction was left to stir for 20 minutes. Aniline (0.51 g, 5.49 mmol) was added, and the reaction was left to stir for 4.5 hours. The reaction was quenched with 10% aqueous HCl (20 mL). The aqueous layer was extracted with ethyl acetate (4 × 20 mL). The combined organic extracts were washed with a saturated NaHCO3 (10 mL) and then dried over anhydrous Na2SO4, filtered, and evaporated. The product was purified by silica flash chromatography (ethyl acetate:hexanes 1:4) to afford 8a (434 mg, 43% over four steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.90 (d, J = 6.4 Hz, 3H), 1.21 (m, 1H), 1.43 (m, 3H), 1.74 (m, 3H), 2.67 (m, 4H), 3.63 (s, 3H), 7.06 (t, J = 7.6 Hz, 1H), 7.28 (t, J = 8.4 Hz, 2H), 7.53 (d, J = 8.8 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 18.15, 22.90, 31.13, 31.50, 31.98, 35.79, 36.71, 50.61, 119.83, 123.69, 128.36, 138.50, 173.12, 174.72. IR: 3302, 3137, 3061, 2953, 2940, 2869, 1736, 1662, 1600, 1542, 1499, 1442 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C16H23NO3H+, 278.18, found 278.17; calculated for [M+Na]+ C16H23O3Na+, 300.16, found 300.10.
4.2.1.8. Synthesis of methyl 4-ethyl-8-oxo-8-(phenylamino)octanoate (8b)
The procedure was similar to the last step of 8a except the following reagents were used: DIPEA (319 mg, 2.47mmol), TBTU (594 mg, 1.85 mmol) and aniline (172 mg, 185 mmol). The extraction was done with dichloromethane (4 × 20 mL). The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:4) to afford 8b (250 mg, 75%). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.87 (t, J = 7.2 Hz, 3H), 1.34 (m, 5H), 1.60 (m, 2H), 1.68 (m, 2H), 2.33 (m, 4H), 3.64 (s, 3H), 7.07 (t, 1H), 7.29 (t, J = 7.2 Hz, 2H), 7.53 (d, J = 7.2 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 9.35, 22.23, 24.83, 27.55, 30.56, 31.63, 36.48, 37.83, 50.32, 119.55, 123.40, 128.06, 138.19, 172.82, 174.49. IR: 3302, 3198, 3137, 3062, 2955, 2930, 2862, 1737, 1663, 1600, 1542, 1499, 1442 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C17H25O3H+, 292.19, found 292.19; calculated for [M+Na]+ CI7H25O3Na+, 314.17, found 314.17.
4.2.1.9. Synthesis of methyl 4-butyl-8-oxo-8-(phenylamino)octanoate (8c)
The procedure was similar to that of 7b except the following reagents were used: 6c (1.2 g, 3.6 mmol) Pd(OH)2 (504 mg of 20 wt. % Pd(OH)2 on carbon, 0.72 mmol). The crude product was used in the following reaction.
The procedure was similar to the last step of 8a except the following reagents were used: Crude 7c from prior reaction, DIPEA (464 mg, 3.59 mmol), TBTU (865 mg, 2.69 mmol) and aniline (250 mg, 2.69 mmol). The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:6) to afford 8c (250 mg, 44% over two steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.89 (t, J = 6.4 Hz, 3H), 1.31 (m, 9H), 1.60 (m, 2H), 1.68 (m, 2H), 2.32 (m, 4H), 2.82 (s, 3H), 7.06 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 8 Hz, 2H), 7.53 (d, J = 8 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 13.07, 22.48, 22.66, 28.29, 28.46, 30.84, 32.37, 32.55, 36.56, 36.77, 50.63, 119.84, 123.69, 128.36, 138.49, 173.10, 174.77. IR: 3302, 3198, 3137, 3041, 2953, 2928, 2859, 1737, 1661, 1600, 1541, 1499, 1441 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C19H29NO3H+, 320.22, found 320.21; calculated for [M+Na]+ C19H29NO3Na+, 342.20, found 342.17.
4.2.1.10. Synthesis of methyl 4-hexyl-8-oxo-8-(phenylamino)octanoate (8d)
The procedure was similar to that of 6b except the following reagents were used: 4 (460 mg, 3.24 mmol) dry THF (20 mL), copper(I)bromide dimethyl sulfide (2 g, 9.73 mmol), n-hexyllithium (8.46 ml of a 2.3 M solution in hexanes, 1.792 g, 19.46 mmol), chlorotrimethylsilane (3.17 g, 29.19 mmol) and methyl pent-4-enoate 4 (461 mg, 3.24 mmol), NaH (259 mg of 60% NaH in mineral oil, 6.48 mmol), and benzyl diethyl phosphonoacetate 5b (1.85 g, 6.48 mmol). The reaction was heated to reflux for 4.5 hours. The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:10). The purified product was used in the following reaction.
The procedure was similar to that of 7b except the following reagents were used: 6d from prior reaction, Pd(OH)2 (228 mg of 20 wt. % Pd(OH)2 on carbon, 0.32 mmol). The crude product was used in the following reaction.
The procedure was similar to that of 8a except the following: 7d from prior reaction, acetonitrile (15 mL), DIPEA (838 mg, 6.48 mmol), TBTU (1.56 g, 4.86 mmol) and aniline (452 mg, 4.86 mmol). The reaction was quenched with 10% aqueous HCl (10 mL). The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:6) to afford 8d (214 mg, 19% over four steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.88 (t, J = 6.8 Hz, 3H), 1.31 (m, 13H), 1.60 and 1.68 (overlapped quartet and quintet, J = 7.6 and 7.6 Hz, 4H), 2.32 (m, 4H), 3.64 (s, 3H), 7.07 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 13.02, 22.30, 22.47, 26.14, 28.32, 29.34, 30.85, 31.61, 32.35, 32.86, 36.57, 36.75, 50.61, 119.86, 123.70, 128.34, 138.47, 173.13, 174.81. IR: 3294, 3138, 3061, 2926, 2856, 1794, 1659, 1599, 1541, 1442 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C21H33NO3H+, 348.25, found 348.26; calculated for [M+Na]+ C21H33NO3Na+, 370.24, found 370.22.
4.2.1.11. Synthesis of methyl 8-oxo-4-phenyl-8-(phenylamino)octanoate (8e)
8-Methoxy-8-oxo-5-phenyloctanoic acid 7e (0.763 g, 2.89 mmol) was dissolved in dichloromethane (25 mL), then aniline (0.32 mL, 3.47 mmol) was added, followed by 4-(dimethylamino)pyridine (0.424 g, 3.47 mmol). The mixture was stirred until 4-(dimethylamino)pyridine was completely dissolved, then dicyclohexyl carbdiimide (0.716 g, 3.47 mmol) was added, and the reaction was stirred for 4 hours at room temperature. The reaction was quenched with 10% aqueous HCl (40 mL), the organic layer was washed with a saturated NaHCO3 solution, and then brine, dried over anhydrous Na2SO4, concentrated, and purified by column chromatography (acetone:petroleum ether 1:6) to give 8e (704 mg, 72%). 1HNMR (400 MHz, CD3OD): 1.46 (m, 2H), 1.65 (m, 2H), 1.80 (m, 1H), 1.98 (m, 1H), 2.11 (m, 2H), 2.28 (m, 2H), 2.53 (m, 1H), 3.55 (s, 3H), 7.05 (t, J = 7.6 Hz, 1H), 7.16 (m, 3H), 7.26 (m, 4H), 7.52 (d, J = 8.4 Hz, 2H); 13CNMR (100 MHz, CD3OD): 23.59, 31.55, 31.58, 35.92, 36.50, 45.05, 50.59, 119.83, 123.71, 126.10, 127.41, 128.21, 128.39, 138.48, 144.07, 172.96, 174.34. IR: 3301, 3197, 3135, 3061, 3027, 2949, 2865, 1734, 1663, 1600, 1543, 1499, 1442 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C21H25NO3H+, 340.2; found 340.2; calculated for [M+Na]+ C21H25O3Na+, 362.2; found 362.2.
4.2.1.12. Synthesis of methyl 4-benzyl-8-oxo-8-(phenylamino)octanoate (8f)
The procedure was similar to that of 6b except the following reagents were used: 4 (500 mg, 3.52 mmol), dry THF (20 mL), copper(I)bromide dimethyl sulfide (2.17 g, 10.56 mmol), benzylmagnesium chloride (21.11 ml of a 1.0 M solution in methyl THF, 3.189 g, 21.12 mmol), chlorotrimethylsilane (3.44 g, 31.68 mmol), NaH (282 mg of 60% NaH in mineral oil, 7.04 mmol) and benzyl diethyl phosphonoacetate 5b (2.02 g, 7.04 mmol). The reaction was heated to reflux for 1 hour 45 minutes. The crude product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:9). The purified product was used in the following reaction.
The procedure was similar to that of 7b except the following: 6f from prior reaction, Pd(OH)2 (247 mg of 20 wt. % Pd(OH)2 on carbon, 0.35 mmol). The reaction was stirred for 4.5 hours under hydrogen, then it was filtered, and the solvent was evaporated. The crude product was used in the following reaction.
The procedure was similar to that of 8a except the following: crude 7f from prior reaction, DIPEA (464 mg, 3.59 mmol), TBTU (865 mg, 3.59 mmol), and aniline (250 mg, 2.69 mmol). The reaction was stirred for 4 hours 45 minutes. The combined organic extracts were dried over anhydrous Na2CO3. The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:3) to afford 8f (340 mg, 27% over four steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 1.35 (m, 2H), 1.60 and 1.72 (overlapped m and m, 5H), 2.34 (m, 4H), 2.55 (d, J = 6.8 Hz, 2H), 3.61 (s, 3H), 7.10 (m, 4H), 7.21 (t, J = 7.6 Hz, 2H), 7.28 (t, J = 7.2 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 22.35, 28.00, 30.84, 31.97, 36.66, 38.87, 39.61, 50.62, 119.90, 123.71, 125.47, 127.86, 128.34, 128.79, 138.44, 140.65, 173.04, 174.65. IR: 3302, 3026, 2929, 2863, 1734, 1661, 1599, 1542, 1498, 1441 cm−1. LRMS (ESI, m/z): calculated for [M+H]+ C22H27NO3H+, 354.21, found 354.22; calculated for [M+Na]+ C22H27NO3Na+, 376.19, found 376.21.
4.2.1.13. Synthesis of N1-hydroxy-4-methyl-N8-phenyloctanediamide (1a)
In an acid-washed flask, hydroxylamine HCl (1.09 g, 15.67 mmol) was dissolved in MeOH (10 mL). KOH (1.76 g, 31.33 mmol) was added at 0°C and allowed to stir for 20 minutes. An alcoholic solution of 7a (434 mg, 1.57 mmol, in 10 mL MeOH) was added, and the reaction was stirred for 4.5 hours at 0°C. The pH of the reaction mixture was adjusted to 6 with concentrated aqueous HCl, followed by dilution with distilled de-ionized water (30 mL). The reaction was extracted with ethyl acetate (3 × 30 mL). The organic extracts were collected together and dried over anhydrous Na2SO4. The product was purified by silica gel flash chromatography (acetone:dichloromethane 1:3) using iron-free silica gel to afford 1a as an oil (312 mg, 72%). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.92 (d, J = 6.0 Hz, 3H), 1.23 (m, 1H), 1.44 (m, 3H), 1.72 (m, 3H), 2.15 (m, 2H), 2.35 (t, J = 7.6 Hz, 2H), 7.07 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 18.19, 22.89, 29.98, 32.01, 32.26, 35.76, 36.64, 119.87, 123.72, 128.35, 138.45, 171.79, 173.21. IR: 3308, 3138, 3063, 3030, 2954, 2930, 2861, 1737, 1695, 1663, 1601, 1543, 1500, 1443 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C15H22N2O3Na+, 301.1528, found 301.1520. HPLC analytical purity analysis 98.4%.
4.2.1.14. Synthesis of 4-ethyl-N1-hydroxy-N8-phenyloctanediamide (1b)
The procedure was similar to that of 1a except the following: hydroxylamine HCl (597 mg, 8.59 mmol), KOH (964 mg, 17.18 mmol), and 7b (250 mg, 0.86 mmol). The reaction was left to stir for 4 hours at 0°C, then at room temperature overnight. The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:4 to 1:3) using iron-free silica gel to afford 1b as an oil (116 mg, 46%). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.87 (t, J = 6.4 Hz, 3H), 1.32 (m, 5H), 1.59 and 1.67 (overlapped m and m, 4H), 2.08 (t, J = 7.2 Hz, 2H), 2.35 (t, J = 7.2 Hz, 2H), 7.06 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.6 Hz, 2H), 7.54 (d, J = 8 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 9.72, 22.52, 25.16, 28.67, 29.82, 31.90, 36.76, 38.14, 119.90, 123.76, 128.39, 138.45, 171.89, 173.25. IR: 3252, 3199, 3061, 2960, 2932, 2872, 1658, 1600, 1546, 1500, 1444 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C16H24N2O3Na+, 315.1685, found 315.1669. HPLC analytical purity analysis 97.5%.
4.2.1.15. Synthesis of 4-butyl-N1-hydroxy-N8-phenyloctanediamide (1c)
The procedure was similar to that of 1a except the following: hydroxylamine HCl (538 mg, 7.74 mmol), KOH (869 mg, 15.49 mmol), and 7c (247 mg, 0.77 mmol). The product was purified by silica gel flash chromatography (acetone:dichloromethane 1:4) using iron-free silica gel to afford 1c as an oil (167 mg, 67%) 1HNMR (400 MHz, CD3OD) δ (ppm): 0.90 (t, J = 7.6 Hz, 3H), 1.33 (m, 9H), 1.60 and 1.69 (overlapped m and quintet, J = 7.2 Hz, 4H), 2.08 (t, J = 7.2 Hz, 2H), 2.35 (t, J = 7.2 Hz, 2H), 7.07 (t, J = 8.0 Hz, 1H), 7.29 (t, J = 7.6 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 13.06, 22.45, 22.68, 28.48, 29.11, 29.76, 32.30, 32.62, 36.62, 36.72, 119.86, 123.73, 128.36, 138.45, 171.87, 173.23. IR: 3288, 2972, 2927, 2872, 1647, 1600, 1545, 1499, 1443 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C18H28N2O3Na+, 343.1998, found 343.1985. HPLC analytical purity analysis 97.7%.
4.2.1.16. Synthesis of 4-hexyl-N1-hydroxy-N8-phenyloctanediamide (1d)
The procedure was similar to that of 1a except the following: hydroxylamine HCl (573 mg, 8.24 mmol), KOH (925 mg, 16.48 mmol), and 7d (286 mg, 0.82 mmol). The reaction was stirred for 2 hours at 0°C, then pre-incubated solution of hydroxylamine HCl (573 mg, 8.24 mmol) and KOH (925 mg, 16.48 mmol) was added followed by stirring for 1.5 hour at 0°C. The product was purified by sequential silica gel flash chromatography (acetone:dichloromethane 1:3 and a second purification with acetone:dichloromethane 1:2) using iron-free silica gel. The compound was further purified by HPLC on a reverse phase HPLC semi-preparative column (YMC America, 250 × 10 mmI.D., 4μm, 8 nm) using a gradient of 60% to 10% of buffer A over 90 minutes (buffer A = 0.1% HPLC grade TFA in water; buffer B = HPLC grade acetonitrile) at a flow rate of 3.0 mL/min at room temperature to yield 1d as an oil (49 mg, 17%). 1HNMR (400 MHz, CD3OD) δ (ppm): 0.88 (t, J = 6.8 Hz, 3H), 1.33 (m, 13H), 1.59 (m, 2H), 1.69 (quintet, J = 7.6 Hz, 2H), 2.08 (t, J = 8.0 Hz, 2H), 2.35 (t, J = 7.6 Hz, 2H), 7.07 (t, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 13.01, 22.30, 22.44, 26.20, 29.14, 29.38, 29.78, 31.63, 32.29, 32.96, 36.66, 36.71, 119.89, 123.71, 128.34, 138.45, 171.85, 173.21. IR: 3254, 3064, 2957, 2927, 2858, 1660, 1601. 1547, 1500, 1444 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C20H32N2O3Na+, 371.2311, found 371.2319. HPLC analytical purity analysis 98.2%.
4.2.1.17. Synthesis of 4-hexyl-N1-hydroxy-N8-phenyloctanediamide (1e)
The procedure was similar to that of 1a except the following: hydroxylamine HCl (1.38 g, 19.91 mmol), MeOH (50 mL), KOH (2.23 g, 39.82 mmol), and 7e (675 mg, 1.99 mmol). The reaction was stirred overnight. The product was purified by silica gel flash chromatography (5% MeOH in dichloromethane) using iron-free silica gel, followed by crystallization from MeOH to afford 1e as an oil (350 mg, 52%). 1HNMR (400 MHz, CD3OD): 1.53 (m, 2H), 1.68 (m, 2H), 1.85 (m, 3H), 2.01 (m, 1H), 2.30 (m, 2H), 2.57 (m, 1H), 7.06 (t, J = 7.6 Hz, 1H), 7.18 (m, 3H), 7.28 (m, 4H), 7.50 (d, J = 7.2 Hz, 2H). 13CNMR (100 MHz, CD3OD): 23.58, 30.50, 32.22, 35.90, 36.43, 45.14, 119.84, 123.69, 126.04, 127.40, 128.21, 128.34, 138.42, 144.19, 171.5, 173.02. IR: 3253, 3199, 3061, 3027, 2929, 2866, 1657, 1600, 1545, 1499, 1444 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C20H24N2O3Na+, 363.1685, found 363.1686. HPLC analytical purity analysis 98.8%.
4.2.1.18. Synthesis of 4-benzyl-N1-hydroxy-N8-phenyloctanediamide (1f)
The procedure was similar to that of 1a except the following: hydroxylamine HCl (600 mg, 8.64 mmol), KOH (970 mg, 17.28 mmol), and 7f (305 mg, 0.86 mmol). The reaction was stirred for 2 hours at 0°C, then another premixed solution of hydroxylamine HCl (600 mg, 8.64 mmol) and KOH (970 mg, 17.28 mmol) was added followed by stirring for 1.5 hour at 0°C. The product was purified by silica gel flash chromatography (acetone:dichloromethane 1:2) using iron-free silica gel to afford 1f as an oil (158 mg, 51%). 1HNMR (400 MHz, CD3OD) δ (ppm): 1.33 (m, 2H), 1.67 (m 5H), 2.12 (m, 2H), 2.29 (m, 2H), 2.56 (m, 2H), 7.15 (m, 6H), 7.28 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 22.28, 28.83, 29.77, 31.72, 36.64, 38.93, 39.57, 119.91, 123.73, 125.46, 127.86, 128.352, 128.846, 138.43, 140.65, 171.70, 173.13. IR: 3253, 3063, 3027, 2972, 2927, 1660, 1600, 1547, 1499, 1444 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C21H26N2O3Na+, 377.1841, found 377.1824. HPLC analytical purity analysis 98.9%.
4.2.2. Synthesis procedures for (R)-1f and (S)-1f
4.2.2.1. Synthesis of (R)-4-benzyl-3-(pent-4-enoyl)oxazolidin-2-one ((R)-9)
The compound was synthesized in a similar way to the reported procedure [59]. Briefly, (R)-8 (1.0 g, 5.64 mmol) was dissolved in dry THF (25 mL) followed by the addition of n-butyl lithium (2.5 mL of 2.5 M solution, 5.64 mmol) drop wise under argon at -78°C. The reaction was stirred at -78°C for 10 minutes, then 4-pentenoyl chloride (0.81 mL, 6.77 mmol) was added drop wise. Stirring was continued for 30 minutes at -78°C. Then the reaction temperature was raised gradually over 30 minutes to room temperature. The reaction was diluted by addition of saturated solution of ammonium chloride (30 mL) followed by a saturated solution of sodium carbonate (30 mL) and stirred for 15 minutes at room temperature. The solution was extracted with ethyl acetate (3 × 40 mL). The organic extracts were combined and evaporated, and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:9-1:3) which yielded the product (R)-9 (954 mg, 65%). 1HNMR (400 MHz, CDCl3) d (ppm): 2.45 (q, J = 6.8 Hz, 2H), 2.61 (dd, J = 3.6 Hz and 13.2, 1H), 3.04 (m, 2H), 3.31 (dd, J = 2.8 and 13.2 Hz, 1H), 4.17 (m, 2H), 4.68 (m, 1H), 5.07 (overlapped d and d, J = 10.4 and 17.2 Hz, 2H), 5.87 (m, 1H), 7.21 (d, J = 7.2 Hz, 2H), 7.31 (m, 3H); 13CNMR (100 MHz, CDCl3) δ (ppm): 28.16, 34.81, 37.92, 55.16, 66.21, 115.74, 127.36, 128.96, 129.42, 135.26, 136.69, 153.46, 172.55. LRMS (LC-SQMS, m/z); found: [M+H], 260.01, calculated for C15H18NO3, 260.13, found: [M+Na], 281.97, calculated for C15H17NO3Na, 282.11. Spectral data were consistent with the reported spectra [59].
4.2.2.2. Synthesis of (R)-4-benzyl-3-((S)-2-benzylpent-4-enoyl)oxazolidin-2-one ((RS)-10)
To compound (R)-9 (951 mg, 3.67 mmol) was added dry THF (20 mL) followed by lowering of the temperature to -78°C. NaHMDS (2.0 mL of 2 M solution, 4.04 mmol) was added drop wise under Argon and the reaction was stirred at -78°C for 30 minutes. Benzyl bromide (0.86 mL, 7.34 mmol) was then added drop wise, and the reaction was stirred at -78°C for 5 hours. The reaction temperature was increased gradually to room temperature overnight. The reaction was then quenched with a saturated ammonium chloride solution (15 mL) and was left to stir at room temperature for 15 minutes. The reaction was extracted with ethyl acetate (2 × 30 mL). The extracts were combined and evaporated, and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:15-1:10) which yielded (RS)-10 as a white solid (730 mg, 57%). 1HNMR (400 MHz, CDCl3) d (ppm): 2.29 (m, 1H), 2.47 (m, 2H), 2.83 (dd, J = 6.8 and 13.6 Hz, 1H), 3.03 (overlapped dd and dd, J = 3.2, 13.2, 8.4, and 13.6 Hz, 2H), 4.05 (dd, J = 3.2, and 9.2 Hz, 1H), 4.11 (t, J = 7.6 Hz, 1H), 4.35 (m, 1H), 4.62 (m, 1H), 5.05 (m, 2H), 5.81 (m, 1H), 7.01 (dd, J = 2.4, and 8.0 Hz, 2H), 7.20 (m, 1H), 7.25 (m, 3H), 7.28 (m, 4H); 13CNMR (100 MHz, CDCl3) δ (ppm): 36.34, 37.56, 38.13, 44.27, 55.05, 65.77, 117.28, 126.47, 127.24, 128.38, 128.88, 129.37, 129.38, 135.18 (2), 138.90, 153.07, 175.26. LRMS (LC-SQMS, m/z); found: [M+H], 349.98, calculated for C22H24NO3, 350.18, found: [M+Na], 371.95, calculated for C22H23NO3Na, 372.16. [α]D23 = -48.40 (c = 1.00, CH2Cl2). Spectral data were consistent with the reported spectra [60]. The diastereomeric ratio of 99:1 was calculated by dividing the integration of each peak of both the major and the minor diastereomers by the sum of the integration of both peaks in the 1H NMR spectrum (See Figure S78).
4.2.2.3. Synthesis of (R)-4-benzyl-3-(3-phenylpropanoyl)oxazolidin-2-one ((R)-12)
The procedure was similar to that of (R)-9 except the following reagents were used: (R)-8 (1.5 g, 8.47 mmol), n-butyl lithium (4.1 mL of 2.5 M solution, 10.16 mmol), and 3-phenyl propanoyl chloride (1.64 mL, 11 mmol). The reaction gave (R)-12 in 90% yield (2.36 g). 1HNMR (400 MHz, CDCl3) d (ppm): 2.75 (dd, J = 9.6 Hz and 13.2, 1H), 3.03 (m, 2H), 3.29 (m, 3H), 4.16 (m, 2H), 4.67 (m, 1H), 7.22 (m, 3H), 7.32 (m, 7H); 13CNMR (100 MHz, CDCl3) δ (ppm): 30.26, 37.13, 37.82, 55.11, 66.18, 126.28, 127.36, 128.48, 128.58, 128.96, 129.42, 135.19, 140.44, 153.41, 172.41. LRMS (LC-SQMS, m/z); found: [M+H], 310.31, calculated for C19H20NO3, 310.14, found: [M+Na], 332.31, calculated for C19H19NO3Na, 332.13. Spectral data are consistent with the reported spectra [61].
4.2.2.4. Synthesis of (R)-4-benzyl-3-((R)-2-benzylpent-4-enoyl)oxazolidin-2-one ((RR-10)
The procedure was similar to that of (RS)-10 except the following reagents were used: (R)-16 (2.35 g, 7.59 mmol), NaHMDS (4.18 mL of 2 M solution, 8.35 mmol), and allyl bromide (1.97 mL, 22.8 mmol). The product was purified by Flash silica-gel chromatography (ethyl acetate:hexanes 1:15) which yielded (RS)-10 as an oily product (1.84 g, 69%). 1HNMR (400 MHz, CDCl3) d (ppm): 2.37 (m, 1H), 2.52-2.67 (overlapped m and dd, J = 10.0 and 13.2 Hz, 2H), 2.84 (dd, J = 6.4 and 13.2 Hz, 1H), 2.96 (dd, J = 8.8 and 13.2 Hz, 1H), 3.23 (dd, J = 3.2, and 13.2 Hz, 1H), 3.82 (t, J = 8.4 Hz, 1H), 4.01 (d, J = 9.2 Hz, 1H), 4.33 (m, 1H), 4.45 (m, 1H), 5.10 (m, 2H), 5.86 (m, 1H), 7.17-7.33 (m, 10H); 13CNMR (100 MHz, CDCl3) δ (ppm): 36.32, 38.03, 38.29, 43.95, 55.49, 65.84, 117.41, 126.41, 127.29, 128.35, 128.90, 129.11, 129.42, 135.05, 135.36, 138.91, 153.01, 175.30. LRMS (LC-SQMS, m/z); found: [M+H], 350.45, calculated for C22H24NO3, 350.18, found: [M+Na], 372.47, calculated for C22H23NO3Na, 372.16. [α]D23 = -121.5 (c = 1.00, CH2Cl2). Spectral data were consistent with the reported spectra [60-61]. The diastereomeric ratio of 97:3 was calculated by dividing the integration of each peak of both the major and the minor diastereomers by the sum of the integration of both peaks in the 1H NMR spectrum (See Figure S85).
4.2.2.5. Synthesis of (S)-2-benzylpent-4-en-1-ol ((S)-11)
A solution of (RS)-10 (901 mg, 2.58 mmol) in dry THF (7 mL) was cooled and stirred at 0°C in ice bath for 15 minutes. Lithium aluminum hydride (295 mg, 7.77 mmol) was added portion wise and the reaction was stirred at 0°C for 2 hours. The reaction was quenched by careful drop wise addition of 1M solution of NaOH until no effervescence was observed. The reaction was then diluted with water (3 mL). Extraction of the aqueous layer was done with ethyl acetate (3 × 40 mL). The organic extracts were combined and evaporated, and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:15-1:10) which afforded (S)-11 (402 mg, 88%). 1HNMR (400 MHz, CDCl3) d (ppm): 1.38 (br s, 1H), 1.93 (m, 1H), 2.14 (t, J = 7.2 Hz, 2H), 2.64 (m, 2H), 3.55 (m, 2H), 5.06 (m, 2H), 5.83 (m, 1H), 7.19 (m, 3H), 7.28 (m, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 35.50, 37.24, 42.37, 64.74, 116.61, 125.97, 128.35, 129.17, 136.83, 140.48. [α]D23 = -13.96 (c = 0.824, CH2Cl2). Compound characterization and specific rotation were consistent with literature [62].
4.2.2.6. Synthesis of (R)-2-benzylpent-4-en-1-ol ((R)-11)
The procedure was similar to that of (S)-11 except the following reagents were used: (RR)-10 (1.83 g, 5.23 mmol) in dry THF (25 mL), Lithium aluminum hydride (595 mg, 15.68 mmol). The reaction afforded (R)-11 in 70% yield (645 mg). 1HNMR (400 MHz, CDCl3) d (ppm): 1.42 (t, J = 5.6 Hz, 1H), 1.92 (m, 1H), 2.15 (m, 2H), 2.65 (m, 2H), 3.55 (m, 2H), 5.07 (m, 2H), 5.83 (m, 1H), 7.21 (m, 3H), 7.28 (m, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 35.49, 37.24, 42.37, 64.71, 116.62, 125.97, 128.35, 129.18, 136.84, 140.50. [α]D23 = +15.80 (c = 1, CH2Cl2).
4.2.2.7. Synthesis of (S)-2-benzylpent-4-en-1-yl (R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate ((S)-11-(R)-MTPA)
Alcohol (S)-11 (35 mg, 0.2 mmol) was dissolved in dry DCM (4 mL), followed by replacement of air with argon and addition of (R)-(+)-a-Methoxy-atrifluoromethylphenylacetic acid ((R)-MTPA, 71 mg, 0.3 mmol), EDCI (116 mg, 0.6 mmol), and 4-(dimethylamino)pyridine (73 mg, 0.6 mmol). The reaction was stirred overnight at room temperature. Solvent was evaporated and the residue was suspended in 1N HCl (5 mL) and extracted with ethyl acetate (2 × 15 mL). The combined extracts were combined and evaporated, and purified by silica gel flash chromatography (ethyl acetate:hexanes 1:15) to afford (S)-11-(R)-MTPA (49 mg, 63%). 1HNMR (600 MHz, CDCl3) δ (ppm): 2.03 (m, 3H), 2.51 (m, 2H), 3.49 (s, 3H), 4.06-4.13 (overlapped dd and dd, J = 2.8 and 7.2 Hz, 2H), 4.99 (m, 2H), 5.68 (m, 1H), 6.96 (d, J = 4.8 Hz, 2H), 7.12 (t, J = 5.2 Hz, 1H), 7.18 (d, J = 5.6 Hz, 2H), 7.35 (m, 3H), 7.46 (m, 2H). 13CNMR (150 MHz, CDCl3) δ (ppm): 35.15, 36.97, 39.26, 55.46, 67.18, 117.45, 121.99, 124.85, 126.23, 127.38, 128.45, 129.07, 129.67, 132.31, 135.48, 139.38, 166.57. 19FNMR (400 MHz, CDCl3) δ (ppm): -71.30. LRMS (LC-SQMS, m/z); found: [M+Na], 415.13, calculated for C22H23F3O3Na, 415.15.
4.2.2.8. Synthesis of (R)-2-benzylpent-4-en-1-yl (R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate ((R)-11-(R)-MTPA)
The procedure was similar to that of (SR)-18 except the following reagents were used (R)-11 (15.8 mg, 0.09 mmol), (R)-(+)-a-Methoxy-atrifluoromethylphenylacetic acid ((R)-MTPA, 46 mg, 0.2 mmol), EDCI (35 mg, 0.27 mmol), and 4-(dimethylamino)pyridine (33 mg, 0.27 mmol). The reaction was stirred at room temperature for 3 hours. The product was purified by silica gel flash chromatography (ethyl acetate:hexanes 1:50) and afforded (R)-11-(R)-MTPA in 89% yield (32 mg). 1HNMR (600 MHz, CDCl3) δ (ppm): 2.03 (m, 3H), 2.54 (m, 2H), 3.48 (s, 3H), 4.01 (dd, J = 2.8 and 7.2 Hz, 1H), 4.20 (dd, J = 2.8 and 7.2 Hz, 1H), 4.97 (m, 2H), 5.66 (m, 1H), 7.01 (d, J = 4.8 Hz, 2H), 7.13 (t, J = 5.2 Hz, 1H), 7.20 (d, J = 5.6 Hz, 2H), 7.35 (m, 3H), 7.46 (m, 2H). 13CNMR (150 MHz, CDCl3) δ (ppm): 34.99, 37.05, 39.15, 55.41, 67.19, 117.48, 121.98, 124.85, 126.26, 127.43, 128.45, 129.10, 129.67, 132.28, 135.42, 139.34, 166.55. 19FNMR (400 MHz, CDCl3) δ (ppm): -71.26. LRMS (LC-SQMS, m/z); found: [M+Na], 415.17, calculated for C22H23F3O3Na, 415.15.
4.2.2.9. Synthesis of dimethyl (R)-2-(2-benzylpent-4-en-1-yl)malonate ((R)-13)
A solution of (S)-11 (400 mg, 2.27 mmol) in dry DCM (10 mL) was cooled to 0°C in ice bath, followed by addition of triethyl amine (265 μL, 3.41 mmol), and then methanesulfonyl chloride (476 μL, 3.41 mmol) drop wise. The reaction was stirred for 10 minutes at 0° C, then for 1 hour at room temperature. The reaction was diluted with water (10 mL), followed by concentration at reduced pressure. The aqueous layer was extracted with ethyl acetate (3 × 20 mL). The organic extracts were combined, dried over anhydrous sodium sulfate, and evaporated. The crude product was used in the following reaction without purification.
In a 2-neck flame dried flask, air was purged with argon, NaH (273 mg of 60% NaH in mineral oil, 6.82 mmol) dissolved in dry THF (20 mL) was added. The reaction was then cooled to 0°C and dimethyl malonate (0.78 mL, 6.82 mmol) was added drop wise with stirring. The reaction was allowed to stir for 15 minutes at 0°C, then the crude product from the previous reaction was added (in 10 mL dry THF). The reaction was heated under reflux for 20 hours, and then another solution of malonate anion (0.78 mL, 6.82 mmol, prepared in the same way as described above) was added to the reaction and reflux was continued for another 20 hours. The reaction was then quenched with a saturated ammonium chloride solution (20 mL) and extracted with ethyl acetate (3 × 30 mL). The extracts were combined and evaporated, and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:9) which yielded (R)-13 in 49% yield (269 mg) over two steps. 1HNMR (400 MHz, CDCl3) d (ppm): 1.67 (m, 1H), 1.83-2.12 (m, 4H), 2.58 (m, 2H), 3.50 (t, J = 7.6 Hz, 1H) 3.69 (s, 3H), 3.73 (s, 3H), 5.05 (m, 2H), 5.76 (m, 1H), 7.13 (d, J = 7.2 Hz, 2H), 7.19 (m, 1H), 7.24 (m, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 32.47, 37.16, 37.37, 39.85, 49.70, 52.48, 52.50, 117.22, 126.02, 128.30, 129.16, 135.70, 140.16, 169.82, 169.89. IR: 3065, 3027, 2953, 2924, 2853, 1733, 1651, 1623, 1592, 1575, 1496, 1436 cm−1. LRMS (LC-SQMS, m/z); found: [M+Na], 313.38, calculated for C17H22O4Na, 313.34.
4.2.2.10. Synthesis of dimethyl (S)-2-(2-benzylpent-4-en-1-yl)malonate ((S)-13)
The procedure was similar to that of (S)-12 except the following reagents were used: (R)-11 (597 mg, 3.34 mmol), triethyl amine (710 μL, 5.09 mmol), methanesulfonyl chloride (394 μL, 5.09 mmol), and the reaction was stirred at room temperature for 3 hours. For the next reaction, the following reagents were used once only, NaH (407 mg of 60% NaH in mineral oil, 10.17 mmol), dimethyl malonate (1.16 mL, 10.17 mmol) and the reaction was refluxed for 20 hours. The reaction afforded (S)-13 in 66% yield (648 mg) over two steps. 1HNMR (400 MHz, CDCl3) d (ppm): 1.74 (m, 1H), 1.88-2.08 (m, 4H), 2.56 (m, 2H), 3.50 (t, J = 7.6 Hz, 1H) 3.69 (s, 3H), 3.72 (s, 3H), 5.05 (m, 2H), 5.75 (m, 1H), 7.13 (d, J = 6.8 Hz, 2H), 7.19 (m, 1H), 7.29 (m, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 32.47, 37.16, 37.37, 39.85, 49.70, 52.48, 52.50, 117.22, 126.02, 128.30, 129.16, 135.70, 140.16, 169.82, 169.89. IR: 3066, 3028, 2953, 2924, 1733, 1657, 1638, 1621, 1605, 1497, 1436 cm−1. LRMS (LC-SQMS, m/z); found: [M+H], 291.37, calculated for C17H23O4, 291.16, found: [M+Na], 313.38, calculated for C17H22O4Na, 313.34.
4.2.2.11. Synthesis of methyl (S)-4-benzylhept-6-enoate ((S)-14)
(R)-13 (269 mg, 0.93 mmol) was dissolved in DMSO (15 mL) followed by addition of LiCl (394 mg, 9.3 mmol) and water (167 μL, 9.3 mmol). The reaction was heated under reflux (150-160 °C) overnight. Water (20 mL) was added to the reaction and the product was extracted with ethyl acetate (2 × 30 mL). The organic layer was evaporated and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:15), which yielded (S)-14 in 75% yield (162 mg). 1HNMR (400 MHz, CDCl3) d (ppm): 1.64 (m, 2H), 1.77 (m, 1H), 2.04 (m, 2H), 2.33 (m, 2H), 2.57 (m, 2H), 3.65 (s, 3H), 5.04 (m, 2H), 5.78 (m, 1H), 7.19 (m, 3H), 7.24 (t, J = 7.2 Hz, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 28.14, 31.61, 37.20, 39.07, 39.91, 51.52, 116.76, 125.89, 128.27, 129.17, 136.29, 140.67, 174.17. IR: 3062, 3027, 2953, 2924, 2855, 1735, 1658, 1640, 1595, 1574, 1511, 1497, 1445 cm−1. LRMS (LC-SQMS, m/z); found: [M+Na], 255.12, calculated for C15H20O2Na, 255.14.
4.2.2.12. Synthesis of methyl (R)-4-benzylhept-6-enoate ((R)-14)
The procedure was similar to that of (S)-14 except the following reagents were used: (S)-13 (617 mg, 2.13 mmol), LiCl (270 mg, 6.38 mmol), and water (115 μL, 6.38 mmol). The reaction afforded (R)-14 in 65% yield (321 mg). 1HNMR (400 MHz, CDCl3) d (ppm): 1.63 (m, 2H), 1.77 (m, 1H), 2.05 (m, 2H), 2.33 (m, 2H), 2.56 (m, 2H), 3.65 (s, 3H), 5.04 (m, 2H), 5.77 (m, 1H), 7.17 (m, 3H), 7.26 (m, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 28.13, 31.61, 37.19, 39.07, 39.91, 51.53, 116.76, 125.89, 128.26, 129.16, 136.29, 140.67, 174.19. IR: 3063, 3027, 2924, 2856, 1736, 1657, 1640, 1596, 1511, 1497, 1436 cm−1. LRMS (LCSQMS, m/z); found: [M+Na], 255.26, calculated for C15H20O2Na, 255.14.
4.2.2.13. Synthesis of N-phenylacrylamide (15)
The compound was synthesized similar to the reported procedure [63]. Briefly, aniline (3.03 mL, 33.15 mmol) and triethyl amine (6 mL, 66.29 mmol) were dissolved in dry DCM (30 mL) and the temperature of the solution was lowered to 0°C. A solution of acryloyl chloride (2.69 mL, 33.15 mmol) in dry DCM (10 mL) was added drop wise. The reaction temperature was increased gradually from 0°C to room temperature and stirring was continued overnight at room temperature. Solvent was evaporated and the residue was suspended in 10% HCl (20 mL) and then extracted with DCM (2 × 30 mL). The combined organic extracts were washed with saturated solution of sodium carbonate (20 mL), dried over anhydrous sodium sulfate, and evaporated to give the product as a yellow solid (4.44 g, 91%). 1HNMR (400 MHz, CDCl3) d (ppm): 5.71 (dd, J = 1.6 and 10.0 Hz, 1H), 6.30 (dd, J = 10.0 and 16.8 Hz, 1H), 6.41 (dd, J = 1.6 and 16.8 Hz, 1H), 7.12 (t, J = 7.6, 1H), 7.28 (m, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.98 (s, 1H); 13CNMR (100 MHz, CDCl3) δ (ppm): 120.15, 124.51, 127.69, 128.99, 131.29, 137.85, 163.86. The spectral data for the synthesized compound was consistent with the reported data in literature [63].
4.2.2.14. Synthesis of methyl (S,E)-4-benzyl-8-oxo-8-(phenylamino)oct-6-enoate ((S)-16)
To a solution of (S)-14 (162 mg, 0.7 mmol) in dry DCM (20 mL) was added 15 (103 mg, 0.7 mmol), and then air was replaced with argon. Grubbs' second generation catalyst (35 mg, 5 mol%) was added and the reaction was heated to 50-60°C for 20 hours . A second addition of Grubbs' second generation catalyst (35 mg, 5 mol%) was done and the reaction was heated to 50-60°C for 28 hours.
The solvent was evaporated and the product was purified by flash silica-gel chromatography (ethyl acetate:hexanes 1:15 followed by 1:7-1:3), which yielded (S)-16 in 46% yield (112 mg). 1HNMR (400 MHz, CDCl3) d (ppm): 1.67 (m, 2H), 1.88 (m, 1H), 2.18 (m, 2H), 2.33 (t, J = 8.0 Hz, 2H), 2.51 (dd, J = 7.6 and 13.6 Hz, 1H), 2.63 (dd, J = 6.4 and 13.6 Hz, 1H), 3.66 (s, 3H), 5.92 (d, J = 15.2 Hz, 1H), 6.91 (m, 1H), 7.12 (m, 3H), 7.21 (t, J = 7.2 Hz, 1H), 7.31 (m, 4H), 7.36 (s, 1H), 7.57 (dd, J = 7.6 Hz, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 28.45, 31.67, 35.61, 38.93, 40.07, 51.65, 119.81, 124.27, 126.00, 126.16, 128.41, 129.01, 129.13, 138.02, 140.03, 143.84, 163.75, 173.97. IR: 3301, 3136, 3062, 3027, 2926, 1733, 1670, 1640, 1600, 1542, 1497, 1441 cm−1. LRMS (LC-SQMS, m/z); found: [M+H], 352.07, calculated for C22H26NO3, 352.19, found: [M+Na], 374.05, calculated for C22H25NO3Na, 374.17.
4.2.2.15. Synthesis of methyl (R,E)-4-benzyl-8-oxo-8-(phenylamino)oct-6-enoate ((R)-16)
A similar to that of (S)-16 was followed except the following reagents were used (R)-14 (321 mg, 1.38 mmol), 15 (204 mg, 1.38 mmol), and Grubbs' second generation catalyst (59 mg, 5 mol%). The reaction afforded (R)-16 in 48% yield (233 mg). 1HNMR (400 MHz, CDCl3) d (ppm): 1.66 (m, 2H), 1.87 (m, 1H), 2.17 (m, 2H), 2.34 (t, J = 8.0 Hz, 2H), 2.52 (dd, J = 7.6 and 13.6 Hz, 1H), 2.62 (dd, J = 6.4 and 13.6 Hz, 1H), 3.65 (s, 3H), 5.93 (d, J = 15.2 Hz, 1H), 6.91 (m, 1H), 7.11 (m, 3H), 7.20 (t, J = 7.6 Hz, 1H), 7.30 (m, 4H), 7.51 (bs, 1H), 7.58 (d, J = 7.2 Hz, 2H); 13CNMR (100 MHz, CDCl3) δ (ppm): 28.43, 31.66, 35.60, 38.92, 40.04, 51.66, 119.85, 124.26, 126.01, 126.15, 128.41, 129.00, 129.13, 138.06, 140.03, 143.79, 163.83, 173.00. IR: 3401, 3062, 3026, 2924, 2856, 1736, 1658, 1640, 1597, 1574, 1512, 1437 cm−1. LRMS (LC-SQMS, m/z); found: [M+H], 352.07, calculated for C22H26NO3, 352.19, found: [M+Na], 374.02, calculated for C22H25NO3Na, 374.17.
4.2.2.16. Synthesis of (R)-4-benzyl-N1-hydroxy-N8-phenyloctanediamide ((R)-1f)
(S)-16 (112 mg, 0.32 mmol) was dissolved in MeOH (15 mL), then Pd (30 mg of 20 wt.% Pd on carbon) was added to the solution. Air inside the flask was purged with argon (three times), then with hydrogen gas (three times). The reaction was stirred under hydrogen overnight. The reaction was filtered, and the solvent was evaporated. The crude product was used in the next reaction without purification.
In an acid-washed flask, hydroxylamine HCl (222 mg, 3.19 mmol) was dissolved in MeOH (10 mL). KOH (358 mg, 6.38 mmol) was added at 0°C and allowed to stir for 10 minutes. An alcoholic solution of the crude product from the previous reaction (in 5 mL MeOH) was added, and the reaction was stirred for 3.5 hours at 0°C. Then another premixed solution of hydroxylamine HCl (222 mg, 3.19 mmol) and KOH (358 mg, 6.38 mmol) was added, followed by stirring for 4 hour at 0°C. The pH of the reaction mixture was adjusted to 6 with concentrated aqueous HCl, followed by dilution with distilled de-ionized water (10 mL). The reaction was extracted with ethyl acetate (2 × 20 mL). The product was purified by silica gel flash chromatography (acetone:dichloromethane 1:4-1:2) using iron-free silica gel to afford (R)-1f as an oil (84 mg, 74% over two steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 1.32 (m, 2H), 1.69 (m 5H), 2.12 (m, 2H), 2.29 (m, 2H), 2.52 (dd, J = 7.2 and 13.2 Hz, 1H), 2.61 (dd, J = 6.4 and 13.6 Hz, 1H), 7.05-7.15 (m, 4H), 7.21 (t, J = 7.2 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 22.28, 28.84, 29.77, 31.72, 36.63, 38.93, 39.57, 119.92, 123.73, 125.46, 127.86, 128.35, 128.85, 138.43, 140.65, 171.70, 173.13. IR: 3230, 3061, 3026, 2925, 2864, 1648, 1598, 1543, 1497, 1443 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C21H26N2O3Na+, 377.1841, found 377.1837. HPLC analytical purity analysis 95.6%.
4.2.2.17. Synthesis of (S)-4-benzyl-N1-hydroxy-N8-phenyloctanediamide ((S)-1f)
A procedure similar to that of (R)-1f was followed, except the following reagents were used: (R)-16 (283 mg, 0.8 mmol), Pd (58 mg of 20 wt.% Pd on carbon). Hydroxylamine HCl (557 mg, 8.02 mmol) and KOH (900 mg, 16.03 mmol) were added once only and the reaction was stirred for 3 hours at 0°C. The product was purified by silica gel flash chromatography (acetone:dichloromethane 1:4-1:2) using iron-free silica gel to afford (S)-1f as an oil (133 mg, 47% over two steps). 1HNMR (400 MHz, CD3OD) δ (ppm): 1.33 (m, 2H), 1.59-1.82 (m 5H), 2.14 (m, 2H), 2.30 (m, 2H), 2.53 (dd, J = 7.2 and 13.6 Hz, 1H), 2.61 (dd, J = 6.8 and 13.6 Hz, 1H), 7.05-7.16 (m, 4H), 7.21 (t, J = 7.2 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H). 13CNMR (100 MHz, CD3OD) δ (ppm): 22.28, 28.83, 29.76, 31.71, 36.62, 38.93, 39.57, 119.91, 123.73, 125.46, 127.86, 128.34, 128.84, 138.43, 140.65, 171.70, 173.13. IR: 3232, 3026, 2926, 2865, 1645, 1598, 1543, 1497, 1443 cm−1. HRMS (ESI-TOF, m/z): calculated for [M+Na]+ C21H26N2O3Na+, 377.1841, found 377.1848. HPLC analytical purity analysis 95.6%.
4.3. Procedures for biological screenings
4.3.1. HeLa cell lysis
Lysates were prepared according to the reported procedure [28]. HeLa-S3 cells (purchased from Biovest) were lysed in lysis buffer (1 × 109 cells in 10 mL lysis buffer; 50 mM Tris-HCl, pH 8.0, 10% glycerol, 150 mM NaCl, 0.5% Triton X-100, and 1X protease inhibitor cocktail (GenDEPOT)) with rotation at 4 °C for 30 min. Cell debris was removed by centrifugation at 12000 rpm at 4 °C for 30 min. Protein concentration of the supernatant was determined using Bio-Rad protein assay (BioRad, Bradford reagent). Lysates were stored at -80 °C.
4.3.2. Global HDAC inhibition
To measure global HDAC inhibition, HeLa cell lysates (1 μg total protein) were mixed with HDACGlo™ buffer in polystyrene 96-well half area white plate (Corning) to a final volume of 12 μL, followed by addition of inhibitors in DMSO (0.5 μL) and incubation for 15 min at room temperature without rocking. An uninhibited control reaction was also included that contained DMSO (0.5 μL). Finally, deacetylase activity was measured using the HDAC-Glo™ assay kit (Promega) as per the manufacturer's protocol. Specifically, the HDAC-Glo™ substrate (1 mL) and developer (1 μL) were first premixed to form the HDAC-Glo™ reagents. Then, to monitor deacetylase activity, HDAC-Glo™ reagent (5μL) and HDAC-Glo™ buffer (7.5 μL) were added to each well (25 μL total volume) and incubated for 35 min at room temperature without rocking. The deacetylase activity was measured as luminescent signal using a GeniosPlus Fluorimeter (Tecan) at optimal gain. The concentrations of inhibitors reported in the dose-dependent studies (Figure S140, and Table S1) are final concentrations after addition of HDAC-Glo™ reagent HDAC-Glo™ buffer. The luminescent signal was first background corrected with the signal from a negative control reaction where no lysates was added to that reaction. Then percent deacetylase activity was calculated by dividing the background corrected signal for each reaction by the background corrected signal of the uninhibited control, then multiplied by 100. IC50 values were calculated by fitting the percent deacetylase activity remaining as a function of inhibitor concentration to a sigmoidal dose-response curve (y = 100/(1+(x/IC50)z), where y = percent deacetylase activity and × = inhibitor concentration) using non-linear regression with KaleidaGraph 4.1.3 software (Tables 1 and S1, and Figure S140).
4.3.3. Inhibitor testing with HDAC isoforms [28]
Individual wells of a high binding polystyrene 96-well white opaque plate (Thermo Scientific) were incubated with binding buffer (100 μL; 0.2 M carbonate/0.2 M bicarbonate buffer, pH 9.4) containing primary HDAC1 antibody (Sigma Aldrich, H3284, 100 μ L of 10 μg/mL), primary HDAC2 antibody (Sigma Aldrich, H3159, 100 μL of 10 μg/mL), HDAC3 antibody (Sigma Aldrich, H3034, 100 μL of 25 μg/mL), or primary HDAC6 antibody (Sigma Aldrich, SAB1404771, 100μL of 2μg/mL) with rocking (3 rpm) for 1 hr at room temperature, or at 4 °C overnight with no rocking. For all reactions, unbound antibody was removed by washing quickly three times with TBST buffer (400 μL; 50mM Tris-HCl, 150 mM NaCl, pH 7.4, 0.05% (v/v) Tween-20), followed by a fourth wash with TBST (400 μL) with 5 minutes incubation and rocking (3 rpm) at room temperature. Blocking of the unbound regions of the well was accomplished with 5% non-fat dry milk in TBST buffer (350 μL) for 1 hr at room temperature with rocking (3 rpm). To affix HDAC enzyme to the plate, HeLa cell lysates (100 μL of 100 μg/mL for HDAC1 and HDAC2, or 100 μL of 1 mg/mL for HDAC3 and HDAC6 in TBST buffer containing 0.1 % (w/v) non-fat dry milk) were added to each well and incubated for 1 h at 4 °C without rocking, followed by washing with TBST, as described previously. HDAC-Glo™ buffer (24 μL) was added to each well, followed by addition of inhibitors in DMSO (1 μL) and incubation for 15 min at room temperature without rocking. An uninhibited control reaction was also included that contained DMSO (1 μL) in HDAC-Glo™ buffer (24 μL). To monitor deacetylase activity, the HDAC-Glo™ reagent (prepared as described earlier, 25μL) was added to each well (50 μL total volume) and incubated for 30-40 min at room temperature without rocking. Deacetylase activity was measured as luminescent signal using a GeniosPlus Fluorimeter (Tecan) at optimal gain. The concentrations of inhibitors reported in the single dose screen (Figure 2 and Table S2) and dose-dependent studies (Tables S3-S9, and Figures S141-S147) are final concentrations after addition of HDAC-Glo™ reagent. For both the single concentration screen and dose-dependent reactions to determine IC50, the luminescent signal was first background corrected with the signal from a negative control reaction where the HDAC antibody was absent in the initial antibody binding step. Then percent deacetylase activity was calculated by dividing the background corrected signal for each reaction by the background corrected signal of the uninhibited control, then multiplied by 100. The mean percent deacetylase activity along with standard error of three independent trials is reported in Figure 2.
Inhibitory activity with HDAC8 was measured using the following procedure. In a half area 96-well white opaque plate, recombinant HDAC8 (75 ng, BPS Bioscience) was incubated in HDAC-Glo™ buffer (39 μL) with inhibitor in DMSO (1 μL), or DMSO alone (1 μL) as a control, for 15 minutes at room temperature. HDAC-Glo™ reagent (10 μL) was added to each reaction and incubated for 30 min at room temperature. Luminescent signal was measured 30 minutes after adding the substrate reagent using a Geniosplus Fluorimeter (Tecan) at optimal gain. To determine IC50, the luminescent signal was first background corrected with the signal from a background control reaction where no HDAC8 enzyme was added.
IC50 values were calculated by fitting the percent deacetylase activity remaining as a function of inhibitor concentration to a sigmoidal dose-response curve (y = 100/(1+(x/IC50)z), where y = percent deacetylase activity and × = inhibitor concentration) using non-linear regression with KaleidaGraph 4.1.3 software (Tables 2, S3-S9, Figures S141-S147).
4.3.4. In cellulo selectivity testing
U937 cells were grown in RPMI media supplemented with 10% fetal calf serum and 1% penicillin/streptomycin under humidified conditions (37 °C, 5% CO2). Cells were added (106 cells/well) to a 12 well plate in RPMI-1640 (no phenol red) media, supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (990 μL final volume). The cells were treated with DMSO (10 μL) or the inhibitor in DMSO (10 μL) and incubated for 18 hours under humidified conditions (37 °C, 5% CO2). The cells were then harvested, washed once with cold DPBS (500 μL, HyClone), and then lysed with lysis buffer (20 μL) containing 1X protease inhibitor for 30 minutes at 0°C. The total protein concentration in the supernatant was then quantified using Bradford assay reagent (Bio-Rad) with bovine serum albumin (BSA) as the standard. Equal quantities of proteins were mixed with β-mercaptoethanol (10% of the final volume) and SDS loading buffer (25% of the final volume, 200 mM Tris-Cl, pH 6.8, 400 mM DTT, 8% SDS, 0.4% bromophenol blue, 40% glycerol) before the proteins were denatured at 95 °C for 3 minutes. The proteins in each sample were separated by 16% SDSpolyacrylamide gel electrophoresis (SDS-PAGE), then transferred to PVDF membrane (Immobilon P, Millipore). The membrane was blocked with 5% (w/v) nonfat milk in TBST buffer (50mM Tris-HCl, 150 mM NaCl, pH 7.4, 0.1% (v/v) Tween-20) at room temperature for 1 h. The blocked membrane was incubated with a primary antibody (anti-GAPDH (Cell Signaling, 5174P); anti-Acetyl-α-tubulin(Lys40) (Cell Signaling, 5335P), or anti-Acetyl-histone H3(Lys9) (Cell Signaling, 9649P) at a 1:1000 dilution in TBST buffer at 4 °C overnight. Finally, the membrane was incubated with HRP-conjugated goat anti-rabbit secondary antibody (Cell signaling, 7074S; 7:10000) at room temperature for 1 h. HRP activity was detected using an enhanced chemiluminescence light-based detection substrate, SuperSignal West Dura Extended Duration Substrate (ThermoFisher Scientific, 34075). Gel images in Figures 3 and S148 are representative of three independent trials.
4.3.5. In vitro cell growth inhibition
Jurkat or U937 cells were grown in RPMI media supplemented with 10% fetal calf serum and 1% penicillin/streptomycin under humidified conditions (37 °C, 5% CO2). Cells were seeded in 96-well cell culture plates with a density of 4×104 cells in 99 μL of media composed of RPMI-1640 (no phenol red), supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. The cells were treated with 1 μM or 10 μM single concentrations or serial dilution (2-fold) of compounds 1c, 1d, or 1f in DMSO (1 μL). DMSO only was used in the no inhibitor control. A negative control was also included where no cells were added. The plate was incubated for 44 hours at 37 °C in humid 5% CO2 atmosphere. A solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in PBS buffer (10 μL of 5 mg/mL) was added to each well. The cells were incubated for another 4 h at 37 °C in humid 5% CO2 atmosphere for development to take place. The resulting purple formazan crystals were dissolved by addition of DMSO (150 μL) and the absorbance was measured at 595 nm using a Geniosplus Fluorimeter (Tecan). For all the wells, the signal was background corrected with the signal from a negative control reaction (media and MTT only) before the percent viable cells was calculated. The percent viable cells was calculated by dividing the absorbance with inhibitor by the absorbance without inhibitor (DMSO, cells, and MTT). The assay was performed at least three independent times. For the single concentrations experiment, the mean percent viable cells, along with standard error of three independent trials, is reported in Figure 3. EC50 values were calculated by fitting the percent viable cells as a function of inhibitor concentration to a sigmoidal dose-response curve (y = 100/(1+(x/EC50)z), where y = percent viable cells and × = inhibitor concentration) using nonlinear regression with KaleidaGraph 4.1.3 software (Tables 2, S12, and Figures S150-S152).
4.4. Docking procedure
The AutoDock 4.2 and Autodock tools programs [52-53] were used to perform the docking studies. HDAC6 catalytic domain 2 (CD2) (PDB: 5G0H) [54] and HDAC3 (PDB: 4A69) [55] crystal structures were downloaded from the protein data bank. PyMOL program (Schrodinger, LLC) was used to delete the co-crystallized inhibitor (S-trichostatin A), ethylene glycol molecules, potassium ions and all water molecules in HDAC6 crystal structure. With HDAC3 crystal, chain A, deacetylase-activation-domain (DAD) (from the SMRT corepressor), glycerol, D-myo-inositol-1,4,5,6-tetrakisphosphate and glycerol molecules, acetate, potassium and sulfate ions, and all water molecules were deleted. Only the zinc atom remained in both crystal structures. AutoDockTools-1.5.4 program [52-53] was used to add all polar hydrogen atoms, modify histidine protonation (H573 and H574 residues of HDAC6, and H134 and H135 of HDAC3) by adding only HD1, compute Gasteiger charges, and merge all non polar hydrogen, followed by generation of the pdbqt output file. The charge of the zinc atom was manually changed from zero to +2. For HDAC6, a grid box with a spacing of 0.375 Å, size of 42 × 40 × 44, and coordinates for the center of the grid box (-13.000, -2.000, -5.000) were used. For HDAC3, a grid box of size 58 X 58 X 54 Å3 with a spacing of 0.375 Å and centered at (8.166, 76.663, 21.318) were used. The map type was set by choosing the ligand and then AutoGrid 4.2 was used to pre-calculate and generate the grid map files required for the docking calculations. All the docked compounds were drawn in ChemBioDraw Ultra 12.0, and MM2 energy minimization was done using Chem 3D Pro 12.0. Then AutoDockTools-1.5.4 program was used to add hydrogens, compute Gasteiger charges, merge non-polar hydrogens, choose torsions, and generate the pdbqt files. All acyclic bonds were made rotatable, except the amide bonds. The AutoDock 4.2 program was used to perform the docking calculations using a genetic algorithm. The generated pdbqt file for the enzymes were set as a rigid macromolecule and the genetic algorithm search parameters were set to 100 GA runs for each ligand with a population size of 150, a maximum number of 2.5 × 105 energy evaluations, a maximum number of 2.7 × 104 generations, a mutation rate of 0.2 and a crossover rate of 0.8. The docking parameters were set to default. All output DLG files were converted to pdbqt format and the results were visualized in PyMOL. Among the 100 docked poses generated, the lowest energy poses displaying optimal distances between the hydroxamic acid group of the inhibitor and the catalytic metal of the protein were discussed in the text.
Supplementary Material
Highlights.
-
-
Analogs of the non-selective FDA-approved drug SAHA (Vorinostat) demonstrated selectivity for HDAC6 and HDAC8 over HDAC1, 2, and 3
-
-
The best compound, (R)-C4-benzyl SAHA, displayed IC50 values of 48 and 27 nM with HDAC6 and HDAC8, respectively.
-
-
(R)-C4-benzyl SAHA maintained 520- to 1300-fold selectivity for HDAC6 and HDAC8.
-
-
The HDAC6 selectivity of C4-benzyl SAHA was reproduced in cancer cells
-
-
Docking studies provided a structural rationale for the HDAC6/HDAC8 selectivity of C4-benzyl SAHA
Acknowledgments
Funding Sources: We thank National Institutes of Health (GM121061) and Wayne State University for funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Lumigen Instrument Center at Wayne State University for NMR and MS instrumentation, Y. Ge for the U937 cell line, K. Honn and Y. Ahn for use of the Alpha Innotech FluorChem imaging systems, and I. Gomes, J. Knoff, D. Nalawansha, and Y. Zhang for comments on the manuscript.
Abbreviations
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
- NAD
Nicotinamide adenine dinucleotide
- DNA
deoxyribonucleic acid
- FDA
Food and Drug Administration
- ELISA
enzyme-linked immunosorbent assay
- TMSCl
Trimethylsilyl chloride
- DCM
Dichloromethane
- THF
Tetrahydrofuran
- NaH
Sodium hydride
- DCC
N, N'-Dicyclohexylcarbodiimide
- DMAP
4-Dimethylaminopyridine
- DIPEA
N,N-Diisopropylethylamine
- TBTU
N,N,N′,N′-Tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
- EDCI
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- NaHMDS
Sodium bis(trimethylsilyl)amide
- LiAlH4
Lithium aluminum hydride
- MSCl
Methanesulfonyl chloride
- LiCl
Lithium chloride
Footnotes
Author Contribution: A. Negmeldin synthesized all analogs and performed all experiments, except for testing tubastatin and BRD-73954 in the HeLa lysate activity assay and determining the cytotoxicity of tubastatin and PCI-34051 using the MTT assay, which were performed by J. Knoff. M. Pflum conceived of the project and assisted in experimental design and interpretation. A. Negmeldin and M. Pflum wrote the manuscript.
Supplementary material: Supplementary material (supporting synthesis scheme, compound characterization data, procedure and data for biological screening, IC50 curves and tables, and docking figures) associated with this article can be found with the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem, Int Ed. 2005;44(45):7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988;7(5):1395–1402. doi: 10.1002/j.1460-2075.1988.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nalawansha DA, Pflum MKH. LSD1 Substrate Binding and Gene Expression Are Affected by HDAC1-Mediated Deacetylation. ACS chemical biology. 2017;12(1):254–264. doi: 10.1021/acschembio.6b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalawansha DA, Gomes ID, Wambua MK, Pflum MKH. HDAC Inhibitor-Induced Mitotic Arrest Is Mediated by Eg5/KIF11 Acetylation. Cell Chemical Biology. 2017;24(4):481–492.e5. doi: 10.1016/j.chembiol.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregoretti I, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Chuang C, Pan J, Hawke DH, Lin SH, Yu-Lee LY. NudC deacetylation regulates mitotic progression. PLoS One. 2013;8(9):e73841. doi: 10.1371/journal.pone.0073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 8.Bartling B, Hofmann HS, Boettger T, Hansen G, Burdach S, Silber RE, Simm A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49(2):145–154. doi: 10.1016/j.lungcan.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S, Iwase H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast cancer research and treatment. 2005;94(1):11–6. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 10.Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other Class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281(19):13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 11.Huang BH, Laban M, Leung CHW, Lee L, Lee CK, Salto-Tellez M, Raju GC, Hooi SC. Inhibition of histone deacetylase 2 increases apoptosis and p21C ip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12(4):395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- 12.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15(1):91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 13.Oehme I, Deubzer HE, Lodrini M, Milde T, Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Invest Drugs. 2009;18(11):1605–1617. doi: 10.1517/14728220903241658. [DOI] [PubMed] [Google Scholar]
- 14.Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe H, Shibahara T, Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006;29(1):117–24. [PubMed] [Google Scholar]
- 15.Bazzaro M, Lin Z, Santillan A, Lee MK, Wang MC, Chan KC, Bristow RE, Mazitschek R, Bradner J, Roden RBS. Ubiquitin Proteasome System Stress Underlies Synergistic Killing of Ovarian Cancer Cells by Bortezomib and a Novel HDAC6 Inhibitor. Clinical Cancer Research. 2008;14(22):7340–7347. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn JS, Lee HY, Park CG, Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep. 2011;25(6):1677–81. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- 17.Rodd AL, Ververis K, Karagiannis TC. Current and emerging therapeutics for cutaneous T-Cell lymphoma: histone deacetylase inhibitors. Lymphoma. 2012;2012 [Google Scholar]
- 18.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 19.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warrell RP, Jr, He LZ, Richon V, Calleja E, Pandolfi PP. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J Natl Cancer Inst. 1998;90(21):1621–1625. doi: 10.1093/jnci/90.21.1621. [DOI] [PubMed] [Google Scholar]
- 21.Grant S, Easley C, Kirkpatrick P. Vorinostat. Nat Rev Drug Discovery. 2007;6(1):21–22. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- 22.Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2003;2(8):721–728. [PubMed] [Google Scholar]
- 23.Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21(21):4767–4773. doi: 10.1158/1078-0432.CCR-15-0530. [DOI] [PubMed] [Google Scholar]
- 24.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409(2):581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 25.Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I Study of an Oral Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid, in Patients With Advanced Cancer. Journal of Clinical Oncology. 2005;23(17):3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Clin Cancer Res. 2002;8:718–728. [PubMed] [Google Scholar]
- 27.Witter DJ, Harrington P, Wilson KJ, Chenard M, Fleming JC, Haines B, Kral AM, Secrist JP, Miller TA. Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18(2):726–731. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 28.Padige G, Negmeldin AT, Pflum MKH. Development of an ELISA-based HDAC activity assay for characterization of isoform-selective inhibitors. J Biomol Screening. 2015;20(10):1277–1285. doi: 10.1177/1087057115598118. [DOI] [PubMed] [Google Scholar]
- 29.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson DE, Wagner FF, Kaya T, Gale JP, Aidoud N, Davoine EL, Lazzaro F, Weiwer M, Zhang YL, Holson EB. Discovery of the first histone deacetylase 6/8 dual inhibitors. J Med Chem. 2013;56(11):4816–4820. doi: 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- 31.KrennHrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg Med Chem Lett. 2007;17(10):2874–2878. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH, Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112(4):981–9. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, Karp JE. Phase 1 and pharmacological study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109(7):2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregoreCortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23(17):3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinsey TA. Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. Journal of molecular and cellular cardiology. 2011;51(4):491–496. doi: 10.1016/j.yjmcc.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Tang G, Wong JC, Zhang W, Wang Z, Zhang N, Peng Z, Zhang Z, Rong Y, Li S, Zhang M, Yu L, Feng T, Zhang X, Wu X, Wu JZ, Chen L. Identification of a Novel Aminotetralin Class of HDAC6 and HDAC8 Selective Inhibitors. Journal of medicinal chemistry. 2014;57(19):8026–8034. doi: 10.1021/jm5008962. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues DA, Ferreira-Silva GÀ, Ferreira ACS, Fernandes RA, Kwee JK, Sant'Anna CMR, Ionta M, Fraga CAM. Design, Synthesis, and Pharmacological Evaluation of Novel N-Acylhydrazone Derivatives as Potent Histone Deacetylase 6/8 Dual Inhibitors. Journal of medicinal chemistry. 2016;59(2):655–670. doi: 10.1021/acs.jmedchem.5b01525. [DOI] [PubMed] [Google Scholar]
- 38.Bieliauskas A, Weerasinghe S, Pflum MH. Structural requirements of HDAC inhibitors: SAHA analogs functionalized adjacent to the hydroxamic acid. Bioorg Med Chem Lett. 2007;17(8):2216–2219. doi: 10.1016/j.bmcl.2007.01.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi SE, Weerasinghe SV, Pflum MK. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C3 position display isoform selectivity. Bioorg Med Chem Lett. 2011;21(20):6139–6142. doi: 10.1016/j.bmcl.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SE, Pflum MKH. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C6 position. Bioorg Med Chem Lett. 2012;22(23):7084–7086. doi: 10.1016/j.bmcl.2012.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negmeldin AT, Pflum MKH. The structural requirements of histone deacetylase inhibitors: SAHA analogs modified at the C5 position display dual HDAC6/8 selectivity. Bioorganic & Medicinal Chemistry Letters. 2017;27(15):3254–3258. 47. doi: 10.1016/j.bmcl.2017.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negmeldin AT, Padige G, Bieliauskas AV, Pflum MKH. Structural Requirements of HDAC Inhibitors: SAHA Analogues Modified at the C2 Position Display HDAC6/8 Selectivity. ACS Medicinal Chemistry Letters. 2017;8(3):281–286. doi: 10.1021/acsmedchemlett.6b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bieliauskas AV, Weerasinghe SVW, Negmeldin AT, Pflum MKH. Structural requirements of histone deacetylase inhibitors: SAHA analogs modified on the hydroxamic acid. Arch Pharm (Weinheim, Ger) 2016;349(5):373–382. doi: 10.1002/ardp.201500472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 45.Hackanson B, Rimmele L, Benkiβer M, Abdelkarim M, Fliegauf M, Jung M, Lübbert M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk Res. 2012;36(8):1055–1062. doi: 10.1016/j.leukres.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Inks ES, Li X, Hou J, Chou CJ, Zhang J, Jiang Y, Zhang Y, Xu W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57(8):3324–3341. doi: 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP. Selective Histone Deacetylase 6 Inhibitors Bearing Substituted Urea Linkers Inhibit Melanoma Cell Growth. Journal of medicinal chemistry. 2012;55(22):9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Senger J, Melesina J, Marek M, Romier C, Oehme I, Witt O, Sippl W, Jung M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J Med Chem. 2016;59(4):1545–1555. doi: 10.1021/acs.jmedchem.5b01493. [DOI] [PubMed] [Google Scholar]
- 49.Evans DA, Ennis MD, Mathre DJ. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of .alpha.-substituted carboxylic acid derivatives. Journal of the American Chemical Society. 1982;104(6):1737–1739. [Google Scholar]
- 50.Dale JA, Dull DL, Mosher HS. alpha.-Methoxy-.alpha.-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. The Journal of organic chemistry. 1969;34(9):2543–2549. [Google Scholar]
- 51.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. A General Model for Selectivity in Olefin Cross Metathesis. J Am Chem Soc. 2003;125(37):11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 52.Sanner MF. Python: a programming language for software integration and development. Journal of molecular graphics & modelling. 1999;17(1):57–61. [PubMed] [Google Scholar]
- 53.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol. 2016;12(9):748–754. doi: 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- 55.Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of HDAC3 bound to corepressor and inositol tetraphosphate. Nature. 2012;481(7381):335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fass DM, Shah R, Ghosh B, Hennig K, Norton S, Zhao WN, Reis SA, Klein PS, Mazitschek R, Maglathlin RL, Lewis TA, Haggarty SJ. Short-chain HDAC inhibitors differentially affect vertebrate development and neuronal chromatin. ACS Med Chem Lett. 2011;2(1):39–42. doi: 10.1021/ml1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gottlieb HE, Kotlyar V, Nudelman A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. The Journal of organic chemistry. 1997;62(21):7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 58.Altendorfer M, Raja A, Sasse F, Irschikc H, Menche D. Modular synthesis of polyene side chain analogues of the potent macrolide antibiotic etnangien by aflexible coupling strategy based on hetero-bis-metallated alkenes. Org Biomol Chem. 2013;11:2116. doi: 10.1039/c2ob26906f. [DOI] [PubMed] [Google Scholar]
- 59.Miller JF, Chong PY, Shotwell JB, Catalano JG, Tai VWF, Fang J, Banka AL, Roberts CD, Youngman M, Zhang H, Xiong Z, Mathis A, Pouliot JJ, Hamatake RK, Price DJ, Seal JW, Stroup LL, Creech KL, Carballo LH, Todd D, Spaltenstein A, Furst S, Hong Z, Peat AJ. Hepatitis C Replication Inhibitors That Target the Viral NS4B Protein. Journal of medicinal chemistry. 2014;57(5):2107–2120. doi: 10.1021/jm400125h. [DOI] [PubMed] [Google Scholar]
- 60.Kim DH, Chung S. Stereochemistry in enzyme inhibition: synthesis and evaluation of enantiomerically pure 2-benzyl-3-formylpropanoic acids as inhibitors of carboxypeptidase A. Tetrahedron: Asymmetry. 1999;10(19):3769–3776. [Google Scholar]
- 61.Tredwell M, Luft JAR, Schuler M, Tenza K, Houk KN, Gouverneur V. Fluorine-Directed Diastereoselective Iodocyclizations. Angewandte Chemie. 2008;120(2):363–366. doi: 10.1002/anie.200703465. [DOI] [PubMed] [Google Scholar]
- 62.Hutchison PC, Heightman TD, Procter DJ. Application of a Recyclable Pseudoephedrine Resin in Asymmetric Alkylations on Solid Phase. The Journal of organic chemistry. 2004;69(3):790–801. doi: 10.1021/jo0354950. [DOI] [PubMed] [Google Scholar]
- 63.Jöst C, Nitsche C, Scholz T, Roux L, Klein CD. Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments. Journal of medicinal chemistry. 2014;57(18):7590–7599. doi: 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.