

Summary
The role of enhancers, a key class of non-coding regulatory DNA elements, in cancer development has increasingly been appreciated. Here, we present the detection and characterization of a large number of expressed enhancers in a genome-wide analysis of 8928 tumor samples across 33 cancer types using TCGA RNA-seq data. Compared with matched normal tissues, global enhancer activation was observed in most cancers. Across cancer types, global enhancer activity was positively associated with aneuploidy, but not mutation load, suggesting a hypothesis centered on “chromatin-state” to explain their interplay. Integrating eQTL, mRNA co-expression, and Hi-C data analyses, we developed a computational method to infer causal enhancer-gene interactions, revealing enhancers of clinically actionable genes. Having identified an enhancer ~140 kb downstream of PD-L1, a major immunotherapy target, we validated it experimentally. This study provides a systematic view of enhancer activity in diverse tumor contexts and suggests the clinical implications of enhancers.
ETOC
Causal enhancer-target-gene relationships are inferred from a systematic analysis of 33 cancer types
Introduction
The biological functions of each cell component are controlled by a Russian-nesting-doll-like, multilevel gene-regulatory hierarchy that includes transcription-factor-promoter interaction, enhancer activation, DNA methylation, microRNA-mediated regulation, translation, and post-translational modification (He and Hannon, 2004; Jaenisch and Bird, 2003; Murakawa et al., 2016). In cancer cells, such regulatory networks are often rewired by molecular aberrations that collectively lead to the cancer phenotype (Chen et al., 2015; Kolch et al., 2015). For example, somatic mutations can modify the functions of both trans and cis elements in a regulatory network, thereby conferring cell behaviors related to tumorigenesis (Garraway and Lander, 2013; Hanahan and Weinberg, 2011). Using high-throughput molecular profiling techniques over large patient cohorts, The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network et al., 2013) has systematically characterized key molecular alterations at different levels in a broad range of cancer types, providing unprecedented insight into oncogenic mechanisms and potential therapeutic approaches.
However, our information on the rewiring of gene regulatory networks in cancer is far from complete, and enhancers represent a missing piece of the jigsaw puzzle (Aran and Hellman, 2013). Enhancers are important non-coding DNA elements that interact spatially with their target promoters to regulate downstream genes (Schmitt et al., 2016). As a major category of regulatory elements in cell development, enhancers also play critical roles in the oncogenic process (Murakawa et al., 2016). Despite recent systematic efforts, including genome-wide profiling of tissue and cell line collections (Encode Project Consortium, 2012; Hnisz et al., 2013; Roadmap Epigenomics et al., 2015) and a pan-cancer analysis of some super-enhancers (Zhang et al., 2016), a global view of enhancer activity in cancer is still lacking. That hole in our understanding is due in part to the technical difficulty of applying high-throughput techniques (e.g., ChIP-seq) to investigate enhancer activity using large patient sample cohorts.
The Functional Annotation of the Mammalian Genome (FANTOM) Project has generated large-scale, high-quality annotation of ~65000 enhancers expressed in the human genome across multiple tissues (Andersson et al., 2014). FANTOM thus provides an alternative solution for studying enhancer activities in cancer. An inactive enhancer is usually well organized by unmodified nucleosomes so it cannot be accessed by either transcription factors or polymerase. When an enhancer is primed for activation in response to signaling, its local chromatin is first modified (often by H3K4Me1) and becomes loose, rendering the motifs on the enhancer available to transcription factors and RNA polymerase. When the bound transcription factors fully activate the enhancer, usually with re-marked by H3K27Ac, the local chromatin is completely open, recruiting RNA polymerase to initiate transcription in both directions (Figure 1A) (Heinz et al., 2015; Li et al., 2016; Murakawa et al., 2016). Thus, the expression level of enhancer RNA molecules represents an essential signature of enhancer activation (Murakawa et al., 2016; Ren, 2010). Indeed, the expression of a substantial proportion of enhancers can be detected by RNA-seq (Andersson et al., 2014; De Santa et al., 2010; Djebali et al., 2012; Murakawa et al., 2016). Using the high-quality expressed enhancer annotations in the FANTOM project, we performed a pan-cancer analysis of enhancer expression using TCGA RNA-seq data on the premise that the expression of an enhancer approximately reflects its activity. We aimed (i) to describe the global pattern of enhancer expression in cancers; (ii) to understand how enhancer activation relates to other genomic aberrations and to the relevant underlying mechanisms; and (iii) to identify key enhancers and explore their potential clinical implications.
Figure 1. Overview of enhancer expression in TCGA RNA-seq data.

(A) When activated, expressed enhancers may generate RNA molecules detectable by RNA-seq. (B) The chromatin status of enhancers, TSSs of protein-coding, and lncRNA genes, as well as their flanking 1-kb regions. The y-axis shows the normalized ChIP-seq signals from the ENCODE bigwig files. (C) Transcription of enhancers and their flanking 2-kb sequences detected in TCGA RNA-seq dataset. The y-axis shows the average reads per million mapped (RPM) to the nucleotide at the relative position from an enhancer, as indicated on the x-axis. Flanking sequences that overlapped with known genes were excluded from the calculation. (D) Numbers of expressed enhancers in different cancer types. An enhancer was considered as expressed in a cancer type if observed in >10% of the samples. (E) Numbers of prognostic enhancers in different cancer types. For each enhancer, its correlation with patients’ survival times in a given cancer type was calculated using Cox regression. The p-value was subjected to multiple-testing correction with FDR = 0.05 as cut-off. (F) Comparison of the proportion of prognostic enhancers and coding genes across cancer types, given the same patient cohorts and FDR cutoffs as in E above. (G) The variation in enhancer expression within and across cancer types. (H) Global enhancer activation in cancer as determined through comparison of matched tumor-normal pairs. Thirteen cancer types with >10 tumor–normal pairs were considered. The y-axis shows changes in global enhancer expression (RPMtumor/RPMnormal −1)%; statistics were performed with paired t-test. See also Figure S1.
Results
Overview of enhancer expression in human cancers
An enhancer’s expression level has been used as an index of its activity (Cheng et al., 2015; Natoli and Andrau, 2012). The FANTOM Project annotated enhancers based on integration of chromatin modification, transcription factor binding, cap analysis of gene expression (CAGE)-seq data (Andersson et al., 2014), and TCGA generated gene expression data using RNA-seq. To ensure the quality of our analysis, we first assessed whether TCGA’s RNA-seq platform could effectively capture the transcriptional signals from the FANTOM enhancers. Through a series of filters, we identified 15808 detectable enhancers out of ~65000 FANTOM-annotated enhancers with a typical length of ~200 bp that do not overlap with other known transcribed elements, thereby avoiding potential signal contamination (Figure S1A, Table S1, STAR Methods). We examined the chromatin status of the flanking 1-kb sequences of those enhancers using several well-established factors, including DNase hypersensitivity, p300 binding, CTCF binding, H3K27Ac, and H3K4Me1 (Heintzman et al., 2007). That analysis revealed chromatin signatures similar to those identified by classical enhancers but in sharp contrast to those of the transcriptional start sites (TSSs) of protein-coding genes or lncRNAs. For example, H3K4Me1, a key marker that distinguishes enhancers from any other TSSs, was substantially enriched in our enhancer set, but showed a clear depletion in the TSSs of protein-coding genes (Figure 1B). The observed chromatin signatures were not affected by genomic annotation (Figure S1B). Those results confirm that our set consists principally of genuine enhancers, rather than other transcribed units or transcriptional noise.
In the TCGA RNA-seq data on 8928 cancer samples of 33 cancer types (Table S2), we observed a substantial number of RNA-seq reads uniquely assigned to the 15808 enhancers. Those mapped reads showed clear peaks centered on the enhancers identified (Figure 1C). That clear pattern confirmed the biological significance of our cross-platform integration and distinguished the signals from transcriptional noise. Another potential issue in our analysis of enhancers arose from the relatively low depth of RNA-seq data (Li et al., 2016). To evaluate the general utility of TCGA RNA-seq data for enhancer expression, we determined the numbers of enhancers with detectable expression in different cancer types. On average, 4591 enhancers were detected in >10% of the samples in a given cancer type, suggesting that enhancer expression signals detected contain considerable sampling power to represent cancer enhancer functions on a global scale (Figure 1D, Table S3). To evaluate further the clinical significance of the enhancer expression, we assessed correlations of the expression levels with patient survival in each of the 25 cancer types that had sufficient sample size and follow-up time. That analysis identified a few dozen to thousands of prognostic enhancers per cancer type, and some of them tended to correlate with prognosis across multiple cancer types (Figure 1E, Figure S1C, D, Table S1, Table S4). Given the same sample cohorts and FDR cutoff = 0.05, we compared the proportions of enhancers and protein-coding genes that showed prognostic significance. Interestingly, in most cancer types, the fraction of enhancers with prognostic power was comparable to, or even higher than, that of protein-coding genes (Figure 1F). Finally, we examined the variation in enhancer expression across cancer types based on global enhancer expression level (summarizing over all of the enhancers surveyed, in reads per million mapped reads [RPM]). Liver hepatocellular carcinoma (LIHC) showed the lowest global enhancer expression level (~100 RPM), and thymoma (THYM) showed the highest (~240 RPM). Within each cancer type, there was also large variation (as large as ~5-fold) among patient samples (Figure 1G). Compared with adjacent normal tissues from the same patients, most cancer types showed global enhancer activation (paired t-test, p < 0.05, for 13 cancer types that had >10 matched tumor-normal pairs, Figure 1H). Collectively, those results present a general picture of enhancer expression in human cancers and suggest that enhancer expression signals detected from TCGA RNA-seq data represent a biological dimension of interest, complementary to other genomic features. The enhancer expression datasets presented here provide a starting point for exploration by the research community.
Global enhancer activation in cancer is positively associated with tumor aneuploidy
Somatic copy-number alteration (SCNA) and point mutation are the two most common types of mutational events that impact the stability of a cancer genome. To explore the relationship between genomic instability and enhancer expression in cancer, we determined the correlations of global enhancer expression level (RPM) with the SCNA and mutation burdens in each cancer type. Specifically, for a given tumor sample, we used the aneuploidy value (defined as the fraction of the genome affected by SCNAs) from Affymetrix SNP6.0 arrays and the total number of silent somatic mutations from whole-exome sequencing to quantify the global SCNA level and mutation burden, respectively. For the majority of cancer types surveyed (19 out of 25 cancer types with ≥80 samples), the aneuploidy level showed a significantly positive correlation with the global enhancer activation level (Figure 2A). In contrast, point mutations showed no correlation or a slight negative correlation (Figure 2B). These results indicate complex relationships between enhancer activation and different types of genomic aberrations in cancer.
Figure 2. Enhancer expression is associated with different types of genomic aberration.

(A, B) Spearman’s correlation coefficient (rho) between global enhancer expression determined by RPM and (A) aneuploidy (measured as the proportion of the genome affected by SCNAs) or (B) mutation burden (measured as the number of silent exonic mutations). Significant correlations are colored. (C, D) Consensus clustering analysis identified three major enhancer expression subtypes. Within each cancer type, the log2RPKM values of 15808 enhancers were scaled to the Z-score before clustering to correct for tissue-specific patterns that would otherwise affect the clustering. Consensus clustering based on 1500 enhancers (~10%) with the highest coefficients of variation identified three major clusters. The Z-score matrix was projected onto the first three dimensions identified in principal component analysis, with colors representing the (C) three clusters or (D) cancer types. (E) Relative global enhancer expression level (RPM) of the three clusters in tumors compared with normal samples. Error bars show mean ± standard error (SE). Statistics were computed using t-test. Absolute RPM levels are shown at the top of each bar. (F) Numbers of enhancers detected in the three clusters (RPKM > 0.5). Error bars show mean ± standard error (SE). Statistics were computed using t-test. (G) Aneuploidy level in the three subtypes; sample proportions of 50% and 75% are in the box and within the limits, respectively. (H) Numbers of silent mutations in the three subtypes; sample proportions of 25% and 50% are in the box and within the limits, respectively. Kolmogorov-Smirnov p-values are shown. (I) Summary of genomic aberration profiles of the three subtypes. See also Figures S2 and S3.
To explore further the global patterns of enhancer expression across cancer types, we performed consensus clustering of tumor samples using the 1500 (~10% of 15808) enhancers with the highest coefficients of variation (CV) across samples (STAR Methods). Enhancer expression levels were Z-normalized within each cancer type to minimize tissue effects. The analysis resulted in three robust subtypes (clusters): C1, C2, and C3 were separated in the space of the top three principal components, and the clusters were not driven by disease type (Figure 2C, D, and Figure S2). Comparison of the enhancer-based subtypes with well-established pathological subtypes further supported the biological significance of the clustering (Figure S3A, B). The most distinct feature among the subtypes was their differential levels of global enhancer expression: the enhancer expression levels of the three subtypes were higher than those of their normal counterparts; subtype C2 showed the highest level of enhancer activation, 1.25 or 1.15-fold higher than that of normal or C1/C3 samples (Figure 2E). Notably, the 15% increase of global enhancer expression was a large effect, indicating >400 more activated enhancers in C2 than in C1/C3 samples, up to be ~30% of the active enhancers that a tumor usually has (Figure 2F). Consistent with the associations between aneuploidy and global enhancer expression within individual cancer types, ~25% of a typical C2 genome was affected by aneuploidy, 1.5-fold more than that of a typical C1 (16%) or C3 (15%) genome (Figure 2G), indicating thousands more genes perturbed by SCNAs. In terms of mutation burden, subtype C1 samples contained slightly more point mutations than subtype C2, and both C1 and C2 showed significantly higher mutation burdens than subtype C3 (Figure 2H). We also saw that some cancer driver genes appeared to be under strong positive selection in subtype C1 (Figure S3C, D). For example, C1 had a 1.5-fold higher TP53 truncation rate than did the others. The decoupling of point mutations and global enhancer activation in the above pan-cancer analysis is compatible with the patterns we observed within cancer types (Figure 2A, B). Taken together, the correlations of enhancer activation with genomic instability can be summarized as a tree (Figure 2I): subtype C1 was enriched with samples having high mutation load and low aneuploidy; subtype C2 was enriched with samples having high mutation load and high aneuploidy; and C3 was “normal-like,” with low mutation load and low aneuploidy. Both the analyses within cancer types and the analyses across cancer types indicate that SCNAs, but not point mutations, are positively associated with enhancer activation.
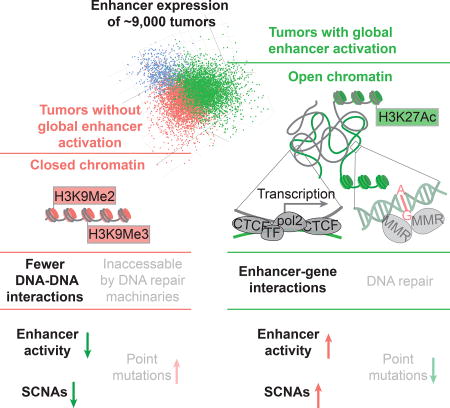
A “chromatin-state”-centered model for the interplay of enhancer activation, SCNAs, and point mutations
The above section raised a question of great interest: why do SCNAs and point mutations correlate with global enhancer activation in cancer differently (or even to some extent inversely in some cancer types)? Variations in chromatin organization of the human genome have been reported to be major determinants of the variation of somatic mutation rates across the genome (Schuster-Bockler and Lehner, 2012); low mutation rate is a feature of open DNA because of their accessibility by the DNA repair machineries (Figure 3A) (Polak et al., 2014). Interestingly, chromatin opening happens to be a prerequisite for enhancer activity (Figure 1A). Upon activation, enhancers loop to, and interact with, their target DNAs, creating topological DNA-DNA interactions (Figure 3A). Meanwhile, long-range DNA-DNA contacts physically increase the chance for loci far apart on the 1D–sequence to meet and rearrange with each other when breaks occur, generating structural alterations (Figure 3A) (Fudenberg et al., 2011). Those observations collectively suggest a molecular mechanism in which SCNAs and point mutations are differently associated with global enhancer activation patterns established through differences in the openness of chromatin. In that model, compact chromatin favors point mutation and keeps enhancers silent; once chromatin opens, enhancers are more likely to be activated, and because unfolded DNA is elongated by 1–2 orders of magnitudes, enabling long-range DNA-DNA interactions, which increase the chance of DNA rearrangements (SCNAs).
Figure 3. A “chromatin-state”-centered mechanistic model for the interplay among enhancer activation, SCNAs, and point mutations.

(A) Hypothetical impacts of chromatin state on the cancer genome. (B) Real correlations between genomic features across genomic regions. The human genome was divided into 2663 1-Mb fragments for correlation analysis. Enhancer activation level was defined as the mean RPKM of all enhancer regions within a fragment. The mutation rate and DNA double-strand break rate were calculated for each fragment using whole-genome data from COSMIC (STAR Methods). DNase hypersensitivity and histone-modifications were obtained from the ENCODE ChIP-seq dataset, and the density of DNA-DNA interactions was determined using Hi-C data (STAR Methods). Spearman’s correlation coefficients between genomic features were plotted as indicated. All correlations were of strong statistical significance (p < 10−16). (C) The top 500 10-kb human genome fragments with the highest breakpoint rates were considered as DSB hotspots, of which 204 and 296, respectively, were found inside and outside of the anchors of DNA loops detected by Hi-C. (D) The distribution of 15808 enhancers inside and outside of DNA loop anchors detected by Hi-C. (E) Hypothetical model demonstrating how chromatin opening favors DNA structural rearrangement. See also Figures S4.
To test that hypothetical model further, we used SCNA and mutation data from whole-genome sequencing to perform the analysis across different genomic regions, thereby providing evidence independent of that from our cross-sample analyses (Figure 3B, STAR Methods). A direct prediction of the model is that loose and compact chromatin regions in the cancer genome are dominated by SCNAs and mutations, respectively. Consistent with that prediction, in the cross-genomic-region analysis (using 1 Mb as a unit), DNA regions that featured markers of open chromatin (DNA hypersensitivity and H3K27Ac) were associated with higher rates of DNA double-strand breaks (DBSs) and lower mutation rates. Also as predicted, the relationship between chromatin state and enhancer expression was the same as that for chromatin state and DSBs. In contrast, closed chromatin, characterized by histone methylation (H3K9Me2 and H3K9Me3), displayed a strikingly opposite pattern (Figure 3B). We also observed a significantly negative association between point mutations and enhancer activation across genomic regions (rho = −0.46, n = 2663, p < 10−16). That correlation was much stronger than those in cross-sample comparisons (only 4 out of 25 cancer types showed significant negative correlations, Figure 2B). Those differences are probably due to genome-wide SCNA and mutation burdens positively correlated across tumor samples of different disease stages (Figure S4). Further, according to the model posited here, the long-range DNA-DNA interactions (Hi-C interactions) in open chromatin should at least partially explain the coincidence of enhancer activation and SCNAs across different genomic regions. To test that possibility, we examined the 500 10 kb-fragments of the human genome with the highest DSB rates. We found that ~40% (n = 204) of them overlapped with anchors of Hi-C loops, representing a >3-fold enrichment (p < 10−3, permutation test, Figure 3C). The same anchor regions also tended to overlap with the enhancers in our study (n = 15808; enrichment = 1.9 fold, p < 10−3; permutation test, Figure 3D). As summarized in Figure 3E, this hypothetical model provides a simple and reasonable, although tentative, explanation for differential associations of enhancer activation with SCNAs and with mutations.
Systematic identification of causal enhancer-cancer gene interactions
To elucidate the molecular functions of individual enhancers in cancer development and assess their clinical utility, it is important to identify their downstream target genes. Although new technologies like Hi-C (Jin et al., 2013) have been used to infer enhancer-gene interactions at the level of chromatin blocks (Dekker et al., 2013), computational methods that can accurately pinpoint target genes are seriously needed. Integrating enhancer and mRNA expression data, we can obtain candidate target genes of an enhancer through co-expression analysis. For a given enhancer-gene combination that is co-expressed, there are at least three possible relationship models: (1) a causal relationship, in which changes in the expression of the enhancer cause differential expression of the gene; (2) a reactive relationship, in which the gene is upstream of the enhancer; and (3) a co-responsive relationship, in which the enhancer and the gene are both responding to other molecular changes (Figure 4A). To distinguish the first (causal) model from the other two, we introduced expression quantitative trait loci (eQTL) information. The rationale was as follows: In the causal model, but not the other two, a single nucleotide polymorphism (SNP) that affects the enhancer’s activity would also affect expression of the enhancer’s downstream target gene, thereby making the SNP (or a nearby, genetically linked SNP) an eQTL of the gene of interest. Finally, for such enhancer-gene pairs, we can use recently available Hi-C data to assess whether the imputed causal relationship is likely (or not) to be direct regulation.
Figure 4. Systematic identification of causal enhancer/cancer-gene interactions.

(A) Three models of enhancer-gene co-expression pairs. (B) Bioinformatic method for inferring causal enhancer-gene interactions. (C) A network view for regulation of cancer genes by enhancers. Each arrow represents an interaction in the causal model in (A). (D) Number of genes in the network that contribute to the set of cancer hallmarks. (E) Number of positive enhancer-gene co-expressions in different steps of (B).
Following the logic above, we developed a computational method to identify potential causal/direct enhancer-gene regulations (Figure 4B). First, we selected enhancers with at least one nearby (< 500 bp) common SNP annotated in the 1000 Genomes Project (minor allele frequency >20%) or GTEx database (1000 Genomes Project Consortium, 2012; Lappalainen et al., 2013). We focused on a set of 822 cancer genes that combined clinically actionable genes (i.e., biomarkers or therapeutic targets) (n = 126), OncoKB (n = 476), and the Cancer Gene Consensus (CGC, n = 567) (Chakravarty et al., 2017; Futreal et al., 2004; Yuan et al., 2016). Second, we performed a co-expression analysis, which revealed ~40,000 associations between the enhancers and the genes in ≥4 of 33 TCGA cancer types (absolute Spearman’s rho > 0.3 and FDR < 10−4; STAR Methods). Third, we inferred casual relationships by examining whether the SNP on a given enhancer was an eQTL of the co-expressed gene in either the 1000 Genome or the GTEx dataset. We then integrated the long-range DNA-DNA interaction data from Hi-C (Rao et al., 2014) to predict whether causal relationships were acting through direct enhancer-gene contacts or through regulatory cascades. Specifically, if an enhancer and its co-expressed gene were located in one of the two anchors of a Hi-C DNA loop, the pair was considered as possible instances of direct regulation if the signal in the eQTL analysis was significant (p < 0.05). For enhancer-gene co-expression pairs without detected Hi-C interactions, we set a more stringent eQTL cutoff at (FDR < 10−4; with multiple comparison correction to account for all SNPs of a given enhancer) and considered them as instances of casual regulation.
Using that approach, we identified 65 such interactions involving 49 enhancers and 47 cancer genes, resulting in a predicted enhancer-gene regulatory network (Figure 4C). In total, 22 and 8 of the cancer genes were annotated, respectively, as oncogenes and tumor suppressor genes (TSG) in CGC, indicating a trend toward oncogene regulations (background Oncogenes: TSGs = 214: 198 in CGC, p = 0.037, chi-squared test, Figure 4C). Those 30 genes are involved in diverse cancer hallmarks with bias toward proliferation and metastasis, according to CGC annotations (Figure 4D). Interestingly, consistent with the bias toward oncogenes and the previously observed global enhancer activation, our network is strongly enriched with positive regulations, 43 of 54 (80%) and 11 of 11 (100%) for causal regulations and direct causal regulations, respectively (p < 0.01 for both enrichments, Figure 4E). Those results provide insight into the way in which global enhancer activation may contribute to tumor progression. They also identify individual enhancer-gene regulations that may be crucial in cancer development or clinical management.
Enhancers of actionable genes show potential clinical relevance
To investigate regulation by the enhancers identified as described above, we analyzed in detail an enhancer on chr22 (chr22:50980817–50981280, henceforth called enhancer-22) and its inferred target SYK. The ENCODE ChIP-seq dataset annotates a large number of protein-DNA interaction peaks within or flanking the enhancer-22 region (Figure 5A), suggesting its role as a hub in a regulatory network. Three genetically-linked SNPs are located in the enhancer, all of which are SYK eQTLs. For example, the T-allele of SNP rs5770772 was associated with higher expression of both enhancer-22 and SYK than the C-allele in the 1000 Genomes Project RNA-seq dataset (Figure 5B, 5C). When we analyzed the TCGA protein expression dataset, we confirmed that higher enhancer activity was associated with higher SYK protein levels (Figure 5D, 5E). SYK is an oncogenic driver that is activated in multiple types of late-stage cancer and is associated with poor clinical outcome (Puissant et al., 2014; Yu et al., 2015). Consistent with that relationship, our own analysis of patient survival times further supported the role of enhancer-22 as a marker of poor prognosis in several cancer types (Figure 5F–K, Figure S5). Since the eQTL, RNA-seq, and protein data were generated from independent platforms and the correlations were observed across multiple tissues of origin, these results provide evidence that enhancer-22 is a prognostic marker, largely through its effect on the downstream gene SYK.
Figure 5. Enhancer-22 as a prognostic marker across cancer types.

(A) Genomic context of enhancer-22 (chr22:50980817-50981280). (B) and (C) SNP rs5770772 is simultaneously a cis-eQTL of enhancer-22 and and trans eQTL of SYK. (D) Co-expression levels between enhancer-22 and SYK in multiple cancer types based on RNA-seq and reverse-phase protein array (RPPA) datasets; P-values calculated by Spearman’s rank correlation and Bonferroni-corrected. (E) Scatter plot showing co-expression between SYK protein level determined by RPPA and enhancer-22 expression level determined as log2RPKM. Kaplan-Meier plots for patient stratification based on enhancer-22 expression in (F) kidney renal cell clear cell carcinoma (KIRC), (G) low-grade glioma (LGG), (H) uveal melanoma (UVM), (I) uterine corpus endometrial carcinoma (UCEC), (J) thymoma (THYM), and (K) pancreatic adenocarcinoma (PAAD). P-values based on log-rank test are shown. See also Figure S5.
Besides prognostic markers, enhancers may serve as predictive markers for therapeutic response. PD-L1 plays a key role in a cancer’s escape from attack by the immune system and thus has been a major target of “check-point inhibition” immunotherapy, most prominently for lung cancer and melanoma (Topalian et al., 2016). In our enhancer-gene regulation network, we found an enhancer (chr9:5580709-5581016, hereafter called enhancer-9) located ~140kb from PD-L1. We observed strong co-expression between the PD-L1 mRNA level and the enhancer expression in multiple cancer types (Figure 6A). We then validated the co-expression in a cohort of 130 lung cancer cell lines from the Cancer Cell Line Encyclopedia database (Figure 6B). A PD-L1 eQTL close to the enhancer suggests the enhancer as an upstream regulator (Figure 6C). The Hi-C dataset from a panel of seven human cell lines further confirmed a direct interaction between the PD-L1 gene and the enhancer (Figure 6D, Figure S6A). Interestingly, out of the 161 transcription factors surveyed in the ENCODE project, NF-κB (measured with RELA/p65 ChIP-seq data) was the only one annotated for enhancer-9. Consistently, ChIP-seq data showed a strong NF-κB binding signal on the enhancer-9 and on the p65 binding motif of PD-L1’s promoter (Figure 6E), strongly suggesting that NF-κB complex is involved in the enhancer/PD-L1 interaction. Consistent with that idea, the NF-κB dimer has recently been reported to be essential to PD-L1 activation (Gowrishankar et al., 2015). To validate the causal effects of enhancer-9 on PD-L1 expression, we designed three pairs of single-guide CRISPR/Cas9 RNAs (sgRNAs) to delete enhancer-9. Using the most effective sgRNA pair, we obtained a stable lung cancer A549 cell line with the homozygous enhancer-9 deletion (Figure 6F, Figure S6B, and STAR Methods). As predicted, knockout of enhancer-9 substantially reduced PD-L1 expression (~10-fold) at both mRNA and protein levels (Figure 6G, H) and largely masked the inductive effect (~80% reduction) of INF-γ (activating NF-κB) on PD-L1 expression (Figure 6H, Figure S6C). Collectively, these results suggest an NF-κB-mediated enhancer-promoter interaction model of PD-L1 activation (Figure 6I). This example highlights the potentially important way in which enhancers can modulate key therapeutic targets.
Figure 6. Enhancer-9 regulates PD-L1, a key target of immunotherapy.

(A) Co-expression levels between enhancer-9 (chr9:5580709-5581016) and PD-L1 in multiple cancer types (RNA-seq); p-values for Spearman’s rank correlations were calculated and Bonferroni-corrected. (B) Scatter plot showing co-expression between PD-L1 mRNA level and enhancer-9 expression level. (C) SNP rs1536927 near enhancer 9 is a PD-L1 eQTL; p-value was calculated using ANOVA. (D) Direct interaction between PD-L1 gene body and enhancer 9 detected by Hi-C. The Hi-C O/E ratio was calculated as the median of O/E ratios of 7 human cell lines. (E) NF-κB ChIP-seq signals of enhancer-9 and the PD-L1 promoter. (F) Experimental design of sgRNA-guided enhancer perturbation by Cas9 protein. Three different sgRNAs were designed for each side of the enhancer. (G) Relative mRNA expression levels of PD-L1 in A549 cells and the same line after homozygous enhancer-9 deletion. Error bars show mean ± SE of results of 4 replicates; the difference was assessed using t-test. (H) PD-L1 protein levels in the control and enhancer-9 deletion cell lines without and with INF-γ stimulation. (I) Cartoon of NF-κB-mediated enhancer/promoter interaction for PD-L1 activation. See also Figure S6.
Discussion
Although the role of enhancers in cancer development has increasingly been recognized, genome-wide profiling studies on enhancer activity using conventional techniques (e.g., ChIP-seq) over large patient sample cohorts have not been done. Using TCGA RNA-seq data, we characterized the enhancer expression landscape in a broad range of cancer types. We observed global enhancer activation positively associated with large SCNAs but not point mutations, and proposed a model in which chromatin state is a key contributor to the observed genomic patterns. In contrast to closed chromatin, which favors point mutations (Polak et al., 2014), open DNA promotes structural rearrangements through long-range DNA-DNA interaction and activates enhancers by exposing them to transcription factors. The model provides insights into mutational landscape and clonal evolution. Epigenetic status, including histone modifications, nucleosome packaging, and DNA methylation could be precisely inherited during cell division (Probst et al., 2009). Variations in chromatin organization in a single tumor progenitor cell could, therefore, create striking differences among tumors if their effects are accumulated for many generations of cell growth. Chromatin organization could be substantially remodeled by histone gene mutations, which are frequently seen in cancers (Yuen and Knoepfler, 2013), or by chromatin remodeling events such as SMURF2 or HMGB1 loss of function (Blank et al., 2012; Celona et al., 2011) that globally loosen the structure of the genome (Schwab, 2009). Such events could create a high across-patient diversity of chromatin organization that may have crucial effects on genomic features such as SCNAs, point mutations, and enhancer activation. Thus, chromatin state could substantially shape the mutational landscape and at least partially explain an interesting but incompletely understood observation that tumors tend to be driven by either mutation (M class) or copy-number alteration (C class) (Ciriello et al., 2013). Due to the polyA selection and relatively low depth of TCGA RNA-seq data, our analysis only covered a proportion of enhancers in the human genome and may contain some noise. Therefore, further efforts using alternative technologies would be required to achieve a more comprehensive picture of enhancer activity in human cancers.
The global enhancer activation we observed in cancer samples (relative to those from normal issues, as shown in Figure 1H, Figure 2E) also provides insights into the clonal evolution of tumors. In contrast with microRNAs, which are dominantly down-regulated in cancer (Lu et al., 2005), enhancers are globally activated. Those up- and down-regulations both serve to up-regulate a large number of protein-coding genes, raising the question of their selective advantages. They may be a handy solution to stress in the short term of cancer evolution (Yona et al., 2012), in which activation of oncogenes and loss of TSGs are favored. However, given the nature of mutations, loss-of-function mutations are much easier to obtain than gain-of-function (activating) ones. As a result, cells with global gene activation could benefit from the advantages of oncogene activation. Meanwhile, the hitchhiked TSGs would be inactivated by loss-of-function mutations, which are more easily generated. Such an alternative may be particularly favored in cancer evolution (Yona et al., 2012).
The co-expression-based enhancer-gene regulatory network we infer here is of high value from the perspectives of both systems and translational research. ChIP-seq and CAGE-seq have proven to be powerful techniques for searching enhancers (Andersson et al., 2014; Park, 2009). A DNA-DNA interaction-based technique, Hi-C, has identified many enhancer-gene interactions (Schmitt et al., 2016). However, these interactions have limitations in that (1) no regulatory relationship behind the interaction is guaranteed, and (2) usually only low-resolution interactions are available. For example, a typical Hi-C result generates interactions at a resolution level of 10–50 kb, whereas a typical enhancer is only ~200 bp in length (Schmitt et al., 2016). Therefore, the method we propose, based on co-expression, eQTL, and Hi-C data integration, may be complementary to the other approaches. Due to limited data from matched tissues across different datasets, we combined signals from different tissues in our pipeline to infer causal enhancer regulations, which may lead to some false positives. Independent experiments are required to validate the proposed enhancer regulations. Our analysis reveals a considerable number of enhancers, including enhancer-9 for PD-L1, that are associated with clinically actionable genes, including the experimentally validated regulation of enhancer-9 for PD-L1. These results suggest a conceptually alternative strategy to inhibit key therapeutic targets, and further efforts are required to investigate the potential of enhancers in clinical applications.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Han Liang (hliang1@mdanderson.org).
Experimental Model and Subject Details
HEK (human embryonic kidney) 293T, HEK 293FT, and A549 cell lines were purchased from American Type Culture Collection (ATCC). HEK 293T and HEK 293FT cells were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) with 10% fetal bovine serum (Invitrogen) at 37 °C and 5% CO2. A549, A549-cas9, and the cell lines established based on A549-cas9 were maintained in RPMI-1640 with 10% fetal bovine serum at 37 °C and 5% CO2. The mycoplasma testing was confirmed to be negative for all the cell lines used in this study.
Method Details
Annotation of expressed enhancers in TCGA RNA-seq data
We obtained the information for 65423 FANTOM enhancers (Andersson et al., 2014) from http://fantom.gsc.riken.jp/5/datafiles/latest/extra/Enhancers/human_permissive_enhancers_phase_1_and_2.bed.gz. For enhancer expression analysis, we first re-annotated the 65423 FANTOM enhancers according to the University of California, Santa Cruz gene annotation file (refgene.txt), FANTOM transcription start sites and alternative polyadenylation sites (Andersson et al., 2014). We removed those that overlapped with known genes or intron regions, resulting in a subset of 15808 enhancers for which we could confidently assign the RNA-seq reads (Table S1). As a further layer of quality control, we extended the definition of known transcription events to all Ensembl transcripts (v90) and identified 3228 out of 15808 enhancers overlapped with at least one Ensembl transcript (Table S1). All annotations were based on the human genome build hg19. We obtained 8928 TCGA RNA-seq BAM files of 33 cancer types from CGHub or the NCI Genomic Data Commons Data Portal. All these files were based on hg19 and processed by the TCGA genomic data analysis center at the University of North Carolina.
Chromatin state analysis of enhancers
We obtained all experimental data of DNase hypersensitivity, p300 binding, CTCF binding, H3K4Me1, and H3K27Ac modification from the ENCODE data portal (https://www.encodeproject.org/matrix/?type=Experiment). All files meeting the following criteria were included in the analysis: (1) Format= BigWig; (2) Genome version= Hg19; (3) Signal type= fold-change over control. The ChIP signal of an interested DNA region was extracted from BigWig files using UCSC software bigWigAverageOverBed. We then chose the output column “mean” to represent the signal intensity. ChIP signals were measured on the whole length of DNA or flanking 25 bp for enhancers or TSSs, respectively. The TSSs loci were obtained from UCSC (refgene.txt). For any element of interest, we also obtained the signal on its flanking 1kb regions (window size: 50 bp; step size: 50 bp). The 41 data points (50 bp ×20 on up-stream sequence, 50 bp ×20 on down-stream sequence, and one of the elements of interest, itself) were normalized as Z-scores. We then calculated the mean signal across all aligned sequences in all cell lines to generate the chromatin status plots in Figure 1B and Figure S1B. To further confirm the quality of our enhancer set, we compared them with all transcripts annotated by Ensembl, which was more comprehensive but also noisier. We identified 3228 out of 15808 (~20%) enhancers overlapping with at least one Ensembl transcript, but detected no differences on chromatin status between the two groups of enhancers; thus, we retained the 3228 enhancers for further analysis.
For the genomic-region-based analysis, we first obtained a human genome benchmark from ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/analysis/NIST_union_callsets_06172013/union13callableMQonlymerged_addcert_nouncert_excludesimplerep_excludesegdups_excludedecoy_excludeRepSeqSTRs_noCNVs_v2.18_2mindatasets_5minYesNoRatio.bed.gz, which excluded the genomic regions that were ambiguous for mutation calling. We divided the human genome into 1Mb-sized fragments and only retained 2663 fragments with >50kb (5% of 1Mb) benchmark sequences. The ChIP signal of each fragment was measured as described above except that only benchmark sequences were considered. For Hi-C data, we obtained the DNA-DNA interaction loops from the Gene Expression Omnibus database (GSE63525)(Rao et al., 2014). For breakpoint and mutation data analysis, we downloaded the files CosmicGenomeScreensMutantExport.tsv.gz and CosmicNCV.tsv.gz from COSMIC FTP and retained only the data annotated as “WGS” (whole genome sequencing) in both files. The mutation rate and breakpoint rate were calculated in the benchmark regions on each 1Mb-sized fragment. For the double strand break (DSB) hotspots, we divided the human genome into 10kb-sized fragments, calculated the DSB rate in the same way as for the 1Mb-sized fragments, and defined the top 500 fragments as DSB hotspots in the human genome, with a minimal DSB rate of 1.6 breakpoints per kb.
Comparison of global enhancer activation between tumor and normal samples
For the tumor vs. normal tissue comparison, we considered a total of 13 cancer types with >10 tumor-normal sample pairs. For each sample, we measured its global enhancer expression level by counting the number of reads per million mapped reads [RPM] on the surveyed enhancers (n = 15808). The y-axis of Figure 1H was defined as (RPMTumor/RPMNormal-1)%, where RPMTumor (or RPMNormal) was the mean RPM cross all tumor (or normal) samples of a given cancer type. Statistics were performed using paired t-test.
Consensus clustering of enhancer expression profile
We calculated the log2RPKM values of the enhancers in TCGA samples as previously described (Li et al., 2015). We excluded 599 samples with a low sequence depth to reduce noise and included 8329 samples in the clustering analysis. Within each of the 33 cancer types, we scaled the expression level (log2RPKM) of each enhancer into a Z-score. The Z-scores across all samples of 33 cancer types were combined to calculate the CV. The 10% (1,500 of 15808) of the enhancers with the highest CVs were subjected to consensus clustering using the R package “ConsensusClusterPlus”. When the number of clusters (k) increased from 2 to 10, we observed little gain of area under the cumulative distribution function curve in consensus clustering after k = 3 (Figure S2B), which indicated that three clusters were identified in our analysis (Stefano Monti 2003).
Integrative analysis of enhancer expression with other molecular and clinical data
For genomic variations, the SCNA level of each tumor was obtained from the cBioportal feature “fraction of copy number altered genome,” while mutations (TCGA PanCanAtlas MC3) were obtained from the NCI Genomic Data Commons Data Portal. We used the number of silent mutations per exome to quantify the overall mutation load since non-silent mutations could be largely affected by positive selection. These statistics were obtained by using the Kruskal-Wallis test. Subtype information was obtained from the TCGA PanCanAtlas Pathway Analysis Working Group. The chi-squared test was used to test the independence of the subtype and enhancer cluster. For prognostic analysis, we first filtered the enhancers without detectable expression in >10% of the samples in each cancer type. For each of the remaining enhancers, we used the Cox regression model and log-rank test to determine the enhancer’s prognostic power. Enhancers associated with either the overall survival time or progression-free interval time (FDR < 0.05, John Storey’s correction, Table S4) were considered as prognostic enhancers.
Inference of causal enhancer–gene regulation
We first constructed a cancer-type–specific enhancer–gene co-expression network. Enhancers with common SNPs (minor allele frequency > 0.2) in the 1000 Genomes Project dataset (1000 Genomes Project Consortium, 2012) or GTEx dataset or within their flanking 500 bp sequences were considered in this analysis. We combined a group of actionable genes (n = 126), OncoKB with the Cancer Gene Consensus (n = 567) to be the cancer gene set in the analysis. In each cancer type, we used Spearman’s rank correlation model with Bonferroni’s correction for multiple comparisons to determine the enhancer-gene co-expression. Co-expression of an enhancer–gene pair was defined as significant co-expressions (absolute Spearman’s rho > 0.3 and FDR < 10−4) in at least four cancer types (>10% of the 33 cancer types). For each enhancer-gene co-expression pair, we then examined whether they contacted each other directly through long-range DNA-DNA loops. If they were located respectively on the two anchors on any DNA loops identified in GSE63525 (Rao et al., 2014), we considered the co-expression as a potential direct regulation, for which, we then tested if the SNP on this enhancer was an eQTL of the paired gene (p < 0.05). For other co-expression pairs without detected interaction through DNA loops, we applied a more stringent cutoff (FDR < 10−4) for the eQTL analysis. For both direct and indirect regulations, the p values in eQTL tests were corrected according to the number of SNPs associated with each enhancer. The RNA-seq and genotype data of the 1000 Genomes Project and GTEx were obtained from http://www.internationalgenome.org/category/rnaseq/ and dbGAP (phg000520), respectively (Lappalainen et al., 2013). For the GTEx dataset, 11 tissues with ≥ 80 samples were subjected to analysis: adipose_tissue, blood, blood_vessel, brain, esophagus, heart, lung, muscle, nerve, skin, and thyroid. For the eQTL analysis, we first artificially assigned the genotype scores of 1, 2, and 3 to individuals with genotypes of 0/0, 0/1, and 1/1, respectively. Spearman’s correlation between the genotype score and the gene’s expression was used to determine if the SNP was an eQTL of the gene. If more than one SNP was associated with an enhancer, the results were subjected to Bonferroni’s correction. Enhancer–gene pairs that survived the eQTL test were considered to have causal regulation.
Computational analysis of enhancer-9
We obtained the raw observation/expectation ratio (O/E ratio) representing the interaction level of distant genomic loci from GSE63525 (Rao et al., 2014). The raw O/E ratio was normalized according to the “readme” description file in the dataset. We then calculated the median O/E ratio of seven human cell lines of different tissue origins as the final interaction level, as presented in Figure 6C. All the ENCODE ChIP-seq bigwig files (including NF-κB ChIP-seq data) were obtained from the ENCODE data portal. The ChIP-seq signal intensities were extracted from bigwig files, and their mean values were used to measure the NF-κB binding affinity on the enhancer-9 or PD-L1 promoter.
CPRISPR/Cas9 genetic perturbation of enhancer-9
The single-guide RNA (sgRNA) sequences were designed using Cas-Designer (http://www.rgenome.net/cas-designer/) within 400 bp sequences flanking the enhancer region (chr9:5580709–5581016). Three gRNAs on either side of the enhancer were selected from the results, generating 9 (3×3) possible combinations of sgRNA pairs (Figure S6B). The upstream and downstream sgRNA sequences were synthesized and cloned into two CRISPR-Cas9 plasmids (Addgene 48138 and 64324) that respectively express GFP and mCherry as reporters. The sgRNA sequences were validated by Sanger sequencing after plasmid construction. The plasmids with upstream and downstream sgRNAs were mixed and then transfected using lipofectamine 3000 into the human embryonic kidney (HEK) 293T cell line grown in Dulbecco’s modification of Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C and 5% CO2. Three independent transfections were carried out for each pair of sgRNAs. Genetic perturbation efficiency was examined using polymerase chain reaction (PCR) six days after transfection. According to their efficiency, we chose sgRNA-L2 and sgRNA-R3 to generate the enhancer-9 homozygous deletion cell line. To obtain a constant cas9 expression cell line, we first packaged the cas9 plasmid (Addgene 73310) into lentivirus in the HEK 293FT cell line (grown in DMEM with 10% FBS), and then infected the human lung cancer cell line A549 (grown in RPMI-1640 with 10% FBS) by the lentivirus. After one week of blasticidin (25 µg/ml) selection, the A549-cas9 cell line was established. On day 0, 500 ng plasmids with sgRNAs or scramble controls were electroporated into A549-cas9 cell line using 100 µl tips at 1230V with 30ms width and 2 pulses (Neon Transfection System, Life Technologies). After three days of puromycin (4 µg/ml) selection, single cells were seeded into 96-well plates. During colony expansion, genotyping was carried out using KAPA Mouse Genotyping Kit (KAPA Biosystems) to screen single-cell clones with the deletion of enhancer-9. We obtained a homozygous enhancer-9 deletion cell line and confirmed the deletion region by Sanger sequencing. The mRNA and protein levels of PD-L1 expression after enhancer-9 deletion were measured by quantitative reverse-transcriptase PCR (qRT-PCR) and western-blot assay, respectively. One day after cell seeding, IFN-γ was added to the medium at the final concentration of 1 ng/ml. The expression level of PD-L1 was quantitated three days later. For mRNA expression level, RNA extraction was performed with RNeasy Plus Mini Kit (Qiagen). The high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) was used to reverse-transcribe 1 µg total RNA into cDNA. The SYBR select master mix (Thermo Fisher Scientific) and the Mastercycler RealPlex4 (Eppendorf) were used to perform qRT-PCR, which used the following primer sequences. For ACTB, the primer sequences were ATTGGCAATGAGCGGTTC/ CGTGGATGCCACAGGACT, and for PD-L1, the primer sequences were GCATTTACTGTCACGGTTCC/ TGCTGAACCTTCAGGTCTTC. For protein expression level, whole-cell lysates were extracted with RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, protease, and phosphatase inhibitor cocktails) and measured concentration using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). For each sample, 30 µg total protein was loaded into 4%-12% SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and depicted with Amersham ECL Western Blotting Detection Reagents (GE Healthcare Life Sciences). The following antibodies were used: PD-L1 (1:1000, Cell Signaling Technology, 13684T), ACTB (1:30,000, Sigma, A3854).
Quantification and Statistical Analysis
The analyses were based on 8928 tumor samples, except for the clustering analysis, which excluded 599 samples with a low read coverage, and the survival analysis, which excluded 8 cancer types with insufficient patient survival data. The definitions of significance for the various statistical tests are described and referenced in the respective Method Details sections.
Data and Software Availability
The TCGA data are available at https://gdc.cancer.gov/about-data/publications/pancanatlas.
Supplementary Material
(A) Processing pipeline of enhancers and tumor samples surveyed in this study. (B) Chromatin status of enhancers with/without overlapping of Ensembl transcripts (n = 3228/12580). Similar patterns were observed for the two groups. As a result, the 3228 enhancers were retained in the analysis. (C) Distribution of enhancers counted as prognostic in cancer types indicated on the x-axis. (D) Results in (C) divided by the expected number of recurrent prognostic enhancers calculated from permutation. Asterisk indicates p < 10−3 in the permutation.
Summary of enhancers detected in various cancer types; Related to Figure 1
Summary of prognostic enhancers in 25 cancer types with sufficient statistical power; Related to Figure 1
(A) Cumulative distribution functions (CDF) of consensus clustering results (k = 2, … , 10) of 8329 tumor samples. (B) Relative change in area under CDF curve when the number of clusters (k) equals 2 to 10. (C) Heatmap of consensus scores of 8329 tumors when k = 3 (k was set to 3 because the area under CDF increased only slightly when k > 3). (D) and (E) 3D scatter plot of the first three PCA components as in Figure 2CD. Each dot represents a tumor. (F) Heatmap of the consensus clustering using the 1500 enhancers with the highest CV across all samples.
(A) Global enhancer expression profile is associated with established cancer subtypes. Seven cancer types with >200 samples having available subtyping information and included in clustering analysis were considered. P-values were calculated by chi-squared test. Multiple comparisons were adjusted using Bonferroni’s correction. Each row on the left panel shows a BRCA sample, with its molecular subtype and enhancer expression cluster indicated by color. (B) Over/under-representation of the BRCA subtypes in the three clusters; p values calculated by chi-squared test. (C) Relative TP53 truncation rate of the three clusters. Absolute TP53 truncation rate was defined as the ratio of the number of TP53 truncations and the number of total silent mutations. Relative rates were normalized to the C2 rate. Error bars show mean ± SE. Statistics were computed using t-test. (D) We selected 7 tumor suppressors (blue) and 11 oncogenes (red) that are most frequently affected by SCNAs in cancer. A thresholded value of −2/2 was considered as deletion/amplification of a given tumor-suppressor or oncogene. Enrichment was calculated using chi-squared test.
(A) Scatter plot of number of point mutations (y) vs. level of SCNAs (x, measured as proportion of the genome affected by SCNAs) for all tumor samples (n = 8928). (B) Spearman’s correlation coefficient between number of point mutations and level of SCNAs in each of the 25 cancer types with ≥80 samples. Significant (p < 0.05) positive/negative correlations are marked by red/ blue, respectively.
This analysis is similar to Figure 5 H–K: progression-free interval (PFI) was considered for (A) PRAD and (B) KIRC; disease-specific survival time (DSS) was considered for (C) PAAD and (D) KIRC. Log-rank p values are shown.
(A) Chromatin interaction of PD-L1 and enhancer-9. This analysis is similar to Figure 6D, except that each of the seven cell types was considered separately. (B) sgRNAs used in the enhancer-9 genetic perturbation. Three sgRNAs were designed for each side of enhancer-9. Nine sgRNAs combinations (3×3) were subjected to efficiency examination. sgRNA L2 and R3 were selected for generating the cell line with the homozygous enhancer-9 deletion. (C) Relative mRNA expression level of PD-L1 in wild-type and the enhancer-9-delection cell line before and after INF-γ stimulation. Error bars show mean ± SE of the results of 4 replicates; significance levels of PD-L1 differential expression from the negative control (the wild-type A549 cell line without INF-γ stimulation) are indicated at the top of each bar; differences were assessed using t-test.
List of enhancers surveyed in this study; Related to Figure 1
Summary of TCGA samples used in this study; Related to Figure 1
Highlights.
Systematic analysis of enhancer expression across ~9,000 samples of 33 cancer types
Global enhancer activation positively correlated with aneuploidy but not mutations
A computational method that infers causal enhancer-target-gene relationships
Enhancers as key regulators of therapeutic targets, including PD-L1
Acknowledgments
This study was supported by grants from the U.S. National Institutes of Health (CA175486 and CA209851 to H.L., and CCSG grant CA016672); a grant from the Cancer Prevention and Research Institute of Texas (RP140462 to H.L.); a University of Texas System STARS award (to H.L.), and a training fellowship from the Gulf Coast Consortia, on the NLM Training Program in Biomedical Informatics and Data Science (T15 LM007093) to H.C, CAS-Sponsored Scholarship Program for Visiting Scholars (2015-40) to C.L. We thank the MD Anderson high-performance computing core facility for computing, Feng Yue for comments, and LeeAnn Chastain for editorial assistance.
Declaration of Interests
H.L. is a shareholder and scientific advisor of Precision Scientific Ltd., and Eagle Nebula Inc. From TCGA Research Network members: Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Andrew D. Cherniack receives research support from Bayer Pharmaceuticals AG. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Charles M. Perou is an equity stock holder, consultant, and Board of Director Member, of BioClassifier LLC and GeneCentric Diagnostics, and he is also listed an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer; is equity holder in, consultant for and Scientific Advisory Board chair for OrigiMed; Inventor on patent for EGFR mutation diagnosisi in lung cancer, licensed to LabCorp. Joshua M. Stuart is founder of Five3 Genomics and shareholder of Nantomics. Kyle R. Covington is an employee of Castle Biosciences Inc. Peter W. Laird is on Scientific Advisory Board for AnchorDx. Anil Sood is on Scientific Advisory Board for Kiyatec and shareholder in BioPath. Beth Y. Karlan is on Advisory Board of Invitae. Ashton C. Berger receives research support from Bayer Pharmaceuticals AG. Christina Yau is a part-time employee/consultant of NantOmics. Galen F. Gao receives research support from Bayer Pharmaceuticals AG.
Secondary author list
Amy Blum, Samantha J. Caesar-Johnson, John A. Demchok, Ina Felau, Melpomeni Kasapi, Martin L. Ferguson, Carolyn M. Hutter, Heidi J. Sofia, Roy Tarnuzzer, Peggy Wang, Zhining Wang, Liming Yang, Jean C. Zenklusen, Jiashan (Julia) Zhang, Sudha Chudamani, Jia Liu, Laxmi Lolla, Rashi Naresh, Todd Pihl, Qiang Sun, Yunhu Wan, Ye Wu, Juok Cho, Timothy DeFreitas, Scott Frazer, Nils Gehlenborg, Gad Getz, David I. Heiman, Jaegil Kim, Michael S. Lawrence, Pei Lin, Sam Meier, Michael S. Noble, Gordon Saksena, Doug Voet, Hailei Zhang, Brady Bernard, Nyasha Chambwe, Varsha Dhankani, Theo Knijnenburg, Roger Kramer, Kalle Leinonen, Yuexin Liu, Michael Miller, Sheila Reynolds, Ilya Shmulevich, Vesteinn Thorsson, Wei Zhang, Rehan Akbani, Bradley M. Broom, Apurva M. Hegde, Zhenlin Ju, Rupa S. Kanchi, Anil Korkut, Jun Li, Han Liang, Shiyun Ling, Wenbin Liu, Yiling Lu, Gordon B. Mills, Kwok-Shing Ng, Arvind Rao, Michael Ryan, Jing Wang, John N. Weinstein, Jiexin Zhang, Adam Abeshouse, Joshua Armenia, Debyani Chakravarty, Walid K. Chatila, Ino de Bruijn, Jianjiong Gao, Benjamin E. Gross, Zachary J. Heins, Ritika Kundra, Konnor La, Marc Ladanyi, Augustin Luna, Moriah G. Nissan, Angelica Ochoa, Sarah M. Phillips, Ed Reznik, Francisco Sanchez-Vega, Chris Sander, Nikolaus Schultz, Robert Sheridan, S. Onur Sumer, Yichao Sun, Barry S. Taylor, Jioajiao Wang, Hongxin Zhang, Pavana Anur, Myron Peto, Paul Spellman, Christopher Benz, Joshua M. Stuart, Christopher K. Wong, Christina Yau, D. Neil Hayes, Joel S. Parker, Matthew D. Wilkerson, Adrian Ally, Miruna Balasundaram, Reanne Bowlby, Denise Brooks, Rebecca Carlsen, Eric Chuah, Noreen Dhalla, Robert Holt, Steven J.M. Jones, Katayoon Kasaian, Darlene Lee, Yussanne Ma, Marco A. Marra, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Karen Mungall, A. Gordon Robertson, Sara Sadeghi, Jacqueline E. Schein, Payal Sipahimalani, Angela Tam, Nina Thiessen, Kane Tse, Tina Wong, Ashton C. Berger, Rameen Beroukhim, Andrew D. Cherniack, Carrie Cibulskis, Stacey B. Gabriel, Galen F. Gao, Gavin Ha, Matthew Meyerson, Steven E. Schumacher, Juliann Shih, Melanie H. Kucherlapati, Raju S. Kucherlapati, Stephen Baylin, Leslie Cope, Ludmila Danilova, Moiz S. Bootwalla, Phillip H. Lai, Dennis T. Maglinte, David J. Van Den Berg, Daniel J. Weisenberger, J. Todd Auman, Saianand Balu, Tom Bodenheimer, Cheng Fan, Katherine A. Hoadley, Alan P. Hoyle, Stuart R. Jefferys, Corbin D. Jones, Shaowu Meng, Piotr A. Mieczkowski, Lisle E. Mose, Amy H. Perou, Charles M. Perou, Jeffrey Roach, Yan Shi, Janae V. Simons, Tara Skelly, Matthew G. Soloway, Donghui Tan, Umadevi Veluvolu, Huihui Fan, Toshinori Hinoue, Peter W. Laird, Hui Shen, Wanding Zhou, Michelle Bellair, Kyle Chang, Kyle Covington, Chad J. Creighton, Huyen Dinh, HarshaVardhan Doddapaneni, Lawrence A. Donehower, Jennifer Drummond, Richard A. Gibbs, Robert Glenn, Walker Hale, Yi Han, Jianhong Hu, Viktoriya Korchina, Sandra Lee, Lora Lewis, Wei Li, Xiuping Liu, Margaret Morgan, Donna Morton, Donna Muzny, Jireh Santibanez, Margi Sheth, Eve Shinbrot, Linghua Wang, Min Wang, David A. Wheeler, Liu Xi, Fengmei Zhao, Julian Hess, Elizabeth L. Appelbaum, Matthew Bailey, Matthew G. Cordes, Li Ding, Catrina C. Fronick, Lucinda A. Fulton, Robert S. Fulton, Cyriac Kandoth, Elaine R. Mardis, Michael D. McLellan, Christopher A. Miller, Heather K. Schmidt, Richard K. Wilson, Daniel Crain, Erin Curley, Johanna Gardner, Kevin Lau, David Mallery, Scott Morris, Joseph Paulauskis, Robert Penny, Candace Shelton, Troy Shelton, Mark Sherman, Eric Thompson, Peggy Yena, Jay Bowen, Julie M. Gastier-Foster, Mark Gerken, Kristen M. Leraas, Tara M. Lichtenberg, Nilsa C. Ramirez, Lisa Wise, Erik Zmuda, Niall Corcoran, Tony Costello, Christopher Hovens, Andre L. Carvalho, Ana C. de Carvalho, José H. Fregnani, Adhemar Longatto-Filho, Rui M. Reis, Cristovam Scapulatempo-Neto, Henrique C.S. Silveira, Daniel O. Vidal, Andrew Burnette, Jennifer Eschbacher, Beth Hermes, Ardene Noss, Rosy Singh, Matthew L. Anderson, Patricia D. Castro, Michael Ittmann, David Huntsman, Bernard Kohl, Xuan Le, Richard Thorp, Chris Andry, Elizabeth R. Duffy, Vladimir Lyadov, Oxana Paklina, Galiya Setdikova, Alexey Shabunin, Mikhail Tavobilov, Christopher McPherson, Ronald Warnick, Ross Berkowitz, Daniel Cramer, Colleen Feltmate, Neil Horowitz, Adam Kibel, Michael Muto, Chandrajit P. Raut, Andrei Malykh, Jill S. Barnholtz-Sloan, Wendi Barrett, Karen Devine, Jordonna Fulop, Quinn T. Ostrom, Kristen Shimmel, Yingli Wolinsky, Andrew E. Sloan, Agostino De Rose, Felice Giuliante, Marc Goodman, Beth Y. Karlan, Curt H. Hagedorn, John Eckman, Jodi Harr, Jerome Myers, Kelinda Tucker, Leigh Anne Zach, Brenda Deyarmin, Hai Hu, Leonid Kvecher, Caroline Larson, Richard J. Mural, Stella Somiari, Ales Vicha, Tomas Zelinka, Joseph Bennett, Mary Iacocca, Brenda Rabeno, Patricia Swanson, Mathieu Latour, Louis Lacombe, Bernard Têtu, Alain Bergeron, Mary McGraw, Susan M. Staugaitis, John Chabot, Hanina Hibshoosh, Antonia Sepulveda, Tao Su, Timothy Wang, Olga Potapova, Olga Voronina, Laurence Desjardins, Odette Mariani, Sergio Roman-Roman, Xavier Sastre, Marc-Henri Stern, Feixiong Cheng, Sabina Signoretti, Andrew Berchuck, Darell Bigner, Eric Lipp, Jeffrey Marks, Shannon McCall, Roger McLendon, Angeles Secord, Alexis Sharp, Madhusmita Behera, Daniel J. Brat, Amy Chen, Keith Delman, Seth Force, Fadlo Khuri, Kelly Magliocca, Shishir Maithel, Jeffrey J. Olson, Taofeek Owonikoko, Alan Pickens, Suresh Ramalingam, Dong M. Shin, Gabriel Sica, Erwin G. Van Meir, Hongzheng Zhang, Wil Eijckenboom, Ad Gillis, Esther Korpershoek, Leendert Looijenga, Wolter Oosterhuis, Hans Stoop, Kim E. van Kessel, Ellen C. Zwarthoff, Chiara Calatozzolo, Lucia Cuppini, Stefania Cuzzubbo, Francesco DiMeco, Gaetano Finocchiaro, Luca Mattei, Alessandro Perin, Bianca Pollo, Chu Chen, John Houck, Pawadee Lohavanichbutr, Arndt Hartmann, Christine Stoehr, Robert Stoehr, Helge Taubert, Sven Wach, Bernd Wullich, Witold Kycler, Dawid Murawa, Maciej Wiznerowicz, Ki Chung, W. Jeffrey Edenfield, Julie Martin, Eric Baudin, Glenn Bubley, Raphael Bueno, Assunta De Rienzo, William G. Richards, Steven Kalkanis, Tom Mikkelsen, Houtan Noushmehr, Lisa Scarpace, Nicolas Girard, Marta Aymerich, Elias Campo, Eva Giné, Armando López Guillermo, Nguyen Van Bang, Phan Thi Hanh, Bui Duc Phu, Yufang Tang, Howard Colman, Kimberley Evason, Peter R. Dottino, John A. Martignetti, Hani Gabra, Hartmut Juhl, Teniola Akeredolu, Serghei Stepa, Dave Hoon, Keunsoo Ahn, Koo Jeong Kang, Felix Beuschlein, Anne Breggia, Michael Birrer, Debra Bell, Mitesh Borad, Alan H. Bryce, Erik Castle, Vishal Chandan, John Cheville, John A. Copland, Michael Farnell, Thomas Flotte, Nasra Giama, Thai Ho, Michael Kendrick, Jean-Pierre Kocher, Karla Kopp, Catherine Moser, David Nagorney, Daniel O’Brien, Brian Patrick O’Neill, Tushar Patel, Gloria Petersen, Florencia Que, Michael Rivera, Lewis Roberts, Robert Smallridge, Thomas Smyrk, Melissa Stanton, R. Houston Thompson, Michael Torbenson, Ju Dong Yang, Lizhi Zhang, Fadi Brimo, Jaffer A. Ajani, Ana Maria Angulo Gonzalez, Carmen Behrens, Jolanta Bondaruk, Russell Broaddus, Bogdan Czerniak, Bita Esmaeli, Junya Fujimoto, Jeffrey Gershenwald, Charles Guo, Alexander J. Lazar, Christopher Logothetis, Funda Meric-Bernstam, Cesar Moran, Lois Ramondetta, David Rice, Anil Sood, Pheroze Tamboli, Timothy Thompson, Patricia Troncoso, Anne Tsao, Ignacio Wistuba, Candace Carter, Lauren Haydu, Peter Hersey, Valerie Jakrot, Hojabr Kakavand, Richard Kefford, Kenneth Lee, Georgina Long, Graham Mann, Michael Quinn, Robyn Saw, Richard Scolyer, Kerwin Shannon, Andrew Spillane, Jonathan Stretch, Maria Synott, John Thompson, James Wilmott, Hikmat Al-Ahmadie, Timothy A. Chan, Ronald Ghossein, Anuradha Gopalan, Douglas A. Levine, Victor Reuter, Samuel Singer, Bhuvanesh Singh, Nguyen Viet Tien, Thomas Broudy, Cyrus Mirsaidi, Praveen Nair, Paul Drwiega, Judy Miller, Jennifer Smith, Howard Zaren, Joong-Won Park, Nguyen Phi Hung, Electron Kebebew, W. Marston Linehan, Adam R. Metwalli, Karel Pacak, Peter A. Pinto, Mark Schiffman, Laura S. Schmidt, Cathy D. Vocke, Nicolas Wentzensen, Robert Worrell, Hannah Yang, Marc Moncrieff, Chandra Goparaju, Jonathan Melamed, Harvey Pass, Natalia Botnariuc, Irina Caraman, Mircea Cernat, Inga Chemencedji, Adrian Clipca, Serghei Doruc, Ghenadie Gorincioi, Sergiu Mura, Maria Pirtac, Irina Stancul, Diana Tcaciuc, Monique Albert, Iakovina Alexopoulou, Angel Arnaout, John Bartlett, Jay Engel, Sebastien Gilbert, Jeremy Parfitt, Harman Sekhon, George Thomas, Doris M. Rassl, Robert C. Rintoul, Carlo Bifulco, Raina Tamakawa, Walter Urba, Nicholas Hayward, Henri Timmers, Anna Antenucci, Francesco Facciolo, Gianluca Grazi, Mirella Marino, Roberta Merola, Ronald de Krijger, Anne-Paule Gimenez-Roqueplo, Alain Piché, Simone Chevalier, Ginette McKercher, Kivanc Birsoy, Gene Barnett, Cathy Brewer, Carol Farver, Theresa Naska, Nathan A. Pennell, Daniel Raymond, Cathy Schilero, Kathy Smolenski, Felicia Williams, Carl Morrison, Jeffrey A. Borgia, Michael J. Liptay, Mark Pool, Christopher W. Seder, Kerstin Junker, Larsson Omberg, Mikhail Dinkin, George Manikhas, Domenico Alvaro, Maria Consiglia Bragazzi, Vincenzo Cardinale, Guido Carpino, Eugenio Gaudio, David Chesla, Sandra Cottingham, Michael Dubina, Fedor Moiseenko, Renumathy Dhanasekaran, Karl-Friedrich Becker, Klaus-Peter Janssen, Julia Slotta-Huspenina, Mohamed H. Abdel-Rahman, Dina Aziz, Sue Bell, Colleen M. Cebulla, Amy Davis, Rebecca Duell, J. Bradley Elder, Joe Hilty, Bahavna Kumar, James Lang, Norman L. Lehman, Randy Mandt, Phuong Nguyen, Robert Pilarski, Karan Rai, Lynn Schoenfield, Kelly Senecal, Paul Wakely, Paul Hansen, Ronald Lechan, James Powers, Arthur Tischler, William E. Grizzle, Katherine C. Sexton, Alison Kastl, Joel Henderson, Sima Porten, Jens Waldmann, Martin Fassnacht, Sylvia L. Asa, Dirk Schadendorf, Marta Couce, Markus Graefen, Hartwig Huland, Guido Sauter, Thorsten Schlomm, Ronald Simon, Pierre Tennstedt, Oluwole Olabode, Mark Nelson, Oliver Bathe, Peter R. Carroll, June M. Chan, Philip Disaia, Pat Glenn, Robin K. Kelley, Charles N. Landen, Joanna Phillips, Michael Prados, Jeff Simko, Jeffry Simko, Karen Smith-McCune, Scott VandenBerg, Kevin Roggin, Ashley Fehrenbach, Ady Kendler, Suzanne Sifri, Ruth Steele, Antonio Jimeno, Francis Carey, Ian Forgie, Massimo Mannelli, Michael Carney, Brenda Hernandez, Benito Campos, Christel Herold-Mende, Christin Jungk, Andreas Unterberg, Andreas von Deimling, Aaron Bossler, Joseph Galbraith, Laura Jacobus, Michael Knudson, Tina Knutson, Deqin Ma, Mohammed Milhem, Rita Sigmund, Andrew K. Godwin, Rashna Madan, Howard G. Rosenthal, Clement Adebamowo, Sally N. Adebamowo, Alex Boussioutas, David Beer, Thomas Giordano, Anne-Marie Mes-Masson, Fred Saad, Therese Bocklage, Lisa Landrum, Robert Mannel, Kathleen Moore, Katherine Moxley, Russel Postier, Joan Walker, Rosemary Zuna, Michael Feldman, Federico Valdivieso, Rajiv Dhir, James Luketich, Edna M. Mora Pinero, Mario Quintero-Aguilo, Carlos Gilberto Carlotti, Jr., Jose Sebastião Dos Santos, Rafael Kemp, Ajith Sankarankuty, Daniela Tirapelli, James Catto, Kathy Agnew, Elizabeth Swisher, Jenette Creaney, Bruce Robinson, Carl Simon Shelley, Eryn M. Godwin, Sara Kendall, Cassaundra Shipman, Carol Bradford, Thomas Carey, Andrea Haddad, Jeffey Moyer, Lisa Peterson, Mark Prince, Laura Rozek, Gregory Wolf, Rayleen Bowman, Kwun M. Fong, Ian Yang, Robert Korst, W. Kimryn Rathmell, J. Leigh Fantacone-Campbell, Jeffrey A. Hooke, Albert J. Kovatich, Craig D. Shriver, John DiPersio, Bettina Drake, Ramaswamy Govindan, Sharon Heath, Timothy Ley, Brian Van Tine, Peter Westervelt, Mark A. Rubin, Jung Il Lee, Natália D. Aredes, Armaz Mariamidze, Anant Agrawal, Jaeil Ahn, Jordan Aissiou, Dimitris Anastassiou, Jesper B. Andersen, Jurandyr M. Andrade, Marco Antoniotti, Jon C. Aster, Donald Ayer, Matthew H. Bailey, Rohan Bareja, Adam J. Bass, Azfar Basunia, Oliver F. Bathe, Rebecca Batiste, Oliver Bear Don’t Walk, Davide Bedognetti, Gloria Bertoli, Denis Bertrand, Bhavneet Bhinder, Gianluca Bontempi, Dante Bortone, Donald P. Bottaro, Paul Boutros, Kevin Brennan, Chaya Brodie, Scott Brown, Susan Bullman, Silvia Buonamici, Tomasz Burzykowski, Lauren Averett Byers, Fernando Camargo, Joshua D. Campbell, Francisco J. Candido dos Reis, Shaolong Cao, Maria Cardenas, Helio H.A. Carrara, Isabella Castiglioni, Anavaleria Castro, Claudia Cava, Michele Ceccarelli, Shengjie Chai, Kridsadakorn Chaichoompu, Matthew T. Chang, Han Chen, Haoran Chen, Hu Chen, Jian Chen, Jianhong Chen, Ken Chen, Ting-Wen Chen, Zhong Chen, Zhongyuan Chen, Hui Cheng, Hua-Sheng Chiu, Cai Chunhui, Giovanni Ciriello, Cristian Coarfa, Antonio Colaprico, Lee Cooper, Daniel Cui Zhou, Aedin C. Culhane, Christina Curtis, Patrycja Czerwińska, Aditya Deshpande, Lixia Diao, Michael Dill, Di Du, Charles G. Eberhart, James A. Eddy, Robert N. Eisenman, Mohammed Elanbari, Olivier Elemento, Kyle Ellrott, Manel Esteller, Farshad Farshidfar, Bin Feng, Camila Ferreira de Souza, Esla R. Flores, Steven Foltz, Mitchell T. Frederick, Qingsong Gao, Carl M. Gay, Zhongqi Ge, Andrew J. Gentles, Olivier Gevaert, David L. Gibbs, Adam Godzik, Abel Gonzalez-Perez, Marc T. Goodman, Dmitry A. Gordenin, Carla Grandori, Alex Graudenzi, Casey Greene, Justin Guinney, Margaret L. Gulley, Preethi H. Gunaratne, A. Ari Hakimi, Peter Hammerman, Leng Han, Holger Heyn, Le Hou, Donglei Hu, Kuan-lin Huang, Joerg Huelsken, Scott Huntsman, Peter Hurlin, Matthias Hüser, Antonio Iavarone, Marcin Imielinski, Mirazul Islam, Jacek Jassem, Peilin Jia, Cigall Kadoch, Andre Kahles, Benny Kaipparettu, Bozena Kaminska, Havish Kantheti, Rachel Karchin, Mostafa Karimi, Ekta Khurana, Pora Kim, Leszek J. Klimczak, Jia Yu Koh, Alexander Krasnitz, Nicole Kuderer, Tahsin Kurc, David J. Kwiatkowski, Teresa Laguna, Martin Lang, Anna Lasorella, Thuc D. Le, Adrian V. Lee, Ju-Seog Lee, Steve Lefever, Kjong Lehmann, Jake Leighton, Chunyan Li, Lei Li, Shulin Li, David Liu, Eric Minwei Liu, Jianfang Liu, Rongjie Liu, Yang Liu, William J.R. Longabaugh, Nuria Lopez-Bigas, Li Ma, Wencai Ma, Karen MacKenzie, Andrzej Mackiewicz, Dejan Maglic, Raunaq Malhotra, Tathiane M. Malta, Calena Marchand, R. Jay Mashl, Sylwia Mazurek, Pieter Mestdagh, Chase Miller, Marco Mina, Lopa Mishra, Younes Mokrab, Raymond Monnat, Jr., Nate Moore, Nathanael Moore, Loris Mularoni, Niranjan Nagarajan, Aaron M. Newman, Vu Nguyen, Michael L. Nickerson, Akinyemi I. Ojesina, Catharina Olsen, Sandra Orsulic, Tai-Hsien Ou Yang, James Palacino, Yinghong Pan, Elena Papaleo, Sagar Patil, Chandra Sekhar Pedamallu, Shouyong Peng, Xinxin Peng, Arjun Pennathur, Curtis R. Pickering, Christopher L. Plaisier, Laila Poisson, Eduard Porta-Pardo, Marcos Prunello, John L. Pulice, Charles Rabkin, Janet S. Rader, Kimal Rajapakshe, Aruna Ramachandran, Shuyun Rao, Xiayu Rao, Benjamin J. Raphael, Gunnar Rätsch, Brendan Reardon, Christopher J. Ricketts, Jason Roszik, Carlota Rubio-Perez, Ryan Russell, Anil Rustgi, Russell Ryan, Mohamad Saad, Thais Sabedot, Joel Saltz, Dimitris Samaras, Franz X. Schaub, Barbara G. Schneider, Adam Scott, Michael Seiler, Sara Selitsky, Sohini Sengupta, Jose A. Seoane, Jonathan S. Serody, Reid Shaw, Yang Shen, Tiago Silva, Pankaj Singh, I.K. Ashok Sivakumar, Christof Smith, Artem Sokolov, Junyan Song, Pavel Sumazin, Yutong Sun, Chayaporn Suphavilai, Najeeb Syed, David Tamborero, Alison M. Taylor, Teng Teng, Daniel G. Tiezzi, Collin Tokheim, Nora Toussaint, Mihir Trivedi, Kenneth T. Tsai, Aaron D. Tward, Eliezer Van Allen, John S. Van Arnam, Kristel Van Steen, Carter Van Waes, Christopher P. Vellano, Benjamin Vincent, Nam S. Vo, Vonn Walter, Chen Wang, Fang Wang, Jiayin Wang, Sophia Wang, Wenyi Wang, Yue Wang, Yumeng Wang, Zehua Wang, Zeya Wang, Zixing Wang, Gregory Way, Amila Weerasinghe, Michael Wells, Michael C. Wendl, Cecilia Williams, Joseph Willis, Denise Wolf, Karen Wong, Yonghong Xiao, Lu Xinghua, Bo Yang, Da Yang, Liuqing Yang, Kai Ye, Hiroyuki Yoshida, Lihua Yu, Sobia Zaidi, Huiwen Zhang, Min Zhang, Xiaoyang Zhang, Tianhao Zhao, Wei Zhao, Zhongming Zhao, Tian Zheng, Jane Zhou, Zhicheng Zhou, Hongtu Zhu, Ping Zhu, Michael T. Zimmermann, Elad Ziv, and Patrick A. Zweidler-McKay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
H.C. and H.L. conceived and designed the study; H.C., C.L., X.P., and H.L. performed data analysis; C.L. and Z.Z. performed experiments; H.C., C.L., J.N.W. and H.L. wrote the manuscript with inputs from other authors; and H.L. supervised the whole project.
References
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11–13. doi: 10.1016/j.cell.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nature medicine. 2012;18:227–234. doi: 10.1038/nm.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celona B, Weiner A, Di Felice F, Mancuso FM, Cesarini E, Rossi RL, Gregory L, Baban D, Rossetti G, Grianti P, et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS biology. 2011;9:e1001086. doi: 10.1371/journal.pbio.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017. 2017 doi: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin F, Xing K, He X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nature communications. 2015;6:6367. doi: 10.1038/ncomms7367. [DOI] [PubMed] [Google Scholar]
- Cheng JH, Pan DZ, Tsai ZT, Tsai HK. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Scientific reports. 2015;5:12648. doi: 10.1038/srep12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS biology. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature reviews Genetics. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Getz G, Meyerson M, Mirny LA. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature biotechnology. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PloS one. 2015;10:e0123410. doi: 10.1371/journal.pone.0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature reviews Genetics. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;(33 Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Halasz M, Granovskaya M, Kholodenko BN. The dynamic control of signal transduction networks in cancer cells. Nature reviews Cancer. 2015;15:515–527. doi: 10.1038/nrc3983. [DOI] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, ‘t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN, Liang H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer research. 2015;75:3728–3737. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Murakawa Y, Yoshihara M, Kawaji H, Nishikawa M, Zayed H, Suzuki H, Fantom C, Hayashizaki Y. Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends in genetics : TIG. 2016;32:76–88. doi: 10.1016/j.tig.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annual review of genetics. 2012;46:1–19. doi: 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nature reviews Genetics. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nature reviews Molecular cell biology. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, Garraway LA, Mirkin S, Getz G, Stamatoyannopoulos JA, et al. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nautre Biotecnology. 2014;32:71–75. doi: 10.1038/nbt.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Fenouille N, Alexe G, Pikman Y, Bassil CF, Mehta S, Du J, Kazi JU, Luciano F, Ronnstrand L, et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer cell. 2014;25:226–242. doi: 10.1016/j.ccr.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B. Transcription: Enhancers make non-coding RNA. Nature. 2010;465:173–174. doi: 10.1038/465173a. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture. Nature reviews Molecular cell biology. 2016;17:743–755. doi: 10.1038/nrm.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Schwab M. Encyclopedia of Cancer (The 2rd edition) 2009. p. 1018. [Google Scholar]
- Stefano Monti PT, Jill Mesirov, Todd Golub. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Machine Learning. 2003;52:91–118. [Google Scholar]
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature reviews Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. Chromosomal duplication is a transient evolutionary solution to stress. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21010–21015. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Gaillard S, Phillip JM, Huang TC, Pinto SM, Tessarollo NG, Zhang Z, Pandey A, Wirtz D, Ayhan A, et al. Inhibition of Spleen Tyrosine Kinase Potentiates Paclitaxel-Induced Cytotoxicity in Ovarian Cancer Cells by Stabilizing Microtubules. Cancer cell. 2015;28:82–96. doi: 10.1016/j.ccell.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Liu L, Chen H, Wang Y, Xu Y, Mao H, Li J, Mills GB, Shu Y, Li L, et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer cell. 2016;29:711–722. doi: 10.1016/j.ccell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nature genetics. 2016;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Processing pipeline of enhancers and tumor samples surveyed in this study. (B) Chromatin status of enhancers with/without overlapping of Ensembl transcripts (n = 3228/12580). Similar patterns were observed for the two groups. As a result, the 3228 enhancers were retained in the analysis. (C) Distribution of enhancers counted as prognostic in cancer types indicated on the x-axis. (D) Results in (C) divided by the expected number of recurrent prognostic enhancers calculated from permutation. Asterisk indicates p < 10−3 in the permutation.
Summary of enhancers detected in various cancer types; Related to Figure 1
Summary of prognostic enhancers in 25 cancer types with sufficient statistical power; Related to Figure 1
(A) Cumulative distribution functions (CDF) of consensus clustering results (k = 2, … , 10) of 8329 tumor samples. (B) Relative change in area under CDF curve when the number of clusters (k) equals 2 to 10. (C) Heatmap of consensus scores of 8329 tumors when k = 3 (k was set to 3 because the area under CDF increased only slightly when k > 3). (D) and (E) 3D scatter plot of the first three PCA components as in Figure 2CD. Each dot represents a tumor. (F) Heatmap of the consensus clustering using the 1500 enhancers with the highest CV across all samples.
(A) Global enhancer expression profile is associated with established cancer subtypes. Seven cancer types with >200 samples having available subtyping information and included in clustering analysis were considered. P-values were calculated by chi-squared test. Multiple comparisons were adjusted using Bonferroni’s correction. Each row on the left panel shows a BRCA sample, with its molecular subtype and enhancer expression cluster indicated by color. (B) Over/under-representation of the BRCA subtypes in the three clusters; p values calculated by chi-squared test. (C) Relative TP53 truncation rate of the three clusters. Absolute TP53 truncation rate was defined as the ratio of the number of TP53 truncations and the number of total silent mutations. Relative rates were normalized to the C2 rate. Error bars show mean ± SE. Statistics were computed using t-test. (D) We selected 7 tumor suppressors (blue) and 11 oncogenes (red) that are most frequently affected by SCNAs in cancer. A thresholded value of −2/2 was considered as deletion/amplification of a given tumor-suppressor or oncogene. Enrichment was calculated using chi-squared test.
(A) Scatter plot of number of point mutations (y) vs. level of SCNAs (x, measured as proportion of the genome affected by SCNAs) for all tumor samples (n = 8928). (B) Spearman’s correlation coefficient between number of point mutations and level of SCNAs in each of the 25 cancer types with ≥80 samples. Significant (p < 0.05) positive/negative correlations are marked by red/ blue, respectively.
This analysis is similar to Figure 5 H–K: progression-free interval (PFI) was considered for (A) PRAD and (B) KIRC; disease-specific survival time (DSS) was considered for (C) PAAD and (D) KIRC. Log-rank p values are shown.
(A) Chromatin interaction of PD-L1 and enhancer-9. This analysis is similar to Figure 6D, except that each of the seven cell types was considered separately. (B) sgRNAs used in the enhancer-9 genetic perturbation. Three sgRNAs were designed for each side of enhancer-9. Nine sgRNAs combinations (3×3) were subjected to efficiency examination. sgRNA L2 and R3 were selected for generating the cell line with the homozygous enhancer-9 deletion. (C) Relative mRNA expression level of PD-L1 in wild-type and the enhancer-9-delection cell line before and after INF-γ stimulation. Error bars show mean ± SE of the results of 4 replicates; significance levels of PD-L1 differential expression from the negative control (the wild-type A549 cell line without INF-γ stimulation) are indicated at the top of each bar; differences were assessed using t-test.
List of enhancers surveyed in this study; Related to Figure 1
Summary of TCGA samples used in this study; Related to Figure 1