

Abstract
A number of prostate cancer (PCa)‐specific genomic aberrations (denominated BRCAness genes) have been discovered implicating sensitivity to PARP inhibition within the concept of synthetic lethality. Recent clinical studies show favorable results for the PARP inhibitor olaparib used as single agent for treatment of metastatic castration‐resistant PCa. Using 2D and 3D cell culture models mimicking the different treatment and progression stages of PCa, we evaluated a potential use for olaparib in combination with first‐line endocrine treatments, androgen deprivation, and complete androgen blockade, and as a maintenance therapy following on from endocrine therapy. We demonstrate that the LNCaP cell line, possessing multiple aberrations in BRCAness genes, is sensitive to olaparib. Additive effects of olaparib combined with endocrine treatments in LNCaP are noted. In contrast, we find that the TMPRSS2:ERG fusion‐positive cell lines VCaP and DuCaP do not show signs of synthetic lethality, but are sensitive to cytotoxic effects caused by olaparib. In consequence, additive effects of olaparib with endocrine therapy were not observable in these cell lines, showing the need for synthetic lethality in combination treatment regimens. Additionally, we show that PCa cells remain sensitive to olaparib treatment after initial androgen deprivation implicating a possible use of olaparib as maintenance therapy. In sum, our preclinical data recommend olaparib as a synthetic lethal treatment option in combination or sequenced to first‐line endocrine therapy for PCa patients with diagnosed BRCAness.
Keywords: combination therapy, endocrine therapy, maintenance therapy, olaparib, PARP inhibition, prostate cancer
Abbreviations
- ADT
androgen deprivation therapy
- CAB
complete androgen blockade
- NGC
normal growth conditions
- RCCS
rotary cell culture system
1. Introduction
Current treatment options for metastatic prostate cancer (mPCa) are based on endocrine therapy and taxane‐based chemotherapy. Although second‐generation drugs such as abiraterone acetate (CYP17A1 inhibitor), enzalutamide (androgen receptor (AR) ligand binding domain inhibitor), and cabazitaxel have been introduced in clinical management of PCa, primary or acquired treatment resistances invariably occur (Attard et al., 2008; Tran et al., 2009). Thus, there is an urgent need to develop novel therapies for mPCa hitting molecular targets different from AR and microtubuli. Such novel treatment options should include (a) single treatment agents, (b) combination treatments with current endocrine and chemotherapy protocols, and (c) maintenance therapies succeeding an initial tumor‐regressive therapy to keep tumor growth and dissemination under control. One compound with the potential to fulfill all three criteria is olaparib (Lynparza™), a first‐in‐class DNA repair inhibitor approved for BRCA‐mutated advanced ovarian cancer with prior three or more chemotherapies (Kim et al., 2015). Clinical efficacy of olaparib in PCa is currently investigated in several trials with encouraging results as, for example, seen in the TOPARP‐A trial (Mateo et al., 2015).
Olaparib inhibits the catalytic activities of key enzymes in single‐strand DNA (ssDNA) break repair, namely poly (ADP‐ribose) polymerases (PARP) 1 and 2 (Menear et al., 2008). This can trap PARP on damaged DNA leading to the formation of cytotoxic DNA‐PARP complexes (Murai et al., 2012). Initially, PARP inhibitors (PARPi) have been developed in the context of synthetic lethality. Inhibition of ssDNA break repair by PARPi leads to accumulation of chromosomal aberrations during DNA replication and consequently to cell death in cells with defects in homologous recombination (HR) repair mechanisms, termed BRCAness (Lord and Ashworth, 2016). Over the past years, a number of molecular aberrations causing BRCAness have been discovered in PCa and other cancer types, including germline (frequency: 5.3%) or somatic BRCA2 (12.7%), ATM (5.3%), PTEN (40.7%), SPOP (10%), and CHD1 (8%) [frequencies in metastatic castration‐resistant PCa (mCRPCa)] aberrations (Boysen et al., 2015; Mendes‐Pereira et al., 2009; Pritchard et al., 2016; Robinson et al., 2015; Shenoy et al., 2017). Moreover, gene fusions with the ETS transcription factor family, such as TMPRSS2:ERG (frequency of ~50% in PCa), have been described to drive double‐strand DNA (dsDNA) break formation and thus sensitize cells to PARPi (Brenner et al., 2011). Rearrangements with TMPRSS2 put ETS family members such as ERG under transcriptional control of the AR signaling axis. In general, PARP‐1 activity is increased in PCa and is required for AR activity and tumor cell growth (Schiewer et al., 2012). Altogether, these molecular aberrations can predict sensitivity to PARPi in PCa patients, and hence, treatment protocols based on the concept of synthetic lethality should encompass selection of the patients based on cancer‐associated BRCAness markers in a personalized medicine approach.
In the present preclinical study, we have addressed the questions whether olaparib, in addition to its usability as a single treatment agent, could be of additive value in a combination treatment with first‐line androgen deprivation (ADT) or complete androgen blockade (CAB) therapy, and whether olaparib could be used as maintenance therapy after initial androgen deprivation.
2. Materials and methods
2.1. Cell lines and culture
Table S1 gives an overview on the prostate cell lines, culture media, and supplements used in this study. All cell lines were purchased from the American Type Culture Collection (ATCC)—LGC Standards (Wesel, Germany), except BPH1 and DuCaP (gifts from JA Schalken, Nijmegen, the Netherlands), EP156T (Kogan et al., 2006), LAPC‐4, and PC3AR (gifts from A Cato, Karlsruhe, Germany). Cell lines were cultured under standard growth conditions with once or twice passaging per week. To mimic ADT and CAB LNCaP, DuCaP and VCaP were cultured in T75 or T175 flasks for ≥ three weeks in +ABL or +BIC conditions, respectively (Table S1), and then seeded into 6‐ or 96‐wells for subsequent experiments. LNCaP‐ABLRes (Culig et al., 1999), LNCaP‐BICRes (Hobisch et al., 2006), and DuCaP‐BICRes were generated by long‐term culture under +ABL or +BIC conditions causing cell cycle arrest/induction of cell death. Upon resuming proliferation, cell lines were considered resistant to +ABL or +BIC conditions and then constantly grown and passaged under +ABL or +BIC conditions, respectively.
2.2. Reagents and chemicals
All three PARP inhibitors rucaparib (S1098), veliparib (S1004), and olaparib (S1060) were purchased from SelleckChem (Eubio, Vienna, Austria) and dissolved in DMSO at a stock concentration of 5 mm. R1881 (methyltrienolone, E3164‐000) was purchased from Serobac (Vienna, Austria) and dissolved in ethanol at 10–100 nm stock concentrations. Bicalutamide (B9061) was ordered from Sigma‐Aldrich (Vienna, Austria) and dissolved in DMSO at 1 mm stock concentration.
2.3. Rotary cell culture assays
The rotary cell culture system RCCS‐4D (Synthecon/Cellon, Bascharage, Luxembourg) with 10‐mL disposable vessels rotated at 45 RPM was used to grow cells in three dimensions. Vessels were inoculated with 3 × 106 cells in suspension in normal growth medium, and rotary cell culture was started. After 24 h, small organoids were observable. Conditioned medium was removed from the vessels at the timepoints two, six, and nine days without disturbing the organoids and replaced with NGC or +ABL medium containing 5 μm olaparib or DMSO. PSA was determined in the conditioned medium as described (Data S1) and normalized on incubation time. On day nine, organoids were removed from the vessels and embedded in coagulated citrate‐plasma (450 μL citrate‐plasma, 45 μL thrombin 120 NIH‐U·mL−1, 11.3 μL 1 m calcium chloride). Organoids were fixed in buffered 4% formaldehyde for 2 h at room temperature, dehydrated in a Tissue‐Tek VIP (Sakura, Vienna, Austria), stained with eosin to visualize organoids, and embedded in paraffin. Two‐micrometer sections were prepared using a microtome, and consecutive sections were stained for MKI67 (anti‐Ki‐67, MIB‐1; 1 : 400, M7240, Dako, Agilent, Vienna, Austria) and cleaved CASP3 (anti‐cleaved caspase 3 (Asp157), 1 : 400, #9661, Cell Signaling, New England Biolabs, Frankfurt a. M., Germany) using a Discovery‐XT staining device (Ventana, Tucson, AZ, USA) with CC1 antigen retrieval conditions. Sections were counterstained with hematoxylin, and images were taken on an Axio Imager Z2 microscope (Carl Zeiss, Vienna, Austria) equipped with a TissueFAXS automated recording system (TissueGnostics, Vienna, Austria) at 20× magnification. Overview images were generated by stitching neighboring 20x images.
For tumor‐initiation experiments in the RCCS‐4D, vessels were inoculated with the indicated number of LNCaP + ABL cells in NGC and treated with DMSO or 5 μm olaparib. After ten days, incubation at 45 RPM PSA in the supernatant was measured and tumor organoids were collected using a 70‐μm cell strainer (BD, Schwechat, Austria). Organoids were documented with a stereomicroscope (Nikon, Vienna, Austria).
2.4. Statistical analysis
prism 5 (GraphPad Software, La Jolla, CA, USA) and STATISTICA (StatSoft, Hamburg, Germany) were used to perform Student′s t‐test, Mann–Whitney test, and Friedman test, as well as one‐way and two‐way ANOVA followed by Bonferroni's multiple comparison test. Figure legends inform on the statistical test used. Data are presented as mean of multiple experiments (n ≥ 3) ± standard error of the mean (SEM) to estimate how variable the means in multiple repeated experiments were, unless indicated otherwise. Statistically significant differences are encoded in the figures as follows: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Data S1 are available online.
3. Results
3.1. Screening of a panel of prostate (cancer) cell lines for PARPi sensitivity
In order to identify prostate cancer cell lines sensitive to PARPi treatment, we performed a screening with a panel of available cell lines cultured under NGC. Immortalized benign cell lines BPH1 and EP156T, as well as AR‐positive LNCaP, LAPC4, VCaP, DuCaP, and 22Rv1 and AR‐negative PC3 and Du145 PCa cell lines, were exposed for 96 h with increasing concentrations (0–20 μm) of three commercially available PARPi: rucaparib, veliparib, and olaparib. Sensitivity to PARPi was assessed by MTT viability (Fig. 1A) and for apoptosis induction using cleaved CASP3/7 activity assays (Fig. 1B). Comparison of the three PARPi showed a more potent activity of rucaparib and olaparib relative to veliparib and on a higher number of cell lines in both viability and apoptosis assays. Statistically significant differences with all three PARPi were found in LNCaP and 22Rv1 in viability and in VCaP in apoptosis assays. Next, we screened the mutation and expression profile of the COSMIC cell line database for a list of BRCAness genes known or susceptible to confer sensitivity to PARPi and/or to be involved in homologous recombination (Forbes et al., 2015). Analysis of the mutational database identified that LNCaP, 22Rv1, and Du145 harbor >25 mutated genes implicated in sensitivity to PARPi including mutations in BRCA1/2 genes (Table 1A, Table S2). Detailed analysis of the pathogenic status of BRCA1/2 genes revealed that LNCaP and 22Rv1 harbor BRCA2 mutations with a high probability to confer PARPi sensitivity, while mutations in BRCA1/2 in Du145 have a low probability (Table S2, pathogenicity of BRCA mutations). Evaluation of the expression database encompassing copy number variations showed differential expression of a number of BRCAness genes among all cell lines (Table 1B, Table S3). Based on this and published data, we decided to further analyze PARPi sensitivity of the androgen withdrawal‐sensitive cell lines LNCaP, VCaP, and DuCaP (the latter two being TMPRSS2:ERG2 fusion‐positive). Among PARPi tested, olaparib showed the highest activity on these cell lines (Fig. 1) and was selected for further investigations.
Figure 1.
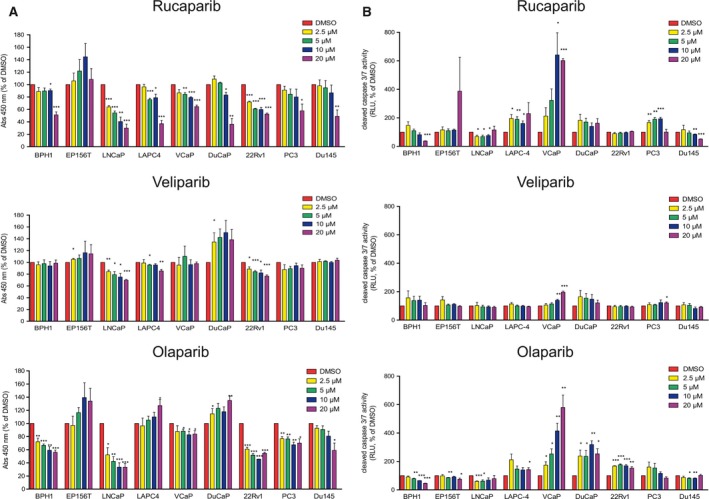
Efficacy of rucaparib, veliparib, and olaparib on various prostate immortalized benign and cancer cell lines. A panel of immortalized benign (BPH1, EP156T) and malignant (LNCaP, LAPC4, VCaP, DuCaP, 22Rv1, PC3, and Du145) prostate cell lines were treated for 96 h with increasing concentrations (2.5–20 μm) of rucaparib, veliparib, or olaparib, or the same volume of vehicle (DMSO), as indicated. (A) Viability was assessed by MTT assay. (B) Induction of apoptosis was assessed by cleaved CASP 3/7 activity assay. (A, B) Statistically significant differences were calculated by Student's t‐test. *P < 0.05; **P < 0.01; ***P < 0.001.
Table 1.
Mutational profile (A) and expression levels (B) of selected genes involved in HR and BRCAness genes in PCa cell lines. The mutations and CNV & Expression COSMIC Cell lines project's databases were screened for known mutations and for expression levels (Z‐scores >2 or <−2 are shown), respectively, using a list of HR and BRCAness genes on the indicated cell line. BRCA2 mutations (indicated with an asterisk) in LNCaP and 22Rv1 have a high probability for pathogenicity. For details, see Tables S2 and S3
| BPH1 | LNCaP | VCaP | 22Rv1 | PC3 | Du145 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | |||||||||||
| None | ABL1 | SSRP1 | AR | GTF2H3 | BRCA1 | ||||||
| AR | ATAD5 | SMARCAL1 | BRCA2 | ||||||||
| ATM | ATM | BRIP1 | |||||||||
| ATR | BARD1 | CDK7 | |||||||||
| BARD1 | BRCA2* | DDB1 | |||||||||
| BRCA2* | CDK7 | FANCB | |||||||||
| BRIP1 | CHD1 | FANCI | |||||||||
| CHD1 | DNMT3A | FLI1 | |||||||||
| CHEK2 | EME1 | IPMK | |||||||||
| DDB1 | FANCM | LIG4 | |||||||||
| DNMT3A | FLI1 | MAPK12 | |||||||||
| ERCC1 | HELLS | MMS22L | |||||||||
| ESCO1 | INO80D | PAPD7 | |||||||||
| ETV1 | KAT5 | POLQ | |||||||||
| FANCA | LIG4 | RAD50 | |||||||||
| FANCE | MCM3 | RBBP8 | |||||||||
| G2E3 | MUM1 | REV3L | |||||||||
| GTF2H3 | NBN | SLX4 | |||||||||
| MMS22L | PALB2 | SMARCA2 | |||||||||
| MUM1 | RAD54L | SSRP1 | |||||||||
| PAPD7 | SHPRH | STK36 | |||||||||
| PLK3 | SLX4 | TP53BP1 | |||||||||
| POLB | SMG1 | UBE2N | |||||||||
| POLH | STK36 | USP1 | |||||||||
| PTEN | TP53BP1 | XRCC2 | |||||||||
| RAD51B | USP1 | ||||||||||
| RAD54L | USP7 | ||||||||||
| RECQL4 | WRN | ||||||||||
| SMARCA5 | XRCC2 | ||||||||||
| SPO11 | |||||||||||
| SSRP1 | |||||||||||
| TOP2B | |||||||||||
| TP53BP1 | |||||||||||
| UBA1 | |||||||||||
| XRCC2 | |||||||||||
| B | |||||||||||
| CHEK2 | −2.283 | AR | 3.51 | AR | 5.181 | AR | 4.466 | BRCA1 | −2.513 | FANCG | 3.231 |
| ETV1 | −2.326 | DDB1 | −2.518 | ATR | −2.619 | INO80D | 2.102 | CDK5 | 2.591 | G2E3 | 2.971 |
| FAM175A | −2.307 | DNASE1L2 | 2.124 | ERG | 3.802 | MSH3 | −4.015 | MUM1 | −3.271 | MUS81 | 2.05 |
| GADD45A | 2.262 | ETV1 | 2.454 | KAT5 | 3.541 | RNF168 | −2.512 | PNKP | 2.18 | XRCC3 | 2.112 |
| LIG3 | −2.599 | PRMT6 | 3.644 | LIG3 | 3.149 | SHPRH | 2.36 | PTEN | −3.164 | ||
| UNG | 2.724 | TMPRSS2 | 3.981 | MUS81 | 2.202 | RAD50 | −2.662 | ||||
| XRCC3 | 2.428 | PALB2 | −3.094 | RECQL4 | 2.449 | ||||||
| POLK | −2.939 | SMG1 | −2.483 | ||||||||
| RAD23B | 3.623 | SPO11 | −2.246 | ||||||||
| SMG1 | −2.385 | ||||||||||
| TMPRSS2 | 2.76 | ||||||||||
| UBE2A | −5.143 | ||||||||||
| USP10 | −5.321 | ||||||||||
3.2. Additive effects of olaparib combined to endocrine therapy
In theory, PARPi could be clinically used in different progression stages with different treatment regimens of PCa, that is, prior (treatment‐naïve), during (combination therapy), and posterior (CRPC) endocrine therapy. These stages of endocrine therapy were mimicked by manipulating androgen concentrations in the culture medium (for culture conditions, see Table S1). To this end, the medium was switched from NGC to +ABL (mimicking ADT) or +BIC (mimicking CAB) (Fig. 2A). To mimic CRPC, cells were cultured under long‐term (up to nine months) +ABL or +BIC conditions. Upon resuming proliferation, cells were considered castration‐resistant and named −ABLRes or −BICRes. Unfortunately, we were unable to generate DuCaP‐ABLRes and VCaP‐ABLRes/‐BICRes cell lines. The effects of olaparib (0–10 μm, 96 h incubation) were assessed under these conditions. While +ABL and +BIC had strong effects on PSA secretion in DuCaP and LNCaP, VCaP cells were less affected by these conditions (Fig. 2B). DuCaP‐BICRes and LNCaP‐BICRes resumed PSA secretion, which was not seen in LNCaP‐ABLRes. Importantly, olaparib did not have any effect on PSA secretion under either stage. Next, the effect of olaparib on cellular proliferation under the different stages was elaborated (Fig. 2C). Culture under +ABL and +BIC conditions resulted in markedly decreased proliferation rates, while proliferation rate was resumed or increased (compared to NGC) in ABLRes and BICRes. Olaparib treatment decreased proliferation of LNCaP under either condition while proliferation of VCaP was not influenced by olaparib treatment. Furthermore, expression levels of AR and ERG, as well as the apoptosis marker cPARP, were assessed by immunoblotting (Fig. 2D). Culture of DuCaP and VCaP under +ABL and +BIC conditions resulted in a stark decrease of the androgen‐regulated TMPRSS2:ERG fusion protein. Interestingly, re‐expression of ERG in DuCaP‐BICRes was not observed. Apoptosis induction by olaparib under NGC was seen in DuCaP and, less pronounced, in VCaP, while almost absent in LNCaP. Contrasting results of olaparib on cPARP levels were also observed under +ABL and +BIC conditions: Apoptosis induction in DuCaP was markedly decreased compared to NGC, while VCAP showed increased levels. No apoptosis induction was observed in LNCAP. In resistant stages, a mild apoptosis induction by olaparib was observed in DuCaP‐BICRes. Finally, we evaluated the induction of dsDNA breaks by γH2AX immunofluorescence (Figs 2E and [Link], [Link], [Link]). Significant increases in γH2AX foci/nucleus were observed in DuCaP and LNCaP in NGC after olaparib treatment, while +ABL precluded induction of dsDNA breaks by olaparib in all cell lines. LNCaP‐ABLRes and ‐BICRes showed increased γH2AX levels after olaparib treatment, which was not observed in DuCaP‐BICRes. Importantly, VCaP showed only a mild, nonsignificant induction of γH2AX foci/cell by olaparib under NGC, which was not seen under +ABL conditions. We therefore concluded that induction of apoptosis by olaparib in VCaP+ABL (Fig. 2D) is likely due to cytotoxic effects and not due to generation of dsDNA damage.
Figure 2.
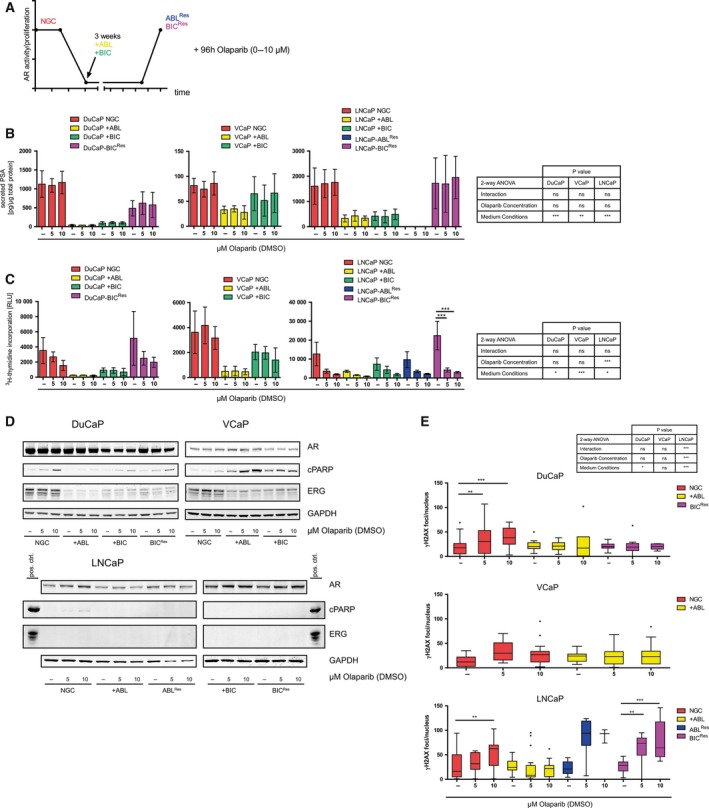
Efficacy of short‐term treatment with olaparib on androgen‐sensitive prostate cancer cell lines mimicking different therapeutic options and disease progression stages. (A) Scheme depicting the various treatment conditions and progression stages and their effects on AR activity and cellular proliferation. Experiments in this figure were conducted with DuCaP, VCaP, and LNCaP that were treated with olaparib for 96 h. (B) Secreted PSA was measured in the supernatant and normalized to total protein content of producing cells, as indicated. (C) Proliferation was measured by 3H‐thymidine incorporation assay. (D) Immunoblotting was performed to determine expression levels of the apoptosis marker cPARP, as well as AR and ERG. GAPDH served as equal loading control. A representative immunoblot of at least three independent experiments is shown. Positive controls for cPARP and ERG have been loaded in adjacent gel lanes in LNCaP immunoblots. (E) Immunofluorescence assay for γH2AX was performed to assess the induction of dsDNA breaks. The number of γH2AX foci per nucleus was assessed, and absolute values are depicted as box‐and‐whisker plots. (B, C, E) Statistically significant differences were calculated by two‐way ANOVA test (table) with Bonferroni post‐test (stars in graph). (A–E) NGC, normal growth conditions; +ABL, androgen deprivation mimicking ADT; +BIC, bicalutamide treated mimicking CAB; ABLR es, resistant to androgen ablation; BICR es, resistant to bicalutamide treatment; RLU, relative light units. *P < 0.05; **P < 0.01; ***P < 0.001.
3.3. Failure of increased ERG expression to sensitize for olaparib treatment
TMPRSS2:ERG fusion could be an important biomarker for PARPi sensitivity due to its high frequency among patients. However, our results showed that ERG expression is not necessary a prerequisite for PARPi sensitivity, as seen, in particular, in VCaP +ABL and +BIC cells (Fig. 2D). To elaborate in more detail the effects of ERG expression on PARPi sensitivity, we treated DuCaP and VCaP with 10 or 100 pm of the synthetic androgen R1881 (Fig. 3A). Although these castrate levels of R1881 were sufficient to increase dose‐dependently ERG expression levels, only minor changes in proliferation indexes could be measured. However, despite increased ERG expression levels, cPARP levels were decreased in samples cotreated with R1881 and olaparib compared to olaparib‐only treated. In addition, stable overexpression of ERG did not increase sensitivity of LNCaP cells to olaparib treatment in either NGC or +ABL conditions, as measured by proliferation and cPARP apoptosis immunoblots (Fig. 3B). To address the effect of ERG expression in the absence of AR expression, PC3 cells were assessed for olaparib sensitivity. PC3 cells stably transfected with a control construct (V5/lacZ) showed decreased proliferation and a very minor induction of apoptosis upon olaparib treatment, which was not affected by AR expression in PC3 stably transfected with AR (PC3AR) (Fig. 3C). Furthermore, ERG overexpression could not increase sensitivity of PC3 to olaparib in neither absence nor presence of R1881‐activated AR.
Figure 3.
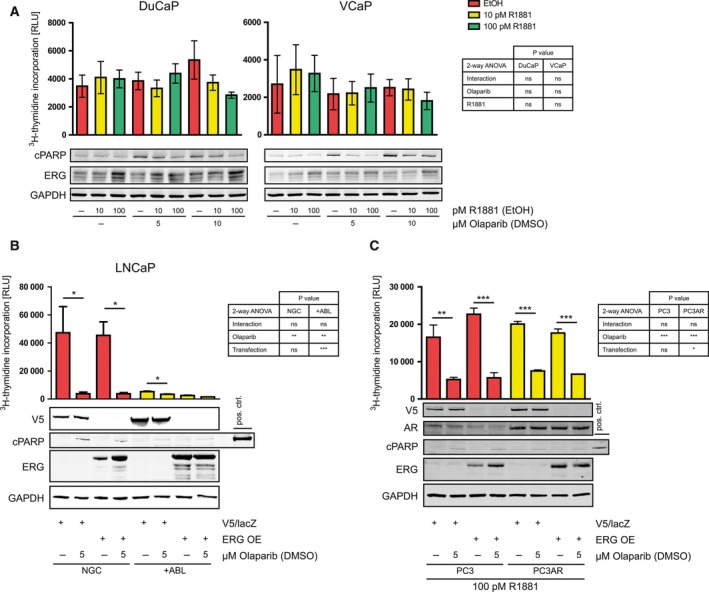
Influence of ERG expression on the sensitivity to olaparib short‐term treatment. (A) DuCaP and VCaP were treated with 10 or 100 pm R1881 or vehicle (EtOH), and 5 or 10 μm olaparib or vehicle (DMSO) for 96 h, as indicated. (B,C) Fusion‐negative LNCaP, PC3, and PC3AR cells were stably transfected with lentiviral constructs to overexpress β‐galactosidase fused to V5 (V5/lacZ, control) or truncated ERG, as found in fusion‐positive cells. LNCaP‐V5/lacZ and –ERG were then treated with 5 μm olaparib or vehicle (DMSO) for 96 h, as indicated. PC3 and PC3AR cells were treated with 100 pm R1881. (A–C) Immunoblotting was performed to determine expression levels of the apoptosis marker cPARP, as well as AR, ERG, or V5/lacZ. GAPDH served as equal loading control. A representative immunoblot of three independent experiments is shown. In parallel, proliferation under those conditions was determined by 3H‐thymidine incorporation assay. (A) No statistically significant differences by two‐way ANOVA test were found. (B, C) Two‐way ANOVA (table) with Bonferroni post‐test (stars in graph) was performed separately for NGC and +ABL conditions (B) and for PC3 and PC3AR (C). RLU, relative light units. *P < 0.05; **P < 0.01; ***P < 0.001.
3.4. Increased induction of apoptosis after long‐term incubations with olaparib in LNCaP, but not in TMPRSS2:ERG‐positive cell lines
In order to determine sensitivity to PARPi with longer incubation times and under clinically more relevant, three‐dimensional conditions we made use of a rotary cell culture system (RCCS). Rotating vessels were inoculated with detached DuCaP, VCaP, and LNCaP cells. Growing cellular aggregates were observable after a few hours resulting in up to ten organoids/disk of up to three millimeter in diameter after nine days. Two days after inoculation, treatments with olaparib under NGC or +ABL conditions were started. PSA in the supernatant was measured 2, 6, and 9 days after inoculation (Fig. 4A). PSA secretion of VCaP organoids was at the lower detection limit of the assay and ranged between 0.4978 ± 0.292 ng/(mL × day) [DuCaP: 22.75 ± 16.37 ng/(mL × day) and LNCaP: 43.48 ± 25.08 ng/(mL × day)]. PSA secretion dropped under +ABL conditions compared to NGC in DuCaP and LNCaP organoids. In DuCaP organoids, olaparib treatment did not have any effect on PSA secretion under either condition. In contrast, olaparib treatment of LNCaP organoids resulted in decreased PSA secretion under NGC and an additive effect of olaparib and +ABL was observable. At day nine after inoculation, organoids were harvested and FFPE sections were stained for cleaved CASP3 (Fig. 4B) and MKI67 (Fig. 4C) as markers for apoptosis and proliferation, respectively. Counterstaining with hematoxylin revealed necrotic areas in the center of organoids likely resulting from a lack of supply with dioxygen and nutrients. DuCaP and VCaP organoids demonstrated a higher degree of cleaved CASP3 staining compared to LNCaP under NGC. When grown under +ABL conditions, the number of apoptotic cells did not markedly change in DuCaP and VCaP organoids, but was increased in LNCaP organoids. Olaparib treatment did not affect apoptosis induction in DuCaP and VCaP organoids under either NGC or +ABL. In contrast, increased levels of cleaved CASP3 staining were observed in LNCaP organoids cultured in NGC after olaparib treatment, which increased further when olaparib was combined to +ABL conditions. Stainings with MKI67 revealed a high proliferative index of DuCaP and VCaP organoids, while a number of cells in LNCaP organoids did not actively cycle. When grown under +ABL conditions, proliferation drastically decreased in all three cell line organoids. Treatment with olaparib was able to decrease MKI67 staining in LNCaP organoids under NGC, but unable in DuCaP and VCaP organoids. Again, a combined effect of +ABL and olaparib treatment in LNCaP organoids was observable, but absent in DuCaP and VCaP organoids.
Figure 4.
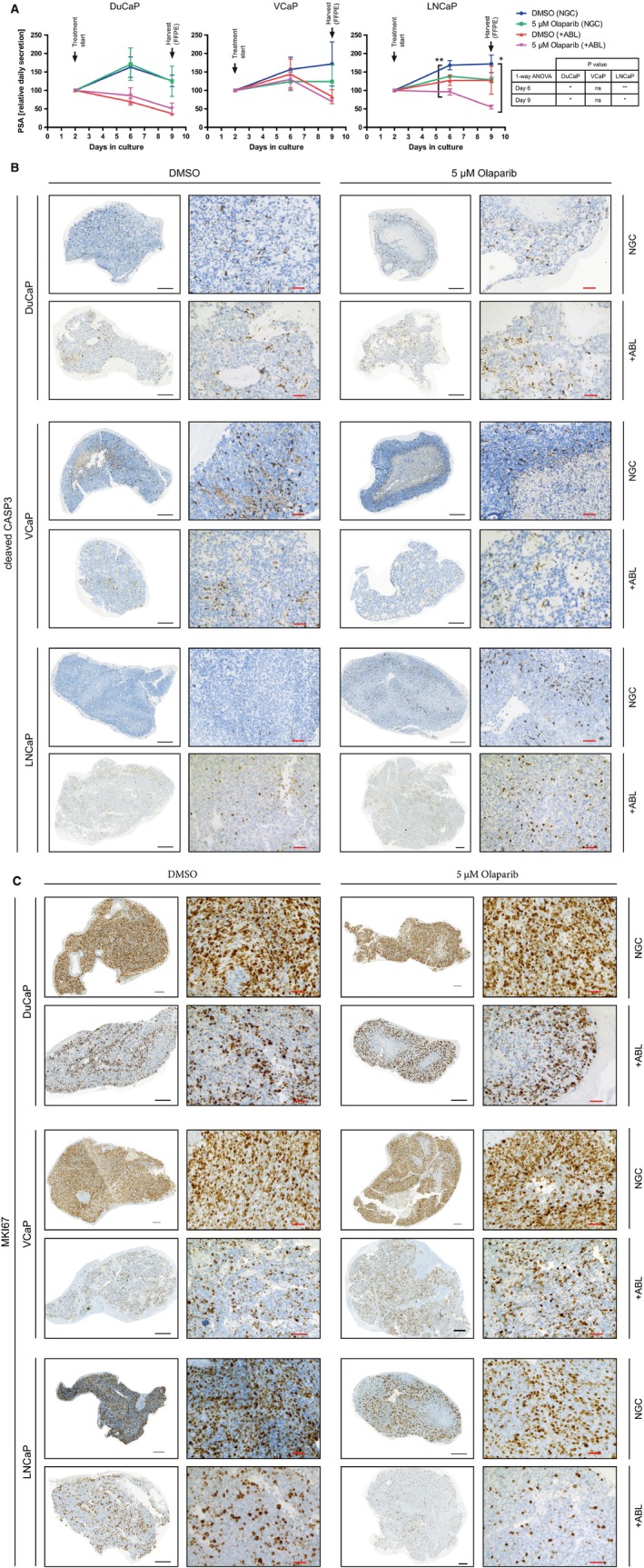
Efficacy of olaparib long‐term treatment on organoids grown in a rotary culture system. Rotary disks were inoculated with dispersed DuCaP, VCaP, and LNCaP cells and incubated at 45 RPM. Organoids formed after 2 days and treatment for 7 days with 5 μm olaparib or vehicle (DMSO) was started, as indicated. Medium was replaced at days 2 and 6. (A) Secreted PSA was measured in the conditioned medium on days 2, 6, and 9, and values are depicted relative to day 2 as secreted PSA per day. One‐way ANOVA (table) with Bonferroni post‐test (stars in graph) was performed to assess statistically significant differences between treatments. (B, C) Nine days after inoculation, organoids were harvested and formalin‐fixed paraffin‐embedded (FFPE) sections were stained for cleaved CASP3 (B) and MKI76 (C). Samples were counterstained with hematoxylin. Representative overview (left) and detail (right) views are shown for each treatment, as indicated. Black scale bar, 200 μm, red scale bar, 50 μm. (A–C) NGC, normal growth conditions; +ABL, androgen deprivation mimicking ADT. *P < 0.05; **P < 0.01.
3.5. Conserved olaparib efficacy after initial ADT
Finally, clonogenic assays with olaparib were performed as an indicator for tumor‐initiation potential. LNCaP NGC, LNCaP‐ABLRes, and DuCaP NGC were able to form colonies, while LNCaP–BICRes, DuCaP‐BICRes, and VCaP did not yield clonogenic growth. LNCaP‐ABLRes had a significantly increased plating efficiency compared to LNCaP NGC (Fig. 5A). LNCaP NGC and LNCaP‐ABLRes were sensitive to low concentrations of olaparib (0.625–2.5 μm), and 1.25 μm was sufficient to completely inhibit clonogenicity of LNCaP NGC (Fig. 5B). Plating efficiency of DuCaP NGC was slightly affected by up to 2.5 μm olaparib. To assess whether olaparib might be used as a maintenance therapy replacing ADT after initial tumor regression, we pretreated LNCaP and DuCaP for three weeks under +ABL conditions followed by clonogenic assays under NGC (Fig. 5C). This +ABL selection step led to increased plating efficiency compared to NGC (Fig. 5A). Importantly, olaparib was able to decrease clonogenic growth in LNCaP under these conditions and also in DuCaP albeit statistically nonsignificant. Finally, we made use again of the RCCS to monitor tumor initiation of +ABL‐pretreated LNCaP under NGC conditions. A minimum number of 10x10³ +ABL‐pretreated LNCaP were necessary to yield organoid growth. Determination of secreted PSA (Fig. 5D) and of organoid size (Fig. 5E) showed that olaparib treatment could inhibit organoid growth after initial +ABL conditions.
Figure 5.
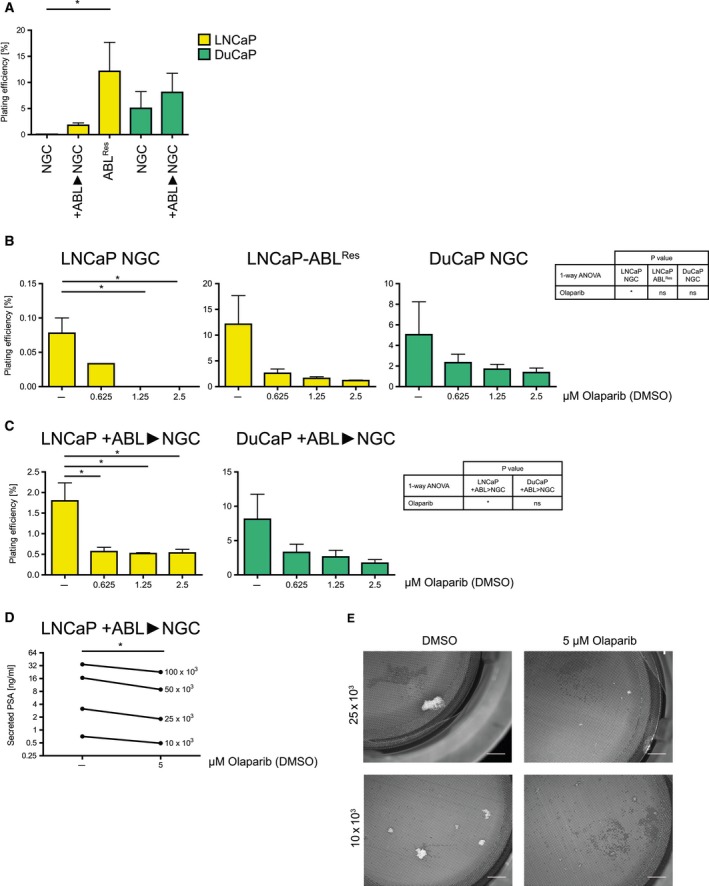
Efficacy of olaparib after initial androgen deprivation. Clonogenic assays were performed with LNCaP, LNCaP‐ABLR es, and DuCaP. In addition, LNCaP and DuCaP were grown for 3 weeks under +ABL conditions and then seeded for clonogenic assay under NGC (indicated as +ABL>NGC). (A) Plating efficiency (ratio of number of colonies per number of seeded cells) is depicted in absolute values. Statistically significant differences were calculated by Mann–Whitney test. (B, C) Clonogenic assays were performed with increasing concentrations of olaparib (0–2.5 μm), as indicated. Plating efficiencies relative to vehicle (DMSO) are indicated, and statistically significant differences were calculated by one‐way ANOVA (table) and Bonferroni post‐test (stars in graphs). (D, E) Tumor‐initiation experiments were performed with 1 × 104–105 LNCaP grown for 3 weeks under +ABL conditions followed by incubation in the RCCS with DMSO or 5 μm olaparib. (D) PSA was measured in the supernatant. Single values resulting from inoculations with different cell numbers are depicted, and statistically significant differences were calculated with Friedman test. (E) Tumor organoids were collected with a cell strainer. Scale bar, 2 mm. (A–D) NGC, normal growth conditions; +ABL, androgen deprivation mimicking ADT. *P < 0.05.
4. Discussion
4.1. Rationales for synthetic lethality by PARP inhibition in PCa
To date, few clinical studies assessing PARP inhibitors in m(CR)PCa are available (Fong et al., 2009; Kaufman et al., 2015; Mateo et al., 2015; Sandhu et al., 2013). Favorable results have been noted, in particular, in patients with defects in DNA repair genes denominated BRCAness genes. In fact, predicting sensitivity to PARPi for personalized cancer therapy is difficult and encompasses routine screening for germline pathogenic mutations in BRCA1/2 genes only. However, molecular findings and retrospective genomic analysis of clinical data show that synthetic lethality with PARPi is not restricted to mutant BRCA1/2 but can also involve other genes. We found that TP53 wild‐type LNCaP cells show an initial proliferative arrest after four‐day olaparib treatment (Fig. 2) and an apoptotic response after seven days (Fig. 4). This shows the necessity for prolonged incubation times to assess synthetic lethality based on generation of DNA damage by PARPi. LNCaP harbor not only a BRCA2 mutation with a high probability of pathogenicity, but also mutations in HR genes RAD51B, RAD54L, ATM, and FANCA (Table 1) showing that multiple genetic aberrations in HR genes could contribute to PARPi sensitivity. Furthermore, LNCaP cells express the fusion TMPRSS2:ETV1, encode mutated CHD1, and are negative for PTEN protein, all of which have been shown to sensitize to PARP inhibition (Brenner et al., 2011; Kari et al., 2016; Mendes‐Pereira et al., 2009; Sharrard and Maitland, 2000). We therefore conclude that the exact mechanism of PARPi sensitization in LNCaP cells is unclear and may likely include more than one single mechanism. Thus, the LNCaP example nicely illustrates the difficulty in stratifying cancer patients that may benefit from PARPi therapy and the need to further investigate synthetic lethality mechanisms with PARPi.
In this work, we focused on the role of TMPRSS2:ERG in mediating synthetic lethality (Brenner et al., 2011). The initial hypothesis was to investigate whether the downregulation of ERG, by suppressing AR activity at the TMPRSS2 promoter, is sufficient to abolish PARPi sensitivity of VCaP and DuCaP cell lines. This may have answered the question whether endocrine therapy in fusion‐positive patients restricts sensitivity to olaparib treatment, thus not supporting such a combination therapy. Instead, we found that the number of γH2AX foci/cell is only moderately increased after olaparib treatment in fusion‐positive VCaP compared to LNCaP under NGC (Fig. 2E). After androgen deprivation, an increase in γH2AX foci/cell was not noted in VCaP (Fig. 2E), while markedly increased apoptosis was observable (Fig. 2D). Detailed analysis by ERG expression level manipulation did also not indicate a contribution of ERG to olaparib sensitivity (Fig. 3), and after longer incubation times, olaparib was not able to induce apoptosis in VCaP and DuCaP cells (Fig. 4). In contrast to the study by Brenner et al. (2011), we were not able to document decreased proliferation rates when PC3 cells were manipulated to overexpress ERG, nor could we find a sensitization after ERG overexpression to olaparib in this cell line (Fig. 3C). Thus, we speculate that the pro‐apoptotic effects observed in VCaP and DuCaP under NGC (Figs 1, 2D) after four days olaparib treatment are due to cytotoxic effects and possibly a result of olaparib trapping PARP to sites of DNA damage resulting in stalled replication forks during DNA replication (Murai et al., 2012). PARP trapping may also contribute to the effects observed in LNCaP and PC3 cells. A higher cytotoxicity of olaparib in VCaP and DuCaP may also be a result of a higher basal apoptotic index as compared to LNCaP (Fig. 4B), which could be caused by genomic instability due to ERG overexpression (Brenner et al., 2011) and/or by a gain‐of‐function mutation in TP53 (p.R248W) (van Bokhoven et al., 2003; Song et al., 2007). In sum, we raise concerns that ERG overexpression in combination with PARPi may not induce synthetic lethality and thus may not serve as biomarker to stratify patients for PARPi therapy. This is underlined by clinical studies (NCT01576172 and NCT00749502) which found no impact of the ETS status on the progression‐free survival (PFS) of patients treated with a combination of abiraterone and veliparib (Hussain et al., 2017) or on clinical benefit with the PARPi niraparib administered as single treatment (Sandhu et al., 2013).
4.2. Combination and maintenance therapies with PARPi for PCa
Published clinical studies have assessed the efficacy of olaparib as a monotherapy in mCRPC only (Fong et al., 2009; Kaufman et al., 2015; Mateo et al., 2015). However, olaparib and possibly other PARPi may be well suited for combination therapies. In particular, combinations with radiotherapy or with DNA‐damaging chemotherapy are considered to add up (Hussain et al., 2014; Reiss et al., 2015). Here, we were interested whether PARP inhibition combined with first‐line endocrine therapy, that is, ADT and CAB could have superior antitumor effects compared to single treatments. The role of androgenic signaling in generation and repair of dsDNA breaks are currently not fully understood. In line with our results in DuCaP and VCaP (Fig. 3A), activation of AR by dihydrotestosterone modestly decreased sensitivity to olaparib in LNCaP cells (Morra et al., 2017). Despite this, it was found that AR reactivation in androgen‐deprived PCa cell lines, including LNCaP and VCaP, induces a transient increase in dsDNA breaks (Hedayati et al., 2016). Furthermore, AR is a direct positive regulator of several DNA repair genes and antiandrogens or chemical castration impair error‐prone nonhomologous end joining repair of dsDNA breaks (Polkinghorn et al., 2013; Tarish et al., 2015). A recent study showed that enzalutamide is also able to decrease expression of selected DNA repair genes involved in HR (Li et al., 2017). Here, we could document additive effects of olaparib combined with androgen deprivation on cellular proliferation and apoptosis, albeit only in a cell line possessing numerous aberrations in BRCAness genes, that is, LNCaP (Fig. 4). The effects of AR inactivation on generation of DNA damage/DNA repair activity might therefore be insufficient to generate sensitivity to PARPi. In sum, we conclude that such a combination therapy could be of benefit for PCa patients with known sensitivity to PARPi and receiving first‐line endocrine therapy (ADT or CAB). Thus, similar to olaparib monotherapy, a careful selection of patients in a personalized medicine approach for BRCAness may show best results. Furthermore, an in‐depth analysis of the role of AR in dsDNA break formation and in regulation of DNA repair is warranted in order to be able to conclude on a mechanistic contribution of the AR signaling axis on synthetic lethality with PARPi.
In general, olaparib is well‐tolerated by patients and shows few, mostly grade I‐II adverse effects (Kim et al., 2015). Therefore, olaparib and, in general, PARPi are candidates for a maintenance therapy protocol that can be sequenced after an initial tumor‐regressive therapy with the aims to prolong PFS and offering an acceptable quality‐of‐life to patients. Indeed, favorable results in PFS have been noted with olaparib applied as maintenance therapy in ovarian cancer patients with a BRCA mutation after partial or complete response to the most recent platinum treatment (Ledermann et al., 2014) without affecting quality‐of‐life (Ledermann et al., 2016), compared to placebo‐treated patients. Here, we have addressed the question whether PCa cells are still sensitive to olaparib treatment after initial androgen deprivation. Potential for colony formation, as an indicator for tumor initiation (Fig. 5A), and tumor growth in the RCCS (Fig. 5B) showed that olaparib is still effective after an initial period of androgen removal. We therefore speculate that PARPi could prolong time to castration resistance of mPCa patients on ADT/CAB by sequencing PARPi after initial endocrine therapy. Our data could therefore serve as a basis to design innovative clinical trials to assess olaparib as a maintenance therapy for PCa.
5. Conclusions
In summary, olaparib is effective as an anticancer treatment in combination with and as maintenance therapy after first‐line endocrine treatment in PCa cells with markers of BRCAness. ERG rearrangements may not cause and may thus not be suitable as a marker for sensitivity to synthetic lethality with PARPi. Therefore, this preclinical study encourages the design of combination and maintenance therapies with olaparib for endocrine‐treated PCa patients stratified for BRCAness.
Data accessibility
Author contributions
GEF, KT, PDL, FG, SP, MH, JB, and FRS acquired and analyzed the data. JV statistically analyzed the data. JB, ZC, and FRS interpreted the data. IS, JB, ZC, and FRS critically revised the manuscript for important intellectual content. FRS performed study design. FRS prepared the manuscript. All authors approved the final version of the manuscript.
Supporting information
Fig. S1. Immunofluorescence for γH2AX foci (red) in DuCaP, VCaP and LNCaP after olaparib treatment. Nuclei were stained with DAPI (blue). White scale bar, 50 μm; red scale bar, 10 μm.
{kind=link}
{kind=link}
{kind=link}
Data S1. Supplementary material and methods.
Table S1. Medium conditions of the PCa cell lines used in this study and supplementary cell culture conditions.
Table S2. Screening for mutations in HR and BRCAness genes using the COSMIC Cell line project′s Mutations database. Determination of BRCA pathogenicity was done with the BRCA mutation and HCI database (University of Utah).
Table S3. Screening for expression levels in HR and BRCAness genes using the COSMIC Cell line project′s CNV & Expression database.
Acknowledgements
We thank the staff of the central routine laboratory of Innsbruck University Hospital (Head Prof. A. Griesmacher) for PSA measurements. This work was funded by the Austrian Cancer Aid Society/Tyrol (to FRS) and the Austrian Science Fund (FWF, P26799 to FRS and P29457 to IS) and in part by the Czech Ministry of Health (AZV, 15‐28628A, to JB) and the Czech Ministry of Education (NPS I, L01304, to JB and JV; Mobility, 7AMB16AT022, to FRS, JB, and ZC).
References
- Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E et al (2008) Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration‐resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26, 4563–4571. [DOI] [PubMed] [Google Scholar]
- van Bokhoven A, Varella‐Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ and Lucia MS (2003) Molecular characterization of human prostate carcinoma cell lines. Prostate 57, 205–225. [DOI] [PubMed] [Google Scholar]
- Boysen G, Barbieri CE, Prandi D, Blattner M, Chae S‐S, Dahija A, Nataraj S, Huang D, Marotz C, Xu L et al (2015) SPOP mutation leads to genomic instability in prostate cancer. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J et al (2011) Mechanistic rationale for inhibition of poly(ADP‐ribose) polymerase in ETS gene fusion‐positive prostate cancer. Cancer Cell 19, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K et al (1999) Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer 81, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui‐Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ et al (2009) Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S et al (2015) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43, D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedayati M, Haffner MC, Coulter JB, Raval RR, Zhang Y, Zhou H, Mian O, Knight EJ, Razavi N, Dalrymple S et al (2016) Androgen deprivation followed by acute androgen stimulation selectively sensitizes AR‐positive prostate cancer cells to ionizing radiation. Clin Cancer Res 22, 3310–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobisch A, Fritzer A, Comuzzi B, Fiechtl M, Malinowska K, Steiner H, Bartsch G and Culig Z (2006) The androgen receptor pathway is by‐passed in prostate cancer cells generated after prolonged treatment with bicalutamide. Prostate 66, 413–420. [DOI] [PubMed] [Google Scholar]
- Hussain M, Carducci MA, Slovin S, Cetnar J, Qian J, McKeegan EM, Refici‐Buhr M, Chyla B, Shepherd SP, Giranda VL et al (2014) Targeting DNA repair with combination veliparib (ABT‐888) and temozolomide in patients with metastatic castration‐resistant prostate cancer. Invest New Drugs 32, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Daignault S, Twardowski P, Albany C, Stein MN, Kunju LP, Robinson DR, Cooney KA, Montgomery RB, Antonarakis SE et al (2017) Abiraterone + prednisone (Abi) +/‐ veliparib (Vel) for patients (pts) with metastatic castration‐resistant prostate cancer (CRPC): NCI 9012 updated clinical and genomics data. J Clin Oncol 35(Suppl.); abstr 5001. [Google Scholar]
- Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, Sirma H, Simon R, Will H, Dobbelstein M et al (2016) Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep 17, 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira‐Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A et al (2015) Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R et al (2015) FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA‐mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 21, 4257–4261. [DOI] [PubMed] [Google Scholar]
- Kogan I, Goldfinger N, Milyavsky M, Cohen M, Shats I, Dobler G, Klocker H, Wasylyk B, Voller M, Aalders T et al (2006) hTERT‐immortalized prostate epithelial and stromal‐derived cells: an authentic in vitro model for differentiation and carcinogenesis. Cancer Res 66, 3531–3540. [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira‐Frommer R, Safra T et al (2014) Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15, 852–861. [DOI] [PubMed] [Google Scholar]
- Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira‐Frommer R, Safra T et al (2016) Quality of life during olaparib maintenance therapy in platinum‐sensitive relapsed serous ovarian cancer. Br J Cancer 115, 1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, Manyam GC, Wu W, Luo Y, Basourakos S et al (2017) Androgen receptor inhibitor‐induced “BRCAness” and PARP inhibition are synthetically lethal for castration‐resistant prostate cancer. Sci Signal 10, pii: eaam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ and Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez‐Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N et al (2015) DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes‐Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim J‐S, Waldman T, Lord CJ and Ashworth A (2009) Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menear KA, Adcock C, Boulter R, Cockcroft X, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S et al (2008) 4‐[3‐(4‐cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐1‐one: a novel bioavailable inhibitor of poly(ADP‐ribose) polymerase‐1. J Med Chem 51, 6581–6591. [DOI] [PubMed] [Google Scholar]
- Morra F, Merolla F, Napolitano V, Ilardi G, Miro C, Paladino S, Staibano S, Cerrato A and Celetti A (2017) The combined effect of USP7 inhibitors and PARP inhibitors in hormone‐sensitive and castration‐resistant prostate cancer cells. Oncotarget 8, 31815–31829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y (2012) Differential trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D et al (2013) Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 3, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R et al (2016) Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med 375, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss KA, Herman JM, Zahurak M, Brade A, Dawson LA, Scardina A, Joffe C, Petito E, Hacker‐Prietz A, Kinders RJ et al (2015) A Phase I study of veliparib (ABT‐888) in combination with low‐dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin Cancer Res 21, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu Y‐M, Schultz N, Lonigro RJ, Mosquera J‐M, Montgomery B, Taplin ME, Pritchard CC, Attard G et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A et al (2013) The poly(ADP‐ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose‐escalation trial. Lancet Oncol 14, 882–892. [DOI] [PubMed] [Google Scholar]
- Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM et al (2012) Dual roles of PARP‐1 promote cancer growth and progression. Cancer Discov 2, 1134–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrard RM and Maitland NJ (2000) Phenotypic effects of overexpression of the MMAC1 gene in prostate epithelial cells. Br J Cancer 83, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, Wang C, Zhang LZ, Wang Y, Gil V et al (2017) CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error‐prone double‐strand break repair. Ann Oncol, 28, 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Hollstein M and Xu Y (2007) p53 gain‐of‐function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 9, 573–580. [DOI] [PubMed] [Google Scholar]
- Tarish FL, Schultz N, Tanoglidi A, Hamberg H, Letocha H, Karaszi K, Hamdy FC, Granfors T and Helleday T (2015) Castration radiosensitizes prostate cancer tissue by impairing DNA double‐strand break repair. Sci Transl Med 7, 312re11. [DOI] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith‐Jones PM, Yoo D, Kwon A et al (2009) Development of a second‐generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunofluorescence for γH2AX foci (red) in DuCaP, VCaP and LNCaP after olaparib treatment. Nuclei were stained with DAPI (blue). White scale bar, 50 μm; red scale bar, 10 μm.
Data S1. Supplementary material and methods.
Table S1. Medium conditions of the PCa cell lines used in this study and supplementary cell culture conditions.
Table S2. Screening for mutations in HR and BRCAness genes using the COSMIC Cell line project′s Mutations database. Determination of BRCA pathogenicity was done with the BRCA mutation and HCI database (University of Utah).
Table S3. Screening for expression levels in HR and BRCAness genes using the COSMIC Cell line project′s CNV & Expression database.