

Abstract
Random mutagenesis has the potential to optimize the efficiency and selectivity of protein catalysts without requiring detailed knowledge of protein structure; however, introducing synthetic metal cofactors complicates the expression and screening of enzyme libraries, and activity arising from free co-factor must be eliminated. Here we report an efficient platform to create and screen libraries of artificial metalloenzymes (ArMs) via random mutagenesis which we use to evolve highly selective dirhodium cyclopropanases. Error-prone PCR and combinatorial codon mutagenesis enabled multiplexed analysis of random mutations, including at sites distal to the putative ArM active site that are difficult to identify using targeted mutagenesis approaches. Variants that exhibited significantly improved selectivity for each of cyclopropane product enantiomers were identified, and higher activity than previously reported ArM cyclopropanases obtained via targeted mutagenesis was also observed. This improved selectivity carried over to other dirhodium catalyzed transformations, including N- H, S-H and Si-H insertion, demonstrating that ArMs evolved for one reaction can serve as starting points to evolve catalysts for others.
The Importance of Random Mutations
Selective catalysis can greatly improve the efficiency of chemical synthesis, but designing and optimizing catalysts with high activity and selectivity rank among the most challenging endeavors in synthetic chemistry.1 The catalytic proficiency of enzymes acting on their native substrates has inspired a range of approaches to engineer enzymes for chemical synthesis.2 The introduction of mutations to improve enzyme activity and selectivity toward substrates and reactions of interest has proven particularly powerful in this regard, but these efforts are complicated by our inability to fully predict how a given mutation will alter enzyme function.3 In the absence of this understanding, and given the astronomical number of mutations that could be introduced into an enzyme, efficient means of introducing and identifying mutations that provide improved function are required.
When available, information regarding enzyme mechanism, structure, or sequence homology has proven useful in helping to identify beneficial mutations in an enzyme, typically introduced within its active site.4,5 Such information may not be available, however, or it may prove insufficient to provide the level of improvement needed for a desired application. More fundamentally, targeted mutagenesis efforts aimed at the active site of an enzyme miss the potential benefits of mutations that are not apparent from available data or that emerge from synergistic mutations throughout the protein scaffold.3 To confront this challenge, Arnold and Stemmer independently developed approaches to engineer enzymes by incorporating random mutations, screening or selecting for improved enzyme variants, and iterating the process until the desired level of improvement is obtained.6,7 Directed evolution, as this process is called, requires no detailed knowledge of enzyme structure or mechanism and can be used to identify beneficial mutations throughout an enzyme sequence.3 While powerful, directed evolution of enzymes with useful properties typically requires many iterations of mutagenesis and screening. Hybrid directed evolution approaches have therefore been developed to combine the benefits of targeting mutations (or sets of mutations) based on available information and the benefits of sampling diverse mutations throughout the enzyme structure.2,8 Regardless of the methods used, introducing diversity throughout enzyme structures has proven critical for engineering efficient enzyme activity toward both native and non-native substrates.
The success of protein engineering for biocatalysis has inspired similar efforts to engineer artificial metalloenzymes (ArMs), hybrid catalysts comprised of protein scaffolds containing synthetic metal cofactors.9 Unlike natural enzymes, however, extensive mechanistic data on these systems are not available (they must be created to begin with), and it is not obvious that the phylogeny of a scaffold protein, which reflects its natural evolution for a native function, will provide insight into mutations that could improve ArM activity.8,10 While structural information can be used, it is not always available, and homology models are imperfect structural references. Even if the structure of a given scaffold is known, solving structures of descendent ArMs may not be possible or informative since the flexibility of many synthetic cofactor linkers (e.g. 1) can lead to poor occupancy in electron density maps. Despite these limitations, crystal structures of parental scaffolds have guided all ArM engineering efforts reported to date. For example, highly selective ArMs have been obtained via structure-guided, targeted mutagenesis of scaffold proteins, including streptavidin,11,12 cyt cb562,13 and myoglobin.14-17
We previously applied a targeted mutagenesis approach to engineer dirhodium ArMs that catalyze enantioselective cyclopropanation of styrenes (Fig. 1A).18 This effort involved replacing the active site serine (S477) of Pyrococcus furiosus (Pfu) prolyl oligopeptidase (POP) with a genetically encoded p-azidophenylalanine (Z) residue to which cofactor 1 was linked via strain-promoted azide-alkyne cycloaddition (SPAAC, Fig. 1B, C). In contrast to the scaffolds noted above, an X-ray crystal structure for Pfu POP had not been reported at the outset of our studies, it does not bind metal cofactors that can be substituted to form ArMs, and its native substrate binding was not used for ArM formation. Instead, Pfu POP was selected on the basis of its high stability19 and the large active site common to POP family enzymes,20 which earlier efforts21 suggested would be necessary to contain and impart selectivity to 1. A previously reported homology model of this enzyme provided some guidance for targeting mutations,22 but a large number of individual mutants had to be cloned and evaluated,18 which proved to be a time-consuming effort due to differences between the actual and homology structures and a complete lack of information regarding cofactor location within the active site.
Figure 1. Model reaction and ArM structure.

a , Model cyclopropanation reaction used for ArM evolution. The dirhodium cofactor catalyzes diazo decomposition to generate a rhodium carbenoid intermediate that can insert into the olefin π bond or (because the reactions are carried out in aqueous solution) the water O-H bond. The protein scaffold provides chemoselectivity and enantioselectivity. Reactions conducted using 22 mM styrene, 4.4 mM diazo, and 1 mol% ArM in 10% v/v THF/50 mM PIPES (pH 7.4) containing 1.75 M NaBr at 4 °C. b, ArM formation via SPAAC19 between bicyclononyne-substituted metal complexes and a genetically encoded 4-l-azidophenylalnine residue, allowing covalent attachment of the cofactor, even in cellular lysate. c, Structure of dirhodium cofactor 1.
The tedious sequential mutagenesis required to generate selective POP ArMs reflected limitations on the extent to which information-based targeted mutagenesis approaches commonly used for engineering natural enzymes,10 would be effective in ArM engineering. We reasoned that random mutagenesis would be a powerful tool for ArM engineering because it does not require structural information, and could reveal beneficial mutations distal to ArM active sites.3 While such mutations have proven highly important for engineering natural enzymes, they are difficult to predict, and their potential to impact ArM catalysis has not been addressed.15 From the perspective of transition metal catalysis, random mutagenesis of ArMs also offers the unique opportunity to probe the effects of structural changes far beyond the metal secondary coordination sphere. ArM evolution via random mutagenesis is complicated, however, by challenges associated with high throughput expression of scaffold libraries, introducing synthetic metal cofactors into these libraries, eliminating background catalysis by free cofactor, and ensuring that cellular lysate components do not interfere with catalysis.12,23,24 To address these challenges, here we used a dirhodium ArM cyclopropanase previously engineered in our laboratory as a platform to explore the potential to improve ArM selectivity via random mutagenesis. Libraries generated via both random mutagenesis of the entire POP β-propeller domain and combinatorial mutagenesis22 of residues projecting into the POP active site were examined. High selectivity for either product enantiomer was achieved from mutations both inside and outside of the ArM active site, and the evolved cyclopropanases had improved selectivity toward a number of additional dirhodium-catalyzed reactions, including N-H insertion, S-H insertion, and Si-H insertion. These results highlight the potential for random mutagenesis to identify mutations in ArMs that would be difficult to predict even with detailed structural data. When combined with existing targeted mutagenesis approaches,8,10 our strategy provides a general framework for ArM evolution analogous to that used for natural enzymes2 and suggests that similar improvements in the efficiency of diverse ArMs should be possible.
Results
ArM Evolution via Error Prone PCR
Our ArM evolution efforts relied on several unique aspects of the Pfu POP scaffold18,22 and the SPAAC bioconjugation method used for cofactor incorporation (Fig. 2)21. At the outset of our efforts, a crystal structure of Pfu POP was not available, so error prone PCR was used to introduce mutations throughout the POP β-propeller domain (Q48 to V335, Fig. 2) that comprises the putative ArM active site18. The high stability of POP allowed the use of high mutation rates (typically 4-5 residue mutations/variant)25 and various manipulations involved in parallel ArM formation. Gibson assembly of the β-propeller variants provided the desired scaffold libraries without the need for introducing restriction sites in the β-propeller domain. Conditions were then optimized for expressing POP in E. coli in high yield in 24 or 96 well plates. The stability of POP also allowed for thermal denaturation of E. coli proteins following cell lysis.26 Centrifugation provided cell lysates containing approximately 50 μM POP based on SDS PAGE analysis (Supplementary Fig. 1).
Figure 2. Overview of ArM evolution protocol.
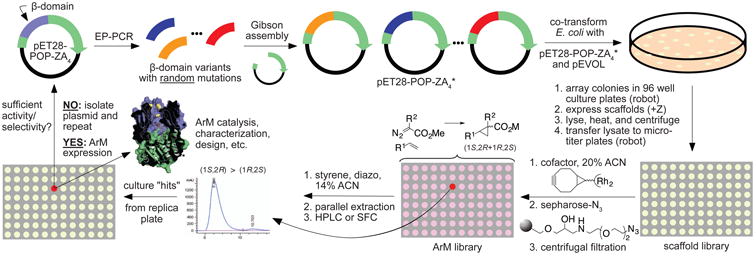
From Top-Left: The gene encoding a POP scaffold (e.g. 0-ZA4) in pET-28 (the POP β-domain is shown in grey-blue in both plasmid and the ArM structure model) is used to generate a library of β-domain variants with random mutations via error prone PCR. The remaining backbone is amplified separately, and the gene library is ligated via Gibson assembly. The gene library is co-transformed into E. coli with pEVOL-pAzF, a plasmid containing an orthogonal tRNA and aaRS for Amber stop codon suppression. A colony picker robot is used to array colonies into 96 well plates, where the POP genes are expressed, and cells are lysed, heated, and centrifuged. Next, a liquid handling robot is used to transfer the lysate to a fresh 96 well plate, cofactor is added to generate POP ArMs, and an azide-substituted resin is added to scavenge unreacted cofactor. The ARM library is screened by HPLC or SFC for increased enantioselectivity of olefin cyclopropanation. Putative hits are isolated and cultured for verification on a larger scale without the presence of cellular lysate debris. If the putative hit is validated, it is used as parent for an additional round of mutagenesis until the desired activity/selectivity is observed.
The high efficiency of SPAAC allowed for rapid bioconjugation of 1 to POP in 96 well plates using a roughly two-fold excess of cofactor (93.75 μM) over the average amount of protein in each well (Supplementary Fig. 1). A commercially available azide-substituted resin could be used to scavenge the excess cofactor following bioconjugation, and a comparable resin was prepared to facilitate this process.27 In the absence of of this step, non-selective reactions catalyzed by free cofactor will compete with ArM catalysis, reducing the observed selectivity. Cyclopropanation reactions were then conducted in deep well plates, the reaction mixtures were extracted with hexanes, and the organic extracts were analyzed by HPLC.
This procedure was used to evolve the selectivity of POP variant 0-ZA4, the starting point for our rational design effort (previously called POP-ZA4-1)18, for the cyclopropanation of 4-methoxystyrene (Fig. 1A). Gratifyingly, only three rounds of mutagenesis and screening (96, 48, and 576 variants/round, respectively, Supplementary Fig. 2) were required to obtain 92% enantiomeric excess (e.e.) for the target cyclopropanation reaction (Fig. 3A, Supplementary Table 2). Variants with improved selectivity relative to the parent enzyme assayed in the same plate were taken to be hits, and these putative hits were validated following purification. The variant with the highest selectivity was selected as the parent for the next round of mutagenesis (Supplementary Fig. 3). The high mutation rates used effectively allowed for multiplexed analysis of several mutations in each variant, and the most improved variant in each round contained four residue mutations (Fig. 3). Analysis of product formation over time in reactions catalyzed by 3-VRVH, 0-ZA4, and HFF (from our previous rational design effort)18 revealed that 3-VRVH possessed significantly higher activity than the unevolved or rationally designed ArMs (Fig. 3B, Supplementary Fig. 6).
Figure 3. Overview of the directed evolution lineages generated and time-course comparison of several catalysts.

a , Enantioselectivities and yields (inset) for cyclopropanation of styrene catalyzed by evolved ArM variants to give 3 (Fig. 1A). Although enantiomeric excess was the screening criteria, yields of the desired product also increased across the lineages. Each variant contains the mutations indicated plus those from the previous round(s) of evolution. The residues identified from deconvolution experiments were cloned into a minimal mutant (1-GSH), which was able to provide equivalent enantioselectivity as the final mutant (3-VRVH) but significantly lower yield. This highlights the complexity of biomolecule scaffolds and the importance of random mutagenesis for ARM engineering. Reactions conducted as shown in Figure 1A using 22 mM styrene, 4.4 mM diazo, and 1 mol% catalyst in 10% v/v THF/50 mM PIPES (pH 7.4) containing 1.75 M NaBr or NaCl at 4 °C for 4h. Enantiomeric excess and yield of 3 were determined by analysis of HPLC chromatograms for crude reaction mixtures relative to internal standards. b, time course experiments for reactions catalyzed by different dirhodium catalysts. The yield and qualitative rate for the evolved variant 3-VRVH exceeds parent 0-ZA4 as well as a previously reported ArM produced via rational design. The reaction in aqueous buffer of the free cofactor is also shown. All data points shown are an average of 2 reactions.
Individually reverting each mutation in each of the improved variants indicated that only three mutations (S301G/G99S/Y326H) were required for most of the improvement in selectivity observed in 3-VRVH, which contains 12 mutations relative to 0-ZA4 (Fig. 3, Supplementary Fig. 7). On the other hand, the product yield obtained for the variant containing only these essential mutations, 1-GSH, was significantly reduced (Fig. 3, inset). This finding suggests that increased selectivity appears to have evolved at the expense of activity, but other mutations throughout the POP structure overcome this limitation, leading to higher conversion for 3-VRVH relative to 1-GSH.
During the course of our evolution efforts, we solved the crystal structure of wt Pfu POP (PDB ID 5T88), which allowed for structural analysis of the mutations identified via directed evolution. Of the mutations in 1-GSH, G99S and Y326H are in the POP active site while S301G is not (Fig. 4). Notably, G99F and L328H were identified as beneficial mutations in our previous engineering effort, but the crystal structure showed that the locations of these residues differed significantly from those suggested by the homology model used.18 The identification of distinct mutations via targeted and random mutagenesis highlights the utility of both approaches for ArM engineering and the range of different active site configurations that can improve ArM selectivity. Moreover, the improvement in selectivity resulting from S301G and the improvements in yield resulting from mutations throughout the POP scaffold clearly attest to the importance of non-active site mutations for ArM optimization and library methods that enable their identification.
Figure 4. Location of mutations in evolved ArMs.
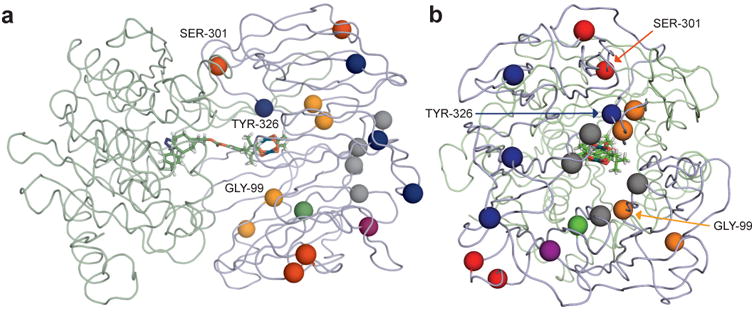
a , side and b, top views of POP ribbon model with spheres showing the location of mutations in variants 0-ZA4 (gray), 1-NAGS (red), 2-NSIA (orange), and 3-VRVH (blue). Mutations with greatest impact on enantioselectivity are labeled. Residues modified via carbene insertion (vide infra) are also shown as spheres (purple: W175; green: W142).
ArM Evolution via Combinatorial Codon Mutagenesis
Despite the efficiency by which directed evolution was able to improve cyclopropanation enantioselectivity, at no point was a variant that provided significant selectivity for the opposite product enantiomer observed. Previous reports have shown that altering the site of cofactor linkage within a scaffold can alter ArM selectivity,28 so we used the Pfu POP crystal structure to identify alternate linkage sites for 1. POP variant F413Z (Fig. 5A, blue sphere) led to an ArM that provided modest selectivity for the desired product enantiomer (30% ee, Fig. 3), opposite that obtained using the S477Z linkage site (Fig. 5A, green sphere). While the Pfu POP crystal structure does not reveal specific residues that could be targeted to further improve the selectivity of variant F413Z,8 it does indicate which residues actually project into the POP active site. We therefore reasoned that random mutagenesis of these active site residues might be a useful approach for rapidly improving the selectivity of variant F413Z.
Figure 5. Combinatorial codon mutagenesis sites and protocol.

a , Ribbon model of POP variant 0-ZA4 showing the location of residue 477 (green sphere), residue 413 (blue sphere), and residues selected for combinatorial codon mutagenesis (remaining spheres). Mutations in 1-RFY (Q98R, G99F, and P239Y) are shown as red, orange, and yellow spheres, respectively. b, Scaffold immobilization and ArM formation on Ni-NTA resin in 96-well plates using libraries of scaffold variants generated via error prone PCR or CCM as shown in Figure 2.
Based on this hypothesis, a combinatorial codon mutagenesis library22 of POP-413Z variants, each containing an average of two degenerate NDT codons at any of the 25 remaining active site residues, was constructed (Fig. 5A). The use of the NDT codon eliminated the possibility of stop codon mutations and allowed for rapid and efficient sampling of random mutations throughout the POP active site. Ninety-two members of this library were screened on-bead using a modified version of the protocol shown in Figure 2 in which POP-413Z variants in cell lysate were immobilized on Ni-NTA resin29 preloaded into 96-well filter plates (Fig. 5B, Supplementary Fig. 8). ArM formation and catalysis using immobilized POP-413Z simplified cofactor removal, reaction set-up, and product isolation relative to the analogous processes in solution. Control reactions using lysate containing POP-413A or lacking POP entirely provided a conversion of 2-6% and negligible enantioselectivity, indicating that properly assembled ArM was responsible for catalysis. One variant identified via this process, 1-RFY (POP-413Z Q98R/G99F/P239Y), possessed significantly improved enantioselectivity, providing the desired cyclopropanation enantiomer in 80% ee (Fig. 3, Supplementary Fig. 9; see Supplementary Fig. 10 for hit validation under screening conditions). Individually reverting each mutation in 1-RFY led to decreased product yield and enantioselectivity (Supplementary Table 6), indicating that all three mutations contributed to the improved selectivity of 1-RFY. Notably, neither Q98 nor P239 was targeted in our earlier more conservative targeted mutagenesis efforts, and no beneficial mutations were identified at these sites during the directed evolution campaign targeting the original enantiomer. These data show how combinatorial codon mutagenesis uniquely balances the benefits of sampling random mutations at a large number of sites with those of restricting those sites to particular regions of an enzyme.10 While more focused mutagenesis strategies can often be used for engineering natural enzymes,8 these require sequence and structural information that is often not available for ArMs; our combined approach used here is an attractive strategy for these enzymes.
In Situ ArM Modification via Carbene Insertion
To begin to shed light on the mechanisms by which the POP scaffolds evolved in this work impart selectivity to synthetic cofactor 1, reaction profiles for the ArMs in the 3-VRVH lineage were examined. These profiles showed significant differences in both cyclopropanation yield and enantioselectivity depending on the reaction times used (Fig. 6). Interestingly, the enantiomeric excess of the cyclopropane formed was observed to decrease during the course of these reactions, and this decrease itself appeared to decrease for variants along the 3-VRVH lineage. While the free energy change associated with the observed decrease in enantioselectivity of 3-VRVH (from 95% ee to 92% ee) is roughly equivalent to that associated with the decrease in enantioselectivity of ZA4 (from 65% ee to 50% ee), 1-GSH undergoes no change in enantioselectivity. Moreover, analysis of reactions catalyzed by 0-ZA4 and 3-VRVH starting immediately after ArM addition (Supplementary Fig. 6) revealed only a negligible decrease in ee for 3-VRVH (from 94% ee to 93% ee) and a larger decrease in ee for 0-ZA4 (from 73% ee to 40% ee). The former shows that minor variation in 3-VRVH selectivity is significantly impacted by error associated with the HPLC measurements used to determine ee, while the latter presumably reflects rapid loss of enantioselectivity at times before those initially analyzed in Fig. 6. Based on these observations and a number of studies on dirhodium catalyzed carbene insertion into peptides,30 we hypothesized that modification of the POP scaffold via carbene insertion could be occurring in certain POP variants. If this was the case, it would provide some rationale by which residues could be targeted to improve ArM efficiency. Indeed, Arnold recently reported that carbene insertion into cytochrome P450 BM3 during cyclopropanation reactions catalyzed by this enzyme led to decreased cyclopropanation activity and selectivity.31
Figure 6. Time course experiments of ArM catalyzed cyclopropanations of styrene with (4-methoxyphenyl) methyldiazoacetate.

(see Supplementary Table 2 for full data). a, Plot of cyclopropane yield versus time, showing increases for each generation of mutants. b, Plot of cyclopropane enantiomeric excess (e.e.) versus time. A decrease in %e.e. over the course of reaction was observed, which decreases along the lineage. The cause of this was investigated but not identified (see Main Text). Reactions were performed in triplicate with standard deviation shown. *2-NSIA reactions were performed in duplicate, prohibiting standard deviation calculation.
To determine if carbene insertion into POP residues was leading to the observed decrease in POP ArM cyclopropanase selectivity (Fig. 6), both 0-ZA4 and 3-VRVH were purified following incubation with styrene and diazo 2 (Fig. 1A), double-digested using cyanogen bromide followed by trypsin, and analyzed by tandem LC-MS/MS (Supplementary Fig. 12). The double-digestion procedure enabled near complete sequence coverage of the thermostable protein (trypsin alone was insufficient), which was exploited for unbiased differential modification searches for carbene-modified residues. These studies identified two modification sites, W175 and W142 (Fig. 4), that were exclusively detected under carbene-generating conditions and not control reactions of scaffold alone (Supplementary Fig. 12). Ion intensities of both the modified and unmodified tryptic peptides indicated a low level of modified W175, which is a surface residue (Supplementary Fig. 12). In contrast, extensive carbene insertion was detected at W142 in both the 1-NAGS and 3-VRVH scaffolds (Supplementary Fig. 12). Both of these residues in variant 0-ZA4 were individually mutated to alanine, but both 0-ZA4-W175A and -W142A provided yield and selectivity profiles similar to those of 0-ZA4 (i.e. similar decreases in ee over time, Supplementary Fig. 12). These data indicate that while scaffold modification occurs during catalysis, modification at W142 or W175 is not responsible for the decrease in POP ArM cyclopropanase selectivity observed.
Comparable levels of modification for evolved variants in the cyclopropanase lineage were also observed based on the relative ion counts of modified and unmodified fragments and intact scaffold observed via LC/MS-MS (Supplementary Fig. 12). This finding does not, however, rule out the possibility that low levels of carbene modification at residues not detected in our analysis could impact ArM selectivity. Given the wide range of residues that can be modified via proximity-driven carbene insertion reactions,32 removing all such residues from an ArM active site will often be challenging and would greatly constrain the range of residues that could be used to improve ArM selectivity. The fact that 3-VRVH suffers from only a minor loss in enantioselectivity over time suggests that if carbene modifications at sites not detected in our analysis impact ArM selectivity, random mutations identified via directed evolution reduced this impact. Indeed, the ability to evolve ArMs via iterative random mutagenesis provides a general framework for improving ArM selectivity even when the molecular origins of selectivity are unknown, just as can be accomplished for natural enzymes.3
Substrate and Reaction Scope of Evolved ArMs
ArM evolution also provides lineages of improved variants that can be studied to provide insight into ArM mechanism and function. Given the wide range of reactions catalyzed by dirhodium complexes, one immediate question that might be asked of the ArM cyclopropanase lineage described in this work is the extent to which improved selectivity for styrene cyclopropanation carries over to other dirhodium-catalyzed reactions. We previously showed that our rationally designed ArM variants provided improved enantioselectivity on several additional styrene/diazo pairs, and this proved to be the case for the variants evolved in the current study (Table 1). Moreover, improved enantioselectivity was also observed for other dirhodium catalyzed reactions, including formal carbene insertion into Si-H, S-H, and N-H bonds (Table 2). 1-RFY provided the opposite enantiomer of each of these products, although lower selectivity was observed (Supplementary Table 3). These results illustrate the power of directed evolution to provide ArMs for reactions beyond the scope of the initial evolution target (i.e. enantioselective cyclopropanation).33 Nonetheless, the general improvement observed for these reactions, which proceed via different mechanisms in solution, suggests that even the evolved 3-VRVH and 1-RFY scaffolds represent only the tip of the iceberg of reactivity that may be accessed using dirhodium ArMs. Further evolution aimed at improving both the selectivity and reaction specificity of these ArMs will shed light on the extent to which proteins can control the reactivity of synthetic organometallic complexes and the mechanism(s) by which this control is imparted.
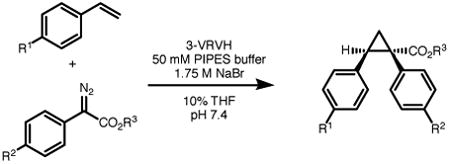
Table 1.
Additional products from 3-VRVH catalyzed (1 mol%) cyclopropanation using conditions from Figure 1a with %yield and (e.e.) shown.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | R1 | R2 | R3 | Yield (%) | e.e. (%) |
| 1 | MeO | MeO | Me | 25 | 86 |
| 2 | H | MeO | Et | 43 | 94 |
| 3 | Cl | MeO | Me | 46 | 86 |
| 4 | H | H | Me | 66 | 88 |
| 5 | H | Cl | Me | 52 | 90 |
| 6 | H | Br | Me | 54 | 92 |
Reactions conducted as shown in Figure 1A using 22 mM styrene, 4.4 mM diazo, and 1 mol% ArM in 10% v/v THF/50 mM PIPES (pH 7.4) containing 1.75 M NaBr at 4 °C for 4 hours. Yields and e.e. were determined by HPLC using an internal standard (1,3,5-trimethoxybenzene).
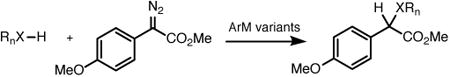
Table 2.
Additional X-H insertion reactions catalyzed by 3-VRVH (1 mol%).
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Substrate | 0-ZA4 | % yield (e.e.) 1-NAGS |
3-VRVH |
| 1 | PhMe2Si–H | 45 (6) | 43 (14) | 35 (64) |
| 2 | PhS–H | 51 (6) | 66 (10) | 64 (32) |
| 3 |
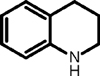
|
55 (8) | 77 (6) | 73 (40) |
Reactions conducted as shown in Figure 1A using 22 mM styrene, 4.4 mM diazo, and 1 mol% ArM in 10% v/v THF/50 mM PIPES (pH 7.4) containing 1.75 M NaBr at 4 °C for 4 hours. Yields and e.e. were determined by HPLC using an internal standard (1,3,5-trimethoxybenzene).
Summary and Conclusion
Random mutagenesis has proven to be a critical tool for engineering natural enzymes for selective catalysis.3 In the absence of a comprehensive understanding of enzyme structure-activity relationships, the ability to create and survey the impact of diversity throughout an enzyme's structure is essential to identifying beneficial mutations that can, collectively, lead to enormous improvements in catalytic efficiency. Many reports have shown that ArM function can be significantly improved via mutations targeted to ArM active sites.15 Prior to this report, however, random mutagenesis had not been used for ArM evolution, and the question of whether ArM function could be further (or even significantly) improved via mutations at sites throughout the ArM structure had not been addressed.
We have shown that iterative random mutagenesis can be used to generate dirhodium ArM variants with improved enantioselectivity for a model styrene cyclopropanation reaction. The evolved ArMs also exhibited improved selectivity toward other dirhodium-catalyzed reactions including Si-H insertion and formal insertion into N-H and S-H bonds. These improvements resulted primarily from higher inherent ArM selectivity, as evidenced by increased enantioselectivity at early reaction times, and may also reflect reduced degradation of enantioselectivity during the course of catalysis (Fig. 4). Importantly, mutations throughout the POP scaffold, including at sites distal to the active site, significantly impacted both ArM activity and selectivity. This finding has broad implications for metalloprotein design. The vast majority of efforts reported to date focus on engineering metal binding sites.34 Using the approach described herein, the primary metal binding site was defined by the cofactor used and only secondary sphere changes were made. Moreover, most of these changes were distal to the pocket in which the metal center resides, clearly showing that a focus on the entire protein, rather than just the primary coordination sphere or even residues proximal to it, can lead to significant improvements over current approaches.
As is frequently found for mutations identified via directed evolution of natural enzymes, the mechanisms by which these distal mutations impact ArM selectivity are not yet clear. LC/MS-MS analysis of ArMs in the selectivity lineage following cyclopropanation reactions revealed significant scaffold modification via carbene insertion, but ArM selectivity still decreased when modified residues were replaced with alanine, ruling out scaffold modification as a major factor for improved ArM selectivity. Distal mutations have often been suggested to affect conformational changes required for enzyme catalysis,35 and, indeed, conformational dynamics are proposed to play a significant role in the native peptide hydrolysis reactions catalyzed by POP.36 The impact of mutations identified in this work on both POP hydrolase and ArM cyclopropanase activity are thus the subject of ongoing efforts in our group. We believe such studies will provide valuable insights into how proteins can control the reactivity of synthetic metal centers, and the POP platform discussed in this work will provide a valuable subject for these studies. Nonetheless, the improved catalytic efficiency of ArM variants resulting from mutations throughout the POP scaffold, including distal to the active site, clearly demonstrate the utility of random mutagenesis for ArM optimization. While we used random mutagenesis to identify these mutations, any number of mutagenesis schemes could ultimately be used to achieve similar results.2 We anticipate that similar efforts with other ArMs, particularly those generated from scaffold proteins that did not evolve in nature to bind metal cofactors,14 will enable the same levels of improvement for these synthetic systems as has been observed for the evolution of natural enzymes.3
Methods
POP scaffold expression and bioconjugation to generate the corresponding ArMs was conducted as previously described.13 Solutions of aryldiazoacetate (25 μL, 96 mM, in THF), styrene (25 μL, 485 mM, in THF), and ArM (500 μL, 48 μM, in 50 mM PIPES buffer, pH 7.4, with 1.75 M NaBr or NaCl additive) were added to a 1.5-ml microcentrifuge tube. The final concentration of the reagents were as follows: 22 mM olefin, 4.4 mM aryldiazoacetate, 44 μM ArM. The resulting mixture was left shaking at 750 r.p.m. on an Eppendorf Thermomixer R at 4 °C for 4 hours. The reaction was quenched by adding 20 μL 1,4-benzodioxole solution (22 mM, in THF) and 600 μL ethyl acetate. The mixture was vortexed and centrifuged (15,000g, 3 min). The top organic layer was collected and the bottom aqueous layer was extracted with 600 μL ethyl acetate twice. The organic layers were combined, evaporated, and re-dissolved in 200 μL 10% v/v 2-propanol in hexane. This solution (5 μL) of the crude product was analysed on NP-HPLC to determine product yield and enantiomeric excess (e.e.). Detailed procedures for the directed evolution method used to improve e.e. and ArM characterization are provided in the supporting information.
Data Availability
Complete experimental procedures, including primer sequences, synthetic procedures, characterization data, library generation and screening protocols are described in the Supplementary Information. Sequencing and library screening data and is available upon request. The crystal structure for POP R464L has been deposited in the Protein Data Bank (PDB) under accession number 5T88.
Supplementary Material
Acknowledgments
This work was supported by, or in part by, the U.S. Army Research Laboratory and the U. S. Army Research Office under contract/grant numbers W911NF-14-1-0334 and 66796-LS-RIP (to J.C.L.), the NSF under CAREER Award CHE-1351991 (to J.C.L.), The David and Lucile Packard Foundation (to J.C.L.), the NIH NCI (R00CA175399 to R.E.M.), the Damon Runyon Cancer Research Foundation (DFS-08-14 to R.E.M.), and the NSF under the CCI Center for Selective C–H Functionalization (CHE-1205646, to J.C.L.).K.E.G. and D.M.U. were funded by an NIH Chemistry and Biology Interface Training Grant (T32 GM008720), and G.L. was supported by the Kwanjeong Educational Foundation. MS data were acquired on instruments purchased using an NSF instrumentation grant (CHE-1048528).
Footnotes
Author Contributions: H.Y. and P.S. developed the soluble ArM evolution procedure.
C.Z. optimized the soluble ArM evolution procedure.
A.M.S. and D.M.U. optimized the soluble ArM evolution procedure and collected conversion and selectivity data for all evolved ArMs.
H.J.P. optimized and executed the immobilized ArM evolution procedure.
K.B. and Y.G. cloned POP amber mutants and the combinatorial codon mutagenesis library.
K.E.G., G.L., and R.E.M. conducted and analyzed the LC-MS/MS experiments.
J.C.L. devised the experiments and procedures, designed the ArM variants and libraries, analyzed data, and wrote the manuscript.
References
- 1.Mahatthananchai J, Dumas AM, Bode JW. Catalytic Selective Synthesis. Angew Chem Int Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- 2.Bornscheuer UT, et al. Engineering the Third Wave of Biocatalysis. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 3.Romero PA, Arnold FH. Exploring Protein Fitness Landscapes by Directed Evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu Y, Berry S, Pfister T. Engineering novel metalloproteins: Design of metal-binding sites into native protein scaffolds. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 5.Gerlt JA, Babbitt PC. Enzyme (re) design: lessons from natural evolution and computation. Curr Opin Chem Biol. 2009;13:10–18. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen K, Arnold FH. Tuning the activity of an enzyme for unusual environments: Sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci USA. 1993;90:5618–5622. doi: 10.1073/pnas.90.12.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stemmer WPC. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 8.Lutz S. Beyond directed evolution-semi-rational protein engineering and design. Curr Opin Biotech. 2010;21:734–743. doi: 10.1016/j.copbio.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis JC. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013;3:2954–2975. [Google Scholar]
- 10.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol. 2009;5:526–529. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 11.Reetz MT, Peyralans JJP, Maichele A, Fu Y, Maywald M. Directed evolution of hybrid enzymes: Evolving enantioselectivity of an achiral Rh-complex anchored to a protein. Chem Commun. 2006:4318–4320. doi: 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- 12.Jeschek M, et al. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature. 2016;537:661–665. doi: 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- 13.Song WJ, Tezcan FA. A designed supramolecular protein assembly with in vivo enzymatic activity. Science. 2014;346:1525–1528. doi: 10.1126/science.1259680. [DOI] [PubMed] [Google Scholar]
- 14.Key HM, Dydio P, Clark DS, Hartwig JF. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature. 2016;534:534–537. doi: 10.1038/nature17968. [DOI] [PubMed] [Google Scholar]
- 15.Hyster TK. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew Chem Int Ed. 2016;55:7344–7357. doi: 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- 16.Sreenilayam G, Moore EJ, Steck V, Fasan R. Metal Substitution Modulates the Reactivity and Extends the Reaction Scope of Myoglobin Carbene Transfer Catalysts. Adv Synth Catal. 2017;359:2076–2089. doi: 10.1002/adsc.201700202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sreenilayam G, Moore EJ, Steck V, Fasan R. Stereoselective Olefin Cyclopropanation under Aerobic Conditions with an Artificial Enzyme Incorporating an Iron-Chlorin e6 Cofactor. ACS Catal. 2017;7:7629–7633. doi: 10.1021/acscatal.7b02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juhász T, Szeltner Z, Polgár L. Properties of the prolyl oligopeptidase homologue from Pyrococcus furiosus. FEBS Letters. 2006;580:3493–3497. doi: 10.1016/j.febslet.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Polgár L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59:349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H, Srivastava P, Zhang C, Lewis JC. A General Method for Artificial Metalloenzyme Formation through Strain‐Promoted Azide–Alkyne Cycloaddition. ChemBioChem. 2014;15:223–227. doi: 10.1002/cbic.201300661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris MN, Madura JD, Ming LJ, Harwood VJ. Kinetic and Mechanistic Studies of Prolyl Oligopeptidase from the Hyperthermophile Pyrococcus furiosus. J Biol Chem. 2001;276:19310–19317. doi: 10.1074/jbc.M010489200. [DOI] [PubMed] [Google Scholar]
- 23.Wilson YM, Dürrenberger M, Nogueira ES, Ward TR. Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts. J Am Chem Soc. 2014;136:8928–8932. doi: 10.1021/ja500613n. [DOI] [PubMed] [Google Scholar]
- 24.Reetz MT, et al. A robust protein host for anchoring chelating ligands and organocatalysts. ChemBioChem. 2008;9:552–564. doi: 10.1002/cbic.200700413. [DOI] [PubMed] [Google Scholar]
- 25.Drummond D, Iverson B, Georgiou G, Arnold FH. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J Mol Biol. 2005;350:806–816. doi: 10.1016/j.jmb.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 26.Dąbrowski S, Kur J. Cloning and Expression in Escherichia coli of the Recombinant His-Tagged DNA Polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein expression and purification. 1998;14:131–138. doi: 10.1006/prep.1998.0945. [DOI] [PubMed] [Google Scholar]
- 27.Punna S, Kaltgrad E, Finn MG. ‘Clickable’ Agarose for Affinity Chromatography. Bioconjugate Chem. 2005;16:1536–1541. doi: 10.1021/bc0501496. [DOI] [PubMed] [Google Scholar]
- 28.Davies RR, et al. Artificial metalloenzymes based on protein cavities: Exploring the effect of altering the metal ligand attachment position by site directed mutagenesis. Bioorg Med Chem Lett. 1999;9:79–84. doi: 10.1016/s0960-894x(98)00684-2. [DOI] [PubMed] [Google Scholar]
- 29.Filice M, et al. Synthesis of a heterogeneous artificial metallolipase with chimeric catalytic activity. Chem Commun. 2015;51:9324–9327. doi: 10.1039/c5cc02450a. [DOI] [PubMed] [Google Scholar]
- 30.Ball ZT. Designing Enzyme-like Catalysts: A Rhodium(II) Metallopeptide Case Study. Acc Chem Res. 2013;46:560–570. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]
- 31.Renata H, et al. Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J Am Chem Soc. 2016;138:12527–12533. doi: 10.1021/jacs.6b06823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popp BV, Ball ZT. Proximity-driven metallopeptide catalysis: Remarkable side-chain scope enables modification of the Fos bZip domain. Chem Sci. 2011;2:690–695. [Google Scholar]
- 33.Lewis JC, Arnold FH. Catalysts on Demand: Selective Oxidations by Laboratory-Evolved Cytochrome P450 BM3. CHIMIA. 2009;63:309–312. [Google Scholar]
- 34.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tokuriki N, Tawfik DS. Protein Dynamism and Evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 36.Kaushik S, Etchebest C, Sowdhamini R. Decoding the structural events in substrate-gating mechanism of eukaryotic prolyl oligopeptidase using normal mode analysis and molecular dynamics simulations. Proteins. 2014;82:1428–1443. doi: 10.1002/prot.24511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete experimental procedures, including primer sequences, synthetic procedures, characterization data, library generation and screening protocols are described in the Supplementary Information. Sequencing and library screening data and is available upon request. The crystal structure for POP R464L has been deposited in the Protein Data Bank (PDB) under accession number 5T88.