

Abstract
Humans have co-speciated with their gut-resident microbes, but it is difficult to infer features of our ancestral microbiome. Here, we examine the microbiome profile of 350 stool samples collected longitudinally for over a year from the Hadza hunter-gatherers of Tanzania. The data reveal annual cyclic reconfiguration of the microbiome, in which some taxa become undetectable only to reappear in a subsequent season. Comparison of the Hadza dataset with data collected from 18 populations in 16 countries with varying lifestyles reveals that gut community membership corresponds to modernization: Notably, the taxa within the Hadza that are the most seasonally volatile similarly differentiate industrialized and traditional populations. These data indicate that some dynamic lineages of microbes have decreased in prevalence and abundance in modernized populations.
The gut microbiota (or microbiome) is an integral part of host biology, influencing immune function and development, metabolism and the central nervous system (1–3). This complex community of microbes must be reassembled each generation since before birth infants lack a gut microbiota. Microbial lineages appear to be vertically transmitted (4, 5) and have been associated with humans for >15M years (6). Microbiota membership is sensitive to diverse perturbations including dietary change and enteric pathogens (7, 8). The resilience of a community is quite individual: in some, a return to starting state after perturbation (9, 10) is observed; in others, new stable states may result (11, 12), which can become pathological. However, most information about human gut microbiota dynamics are collected in the context of responses to antibiotic treatments and described in humans living in an urban setting, which accumulating evidence suggests, have decreased diversity and different microbiota membership as compared to the gut communities of populations living traditional lifestyles (13–18). Indeed, a previous report of 27 Hadza stool microbiomes at a single time point revealed a high level of bacterial diversity (19). Here, we have performed an in-depth, longitudinal analysis of the Hadza hunter-gatherer microbiome to provide insight into the characteristics of gut dynamics of a diverse microbiota in a non-industrial setting.
The Hadza of the central Rift Valley of Tanzania are among the last remaining populations in Africa that live a hunter-gatherer lifestyle (20). Today there are fewer than 200 Hadza that adhere to this traditional way of life. They live in camps that number approximately 5-30 people per camp, although camp numbers vary depending on the season and available resources (21). As a result of encroachment on limited land and rapid transculturation, including increasing exposure to medicines and processed foods, the Hadza way of life is disappearing (20). We collected 350 fecal samples with informed consent from two culturally and geographically similar camps located within 7 km of each other during a 12-month time period spanning 5 sub-seasons (Fig. S1), representing 188 recorded unique individuals (Table S1). To overcome potential biases that repeated sampling might introduce, we limited all analyses in this study to a single sample from each individual, unless otherwise noted. On collection, the samples were immediately stored in liquid nitrogen, and maintained frozen during all transport and storage until processing for analysis.
The Hadza’s activities are largely based around food acquisition. They are affected by the local environment and are subject to two distinct seasons: Wet (Nov-Apr) and Dry (May-Oct). For example, berry-foraging and honey consumption are more frequent during the Wet season whereas hunting is most successful during the Dry. Consumption of fiber-rich tubers and baobab occurs year-round (19, 20). We applied principal coordinates analyses (PCoA) to UniFrac distances of 16S rRNA amplicon profiles generated from samples collected from two Dry and one Wet season (Fig. 1A). Differences in microbiome composition between two seasons have been observed in the agricultural Hutterites of the USA (6, 22). The microbiomes of individual Hadza, when plotted by season revealed cyclical features: microbiotas from the Dry seasons in sequential years were indistinguishable from one another yet were distinguishable from the intervening Wet season microbiota (p<3e-15 and p<3e-16, Wilcoxon; Fig. 1A).
Fig. 1. Hadza gut microbial community compositions are cyclic and can be differentiated by season.
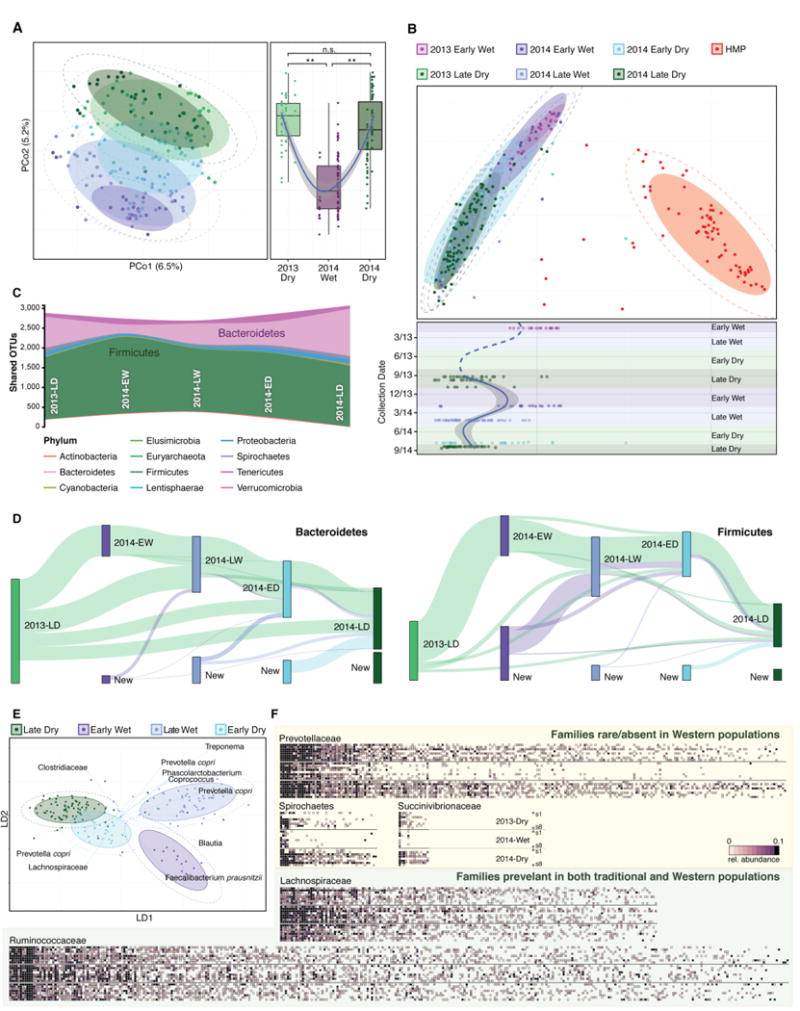
(A) Individual Hadza gut microbiota compositions in 2013-Late-Dry (n=41, light green), 2014-Early-Wet (n=19, purple), 2014-Late-Wet (n=58, light purple), 2014-Early-Dry (n=30, light blue) and 2014-Late-Dry (n=40, dark green) sub-seasons plotted on an unweighted UniFrac PCoA plot (left panel). Samples collected in the Dry season are distinct from Wet season samples (p<3e-15 and p<3e-16, Wilcoxon), while Dry seasons are indistinct (p=0.15, Wilcoxon) (right panel). (B) Individual Hadza gut microbiota compositions from A (n=188), samples collected in 2013-Early-Wet in a previous Hadza study (19) (n=20, violet) and the Human Microbiome Project (HMP) (n=71, red) are shown on a PCoA plot according to their Bray-Curtis dissimilarity at the family taxonomic level (top panel). The Hadza samples across both studies representing 1.75 years are plotted according to their collection date on the y-axis and their position on the first principal coordinate of the Bray-Curtis PCoA in the top panel on the x-axis. The sub-seasons are labeled and indicated by shading, and Loess regression was applied to these points using the collection date and PCo1 coordinates, and the curve was plotted in blue with a 95% pointwise confidence interval band in gray on the plot using the data within this study. The dashed blue line is a continuation of the regression curve yet is an implied regression curve assuming the appropriate inflection points are captured with data from our study. (C) OTUs that are shared by at least 10 percent of the population within each season are tracked using Sankey plots in both the Bacteroidetes and Firmicutes. The heights of the rectangles indicate the relative number of OTUs and each sub-season has a distinct color. The lines represent the transfer of OTUs between seasons and are colored by the first season of appearance. (D) Linear discriminant analysis (LDA), a supervised learning approach that utilizes a linear combination of features to maximize the separation of classes, successfully separates the sub-seasons. The length and direction of the arrows indicate the normalized scalings for each of the features (OTUs). (E) Heatmaps represent microbiotas from all individuals (n=8) that were sampled across the Wet and both Dry seasons. Along the y-axis of each heatmap individuals are ordered similarly across all three seasons. The top eight rows correspond to the individuals’ microbiotas in 2013-Dry; middle, 2014-Wet; bottom, 2014-Dry. Along the x-axis are unique OTUs that are found in at least 0.1% of the OTUs across the eight individuals and are sorted (left-to-right) by their prevalence across all seasons and are shaded according to the relative abundance of OTUs. The shaded ellipses in all plots represent the 80% confidence interval, the dotted ellipse borders represent the 95% confidence interval. All boxplot distributions are tested using the non-parametric two-sided Wilcoxon rank sum test with Holm correction for multiple hypothesis testing, center values indicate the median and error bars the standard deviation (SD)*, P values < 0.05, ** < 0.01.
We compared our dataset with microbiome profiles previously reported for the Hadza (19) and US residents (Human Microbiome Project; HMP) (23). This analysis revealed commonalities in the taxonomic representation of bacteria within the two Hadza datasets, which, independent of season, segregated from the microbiome of US residents (Fig. 1B, top panel). Notably, the previously reported single season collection from the Hadza fit the cyclic pattern of microbiome reconfiguration (Fig. 1B, bottom panel). Both higher phylogenetic diversity and greater numbers of unique OTUs were observed in the Dry seasons as compared with the Wet season (Fig. S2A).
To understand what might be driving the cyclical pattern, we examined the OTUs that are maintained in the Hadza across the phylogenetic shifts through the seasons. Firmicutes composition remained relatively stable throughout the sampling period, whereas Bacteroidetes OTUs, primarily those of the Prevotellaceae, declined significantly in the Wet season (Fig. S2B). Examining commonly shared OTUs, present in at least 10% of the individuals, season-by-season revealed a pronounced constriction of Bacteroidetes in the Early-Wet season (62.8% decrease in shared OTUs for Late-Dry-2013 to Early-Wet-2014, representing 4.4 standard deviations from the means of all other seasons; Fig. 1C). By contrast the shared number of Firmicutes, remained relatively stable across the seasons (0.21 standard deviation; Fig. S2C).
Tracking individual OTUs within different phyla revealed distinct temporal dynamics within the Bacteroidetes and Firmicutes. Many of the Bacteoidetes OTUs display seasonal volatility, with 70.2% disappearing between 2013-Late-Dry and 2014-Early Wet; 78.2% of those that disappeared, reappeared at later time points (Fig. 1D, left panel). A smaller proportion of the Firmicutes OTUs showed this seasonal cyclic pattern. The greatest number of Firmicutes OTUs disappear between 2014-Late-Wet and 2014-Early Dry (62%); 76% of those are detected at other time points (Fig. 1D, right panel). A supervised learning approach that specifically attempts to distinguish groups by integrating linear combination of OTUs was unable to differentiate the same season (Dry) in sequential years, supporting the cyclic nature of the reconfiguration (Fig. 1E, Fig. S3A; Table S2).
We extended our analysis to determine if other taxa were seasonally volatile or stable. Examining OTUs in the eight Hadza individuals that were sampled across three seasons (Fig. S3B), revealed that the Succinivibrionaceae, Paraprevotellaceae, Spirochaetaceae, Prevotellaceae families were among the most variable across seasons (Fig. 1F, Fig S3C).
Systematic seasonal differences in the Hadza microbiota led us to hypothesize that seasonal dietary changes might lead to related changes in the functional capacity of the microbial community. Previous reports were limited to a single season (19, 24), we therefore selected 35 samples across the seasons (Fig. S4A) and performed both shotgun metagenomic sequencing and untargeted metabolomics to gain insight into community functionality.
Comparison of carbohydrate active enzymes (CAZymes) encoded in Hadza gut metagenomes to those of healthy American subjects identified a more diverse repertoire for utilizing carbohydrates in the Hadza (Fig. 2A, 2B). When comparing the Hadza microbiome across time points, we found no significant differences between the Dry season microbiotas, while the Wet season microbiotas possess a significantly less diverse CAZyome compared to the Dry season (p<0.05, Wilcoxon). The Hadza microbiotas show greater functional capacity for utilization of plant carbohydrates than Americans (Fig. 2D; p<2e-16, Wilcoxon). Our metagenomic data suggest that the microbiotas of healthy Americans have a greater mucin-utilization capacity (indicating less plant material in the diet) than those of the Hadza (Fig. 2C, 2D; p<2e-16, Wilcoxon). In the Dry seasons the Hadza consume more meat, which corresponds with the enrichment of CAZYmes related to animal carbohydrate (Fig. 2D; p=0.03, p=0.04, Wilcoxon). Fructan utilization is enriched in the Wet season (Fig. S4B; p=0.02, Wilcoxon) coincident with berry consumption. Overall, the Wet-season Hadza microbiota has fewer plant, animal, and mucin CAZymes compared with the Dry-season (Fig. S4B; p=0.003, p=0.02, p=0.01, respectively; Wilcoxon). Analyses of KEGG functional groups that rely on nucleotide sequence similarities showed analogous patterns, including consistent representation across the Dry seasons, despite limitations of this approach in identifying novel genetic sequences (Tables S3, S4). Notably, the repertoires of antibiotic resistance genes found in the Hadza were distinct from those of US gut metagenomes (25) (Fig. S4C, S5) and less diverse regardless of season (Fig. 2E; p<0.05, Wilcoxon), demonstrating that the increased diversity of Hadza microbiome composition does not necessarily result in an enrichment of diversity in all functional classes of genes.
Fig. 2. Hadza gut microbiome functional capacities are cyclic and differentiable by season.
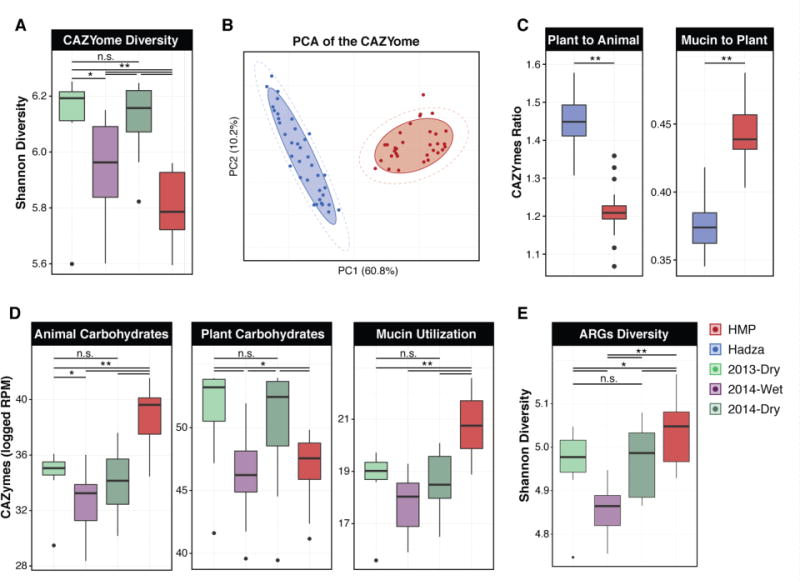
(A) Shannon diversity metric applied to CAZYome representation in the metagenomic datasets of Hadza by season and for a healthy American cohort (Human Microbiome Project; HMP). (B) Principal component analysis (PCA), an unsupervised learning approach that utilizes a linear combination of features to maximize the variance of the data in a reduced multivariate space, applied to CAZYomes of Hadza and Americans (HMP). The shaded ellipses represent the 80% confidence interval, the dotted ellipse borders represent the 95% confidence interval. (C) The ratio of CAZYmes represented within the metagenomes related to plant and animal carbohydrate utilization (left) or the ratio of mucin glycan- to plant carbohydrate-utilization (right) in the Hadza and Americans. (D) Representation of CAZYmes in metagenomic datasets related to multiple classes of polysaccharides are plotted by their respective distributions. (E) The distributions of Shannon diversities for antibiotic resistance families across populations identified in metagenomic data. The color key at the top right of the figure applies to all panels. All boxplot distributions are tested using the non-parametric two-sided Wilcoxon rank sum test with Holm correction for multiple hypothesis testing, center values indicate the median and error bars the SD * P values < 0.05, ** < 0.01.
Therefore, data from the Hadza show both enrichment of function for major dietary components across seasons and conservation of function for two sequential Dry seasons. We employed untargeted metabolomics, a sequencing-independent approach to generate a high dimensionality “fingerprint” of community functionality (26). These data also perfectly differentiated between the seasons using unsupervised learning methods (Fig. S4D) yet did not differentiate between the two Dry seasons.
We wondered how our microbiota profiles from the 350 Hadza stool samples we collected compared with other traditional and industrialized populations. We analyzed compositional data from 18 populations across 16 countries derived from 26 cohorts using taxonomic assignments (Table S5). The 18 populations separated along the first principal coordinate corresponding to modernization (Fig. 3, S1, S6A, S6B).
Fig. 3. Gut microbiotas across geography are distinguishable by lifestyle.
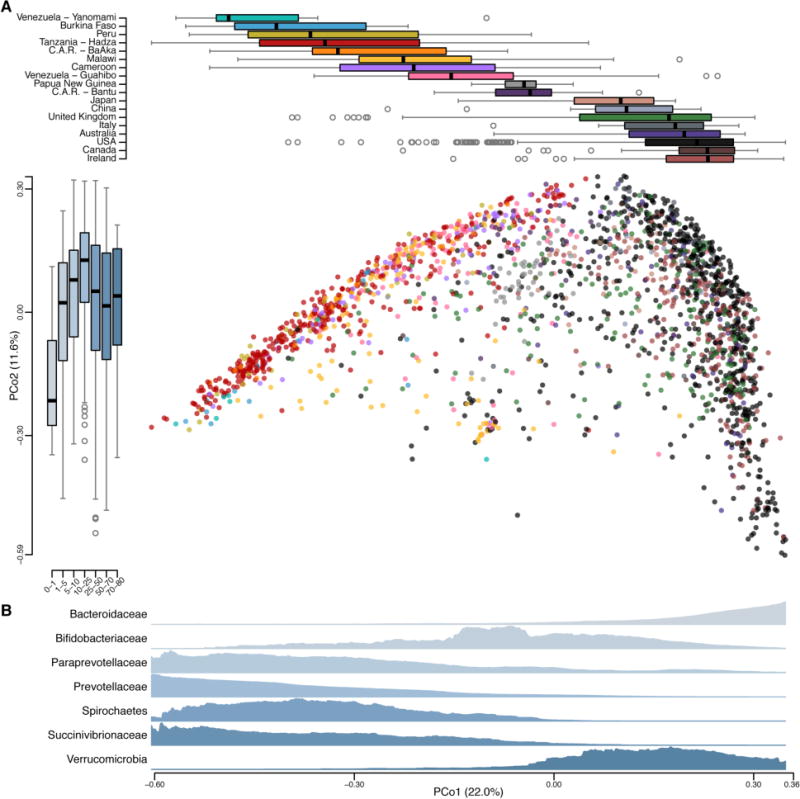
(A) Bray-Curtis dissimilarity PCoA (center panel) based on 2064 microbial community compositions described at the family taxonomic level across populations, including the 350 samples from this study. Each circle represents the placement of a microbial community projected in a subspace that maximizes the variance of the underlying taxonomic data; colors correspond to populations in the top panel. Boxplots (top panel) indicate the distribution of each population along the first principal coordinate (PCo1). The boxplots on the left panel depict the distribution of ages (indicated in years) according to their gut microbial community placement on the second principal coordinate (PCo2). Boxplot center values represent the median and error bars represent the SD. (B) Density plots of seven taxa were generated by using a moving average of the abundance of the families within the communities along PCo1, with a scale from zero to the maximum moving average. These seven families were chosen based on a notable trend along PCo1 or basis in the literature.
In addition to the striking separation of cultures, there were additional features in the data. First, during the cyclic disappearance of taxa, the Hadza microbiota shifts to a state with increased similarity to industrialized microbiotas (Fig. S1). Conversely, some OTUs within microbial families common to both traditional and industrialized populations are less seasonally volatile (Fig. 1F, S3C, S3D; p=7e-13, Wilcoxon). Second, we identified Prevotellaceae as a common family in the Hadza microbiota, leading us to wonder about its relationship to the Bacteroidaceae, a dominant family in industrialized populations; which are both in the Bacteroidetes phylum. A continuous variable contributing to separation along the first principal coordinate is the trade-off between Bacteroidaceae and Prevotellaceae, consistent with previous findings (13, 27, 28) (Fig. S6C, S6D). Industrialized populations have microbiotas that are dominated by Bacteroidaceae (mean 20.9% vs. 0.8% in traditional), whereas traditional populations across African, Asian and South American continents, which include a range of lifestyles from rural agriculturalists to hunter-gatherers, have microbiotas that are in part distinguished by their abundances of Prevotellaceae (mean 29.8% vs. 7.6% in industrialized). Third, Spirochaetaceae and Succinivibrionaceae, two prevalent families within the Hadza and other traditional groups are rare or undetected in Industrialized guts (p<2e-16 and p<2e-16, respectively; Wilcoxon) (Table S5). For example, in one comprehensive study (13) of 299 US residents, 15% of the individuals possessed Succinivibrionaceae, with an average relative abundance of only 0.006%, and Spirochaetaceae were undetected in all samples. By contrast, all Malawians and Venezuelans possess Succinivibrionaceae, with an average relative abundance of 3.2%, and 67% of these people harbor Spirochaetaceae at an average relative abundance of 0.6% (Fig. S7A–C). A fourth feature in the data reveals that Industrialized guts are enriched in Verrucomicrobia, a group of mucin-degrading bacteria that are rare in traditional populations’ guts (p<2e-16; Wilcoxon).
Together, our data show that in Hadza individuals living a traditional hunter-gatherer lifestyle the gut microbiota follow a cyclic succession of species that correspond with enrichment of seasonally associated functions. We show that the abundance of many taxa drop below our ability to detect them and then reappear in other seasons. The taxa that are driven to undetectable levels in the Hadza microbiota correspond to taxa that are rare or absent, regardless of season, in industrialized populations. Our observations reveal industrialized-microbiome-enrichment of mucin-utilizing glycoside hydrolases and the prevalence of Verrucomicrobia, findings that mirror the microbiota response in mouse models deprived of dietary fiber (29, 30). Together, these data indicate the modernized microbiota is characteristic of a diet limited in the plant-derived complex carbohydrates that fuel gut microbiota metabolism and maintain resident bacterial populations (12), although numerous factors associated with modernization could be affecting the microbiota of people from higher income countries. The ecological role and potential functional contributions of species with which humans co-evolved that are now apparently underrepresented or missing in modernized populations remain to be explored.
Supplementary Material
Acknowledgments
We are indebted to the participants in this study. We thank Teren Williams, Gregory Grymes, Aziz Omar, Belinda Bonnen, Mariamu Anyawire and Sierra Ward for assistance in collecting samples. Daudi Peterson of Dorobo Safaris and Chris and Nani of Kisema Ngeda provided logistical support during fieldwork. We also thank David Schneider and Susan Holmes for their insights that led to novel data analyses. This work was funded by grants from the Emch Family Foundation and Forrest & Frances Lattner Foundation, C&D Research Fund, grants from National Institutes of Health NIDDK (R01-DK085025 to JLS; R01-DK090989 to MGDB), a Discovery Innovation Fund Award (JLS), NSF Graduate Fellowship (SAS), and a Smith Stanford Graduate Fellowship (SAS).
The 16S rRNA amplicon sequence data and shotgun metagenomic data has been deposited in the Sequence Read Archive (SRA) under the project IDs: PRJNA392012, PRJNA392180 (http://www.ncbi.nlm.nih.gov/sra). Metabolomics data have been uploaded to Global Natural Products Social Molecular Networking (31) (http://gnps.ucsd.edu), accession number MSV000081199.
Footnotes
References and Notes
- 1.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonnenburg JL, Backhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. doi: 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. The Journal of Neuroscience. 2014;34:15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research. 2016;44:D471–480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352:532–535. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moeller AH, et al. Cospeciation of gut microbiota with hominids. Science. 2016;353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host & Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Lichtman JS, et al. Host-Microbiota Interactions in the Pathogenesis of Antibiotic-Associated Diseases. Cell Reports. 2016;14:1049–61. doi: 10.1016/j.celrep.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonnenburg ED, et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clemente JC, et al. The microbiome of uncontacted Amerindians. Science Advances. 2015;1:e1500183. doi: 10.1126/sciadv.1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obregon-Tito AJ, et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nature Comm. 2015;6:6505. doi: 10.1038/ncomms7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez I, et al. The gut microbiota of rural papua new guineans: composition, diversity patterns, and ecological processes. Cell Reports. 2015;11:527–538. doi: 10.1016/j.celrep.2015.03.049. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki TA, Worobey M. Geographical variation of human gut microbial composition. Biol Lett. 2014;10:20131037. doi: 10.1098/rsbl.2013.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnorr SL, et al. Gut microbiome of the Hadza hunter-gatherers. Nature Comm. 2014;5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marlowe F. The Hadza: hunter-gatherers of Tanzania Origins of human behavior and culture. University of California Press; Berkeley: 2010. [Google Scholar]
- 21.Blurton Jones NG, Smith LC, O’Connell JF, Hawkes K, Kamuzora CL. Demography of the Hadza, an increasing and high density population of Savanna foragers. American Journal of Physical Anthropology. 1992;89:159–181. doi: 10.1002/ajpa.1330890204. [DOI] [PubMed] [Google Scholar]
- 22.Davenport ER, et al. Seasonal variation in human gut microbiome composition. PLoS ONE. 2014;9:e90731. doi: 10.1371/journal.pone.0090731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.C. Human Microbiome Project. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rampelli S, et al. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr Biol. 2015;25:1682–1693. doi: 10.1016/j.cub.2015.04.055. [DOI] [PubMed] [Google Scholar]
- 25.Peterson J, et al. The NIH Human Microbiome Project. Genome Research. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcobal A, et al. A Metabolomic View of How the Human Gut Microbiota Impacts the Host Metabolome Using Humanized and Gnotobiotic Mice. The ISME Journal. 2013;7:1933–43. doi: 10.1038/ismej.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaconis P, Goel S, Holmes S. Horseshoes in multidimensional scaling and local kernel methods. Ann Appl Stat. 2008:777–807. [Google Scholar]
- 28.Gorvitovskaia A, Holmes SP. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4:15. doi: 10.1186/s40168-016-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonnenburg JL, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 30.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host & Microbe. 2008;4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nature Biotechnology. 2016;34:828–837. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilbert JA, Jansson JK, Knight R. The Earth Microbiome project: successes and aspirations. BMC Biology. 2014;12:69. doi: 10.1186/s12915-014-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME Journal. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England) 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 36.DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pedregosa F, et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 2011;12:2825–2830. [Google Scholar]
- 40.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 41.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE. 2011;6:e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rho M, Tang H, Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Research. 2010;38:e191. doi: 10.1093/nar/gkq747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abubucker S, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Computational Biology. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome informatics International Conference on Genome Informatics. 2009;23:205–211. [PubMed] [Google Scholar]
- 46.Yin Y, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Research. 2012;40:W445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. The ISME Journal. 2015;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cantarel BL, Lombard V, Henrissat B. Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE. 2012;7:e28742. doi: 10.1371/journal.pone.0028742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett FJ, Barnicot NA, Woodburn JC, Pereira MS, Henderson BE. Studies on viral, bacterial, rickettsial and treponemal diseases in the Hadza of Tanzania and a note on injuries. Human Biology. 1973;45:243–272. [PubMed] [Google Scholar]
- 50.Black FL, et al. Evidence for persistence of infectious agents in isolated human populations. American Journal of Epidemiology. 1974;100:230–250. doi: 10.1093/oxfordjournals.aje.a112032. [DOI] [PubMed] [Google Scholar]
- 51.Brito IL, et al. Mobile genes in the human microbiome are structured from global to individual scales. Nature. 2016;535:435–439. doi: 10.1038/nature18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claesson MJ, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 53.Morton ER, et al. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genetics. 2015;11:e1005658. doi: 10.1371/journal.pgen.1005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. The ISME Journal. 2015;9:1979–1990. doi: 10.1038/ismej.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakayama J, et al. Diversity in gut bacterial community of school-age children in Asia. Scientific Reports. 2015;5:8397. doi: 10.1038/srep08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.