

Abstract
Purpose
Transforming growth factor-β (TGF-β) production in the tumor microenvironment is a potent and ubiquitous tumor immune evasion mechanism that inhibits the expansion and function of tumor-directed responses; therefore, we conducted a clinical study to discover the effects of the forced expression of a dominant-negative TGF-β receptor type 2 (DNRII) on the safety, survival, and activity of infused tumor-directed T cells.
Materials and Methods
In a dose escalation study, eight patients with Epstein Barr virus–positive Hodgkin lymphoma received two to 12 doses of between 2 × 107 and 1.5 × 108 cells/m2 of DNRII-expressing T cells with specificity for the Epstein Barr virus–derived tumor antigens, latent membrane protein (LMP)-1 and LMP-2 (DNRII-LSTs). Lymphodepleting chemotherapy was not used before infusion.
Results
DNRII-LSTs were resistant to otherwise inhibitory concentrations of TGF-β in vitro and retained their tumor antigen–specific activity. After infusion, the signal from transgenic T cells in peripheral blood increased up to 100-fold as measured by quantitative polymerase chain reaction for the transgene, with a corresponding increase in the frequency of functional LMP-specific T cells. Expansion was not associated with any acute or long-term toxicity. DNRII-LSTs persisted for up to ≥ 4 years. Four of the seven evaluable patients with active disease achieved clinical responses that were complete and ongoing in two patients at > 4 years, including in one patient who achieved only a partial response to unmodified tumor-directed T cells.
Conclusion
TGF-β–resistant tumor-specific T cells safely expand and persist in patients with Hodgkin lymphoma without lymphodepleting chemotherapy before infusion. DNRII-LSTs can induce complete responses even in patients with resistant disease. Expression of DNRII may be useful for the many other tumors that exploit this potent immune evasion mechanism.
INTRODUCTION
The cellular immune response makes a major contribution to the control and eradication of human lymphomas. Both the infusion of antibodies that block inhibitory checkpoint signals to T cells and the adoptive transfer of T cells that express tumor-directed native or chimeric antigen receptors (CARs) have produced remarkable benefits in patients with chemorefractory lymphoma.1-10
Nonetheless, a significant proportion of patients still achieve poor responses, in part, because, as with most other malignancies, lymphomas exploit a broad range of active immune evasion strategies, of which transforming growth factor β (TGF-β) production is one of the most ubiquitous.
TGF-β is secreted by malignant cells and their stroma. It promotes tumor growth and metastasis, but potently inhibits antigen-presenting cells and the growth and effector function of T cells and NK cells.11 The fate of a T cell at the tumor site is determined by a combination of positive and negative signals; thus, the elimination of even a single inhibitory signal may enhance therapeutic benefit, as evidenced by the effects of checkpoint antibodies. We therefore forced the expression of a dominant-negative TGF-β type 2 receptor (DNRII) that inhibits TGF-β receptor signaling and allows effector T cells to resist otherwise inhibitory concentrations of TGF-β, which leaves their proliferation, cytokine secretion, and cytolytic functions unchanged.12
To analyze the safety and efficacy of DNRII-modified T cells, we infused them into patients with Epstein Barr virus (EBV)–associated lymphomas. Up to 40% of Hodgkin lymphomas (HLs) carry EBV genomes and express the type 2 latency proteins, latent membrane protein (LMP)-1, LMP-2, BARF1, and EBNA1.13 We have previously shown that T cells that are directed to the EBV latency-associated antigens, LMP-1 and LMP-2 (LSTs), are safe and have antitumor activity in patients with lymphoma.4 Here, we report the effects of the administration of such T cells that we additionally engineered to express a DNRII (DNRII-LSTs) to eight patients with relapsed EBV-positive HL.
MATERIALS AND METHODS
Patients and LMP Status of the Tumors
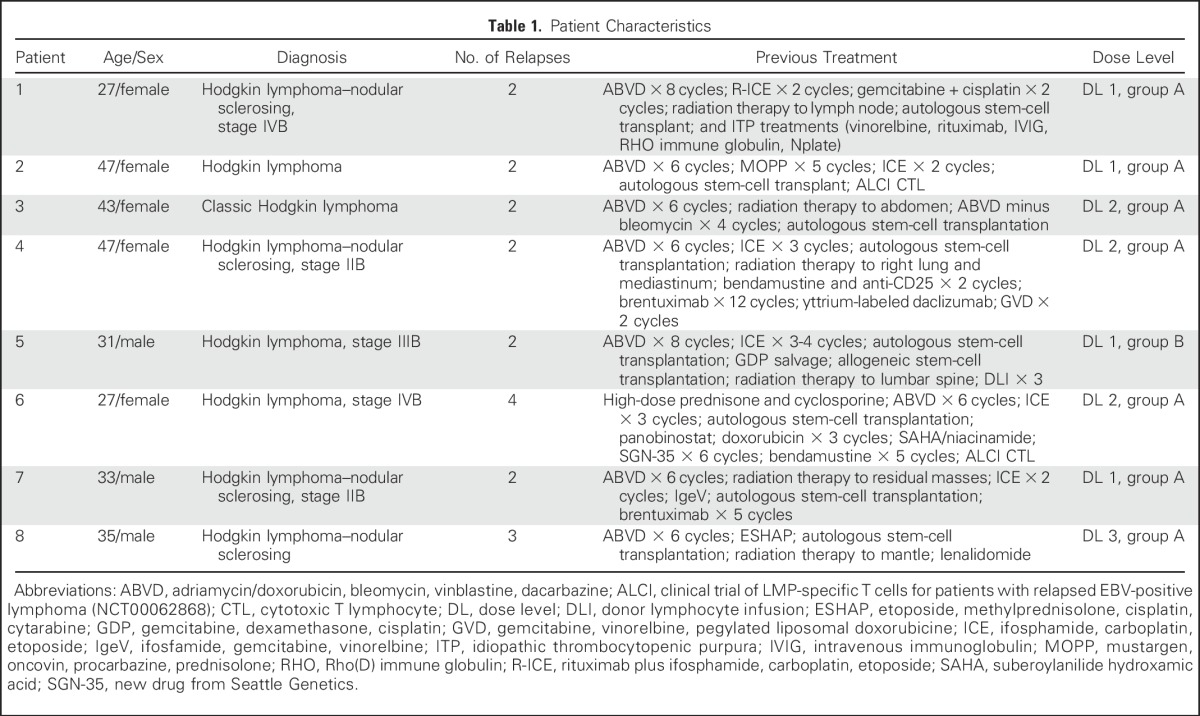
Patients were eligible for this institutional review board–, institutional biosafety committee–, US Food and Drug Administration–approved protocol if they had relapsed EBV-positive HL, including after allogeneic hematopoietic stem-cell transplantation (HSCT), or were considered high-risk for relapse defined as relapse that required high-dose chemotherapy and autologous stem-cell rescue. Patients received two infusions 2 weeks apart (dose range, 2 × 107 cells/m2 to 1.5 × 108 cells/m2 per dose). If patients achieved a partial response (PR) or had stable disease 8 weeks after DNRII-LSTs, they were eligible to receive up to 10 additional T-cell infusions at the same dose level as the initial infusion (Table 1 and Appendix, online only). Analysis of disease response to DNRII-LST therapy was performed using the Lugano Criteria for Response Assessment on computed tomography and 18F-labeled fluorodeoxyglucose positron emission tomography (PET)/computed tomography.14 Scans were reviewed by an independent radiologist.
Table 1.
Patient Characteristics
The dominant-negative receptor for TGF- β (TGFβRIIdcyt) was a gift of Joan Massague (Memorial Sloan Kettering Cancer Center, New York, NY).11,12 Clinical-grade DNRII retroviral vector (SFG-TGFβRIIdcyt) was generated from PG13 producer lines15,16 (Appendix).
Generation and Transduction of DNRII-LSTs
LSTs were generated as previously described.4 After two stimulations, LSTs were transduced with retroviral supernatant and expanded in rhIL-2 (Appendix).
Immunophenotyping
DNRII-LSTs were stained for T-cell markers (Becton Dickinson, San Jose, CA) and the TGFβRII antibody (R&D Systems, Minneapolis, MN; Appendix).
[3H]Thymidine Uptake Assay
The effects of TGF-β on the proliferation of nontransduced and DNRII-transduced LSTs were evaluated using [3H]thymidine uptake assays12 (Appendix).
LMP Multimers and Peptides and Enzyme-Linked Immunospot Assay
To characterize the LST products, soluble HLA peptide tetramers and overlapping peptide pools were used as previously described.4,16-18 Enzyme-linked immunospot (ELISPOT) analysis was used to determine the frequency and function of T cells that secrete interferon-gamma (IFN-γ) in response to LMP-1 and LMP-2 pepmixes19 (Appendix).
Quantification of the Transduction Rate by Real-Time PCR
Real-time polymerase chain reaction (PCR) was performed to quantify the transduction of DNR-transduced LSTs (Appendix).
Statistical Analysis
Descriptive statistics, such as mean, standard deviations, and medians, were used to summarize data (Appendix).
RESULTS
Phenotype and Antigen Specificity of DNRII-LSTs and Resistance to the Antiproliferative Effects of Exogenous TGF-β1
A mean of 62% of CD3+ T cells that were transduced with the SFG:DNRII vector expressed DNRII as determined by flow cytometry (Figs 1A and 1B). Transgene levels were also measured by quantitative PCR (Fig 1B). DNRII-LSTs displayed a predominantly effector memory phenotype (mean, 61%; range, 40% to 76%) with populations demonstrating a CD45RAneg/CCR7neg/CD62Lpos phenotype that was intermediate between central memory (CCR7+CD45RA−) and effector memory (mean, 31.03%; Fig 1C). Exhaustion markers, TIM3 and programmed death 1 (PD1), were expressed on a mean of 20% and 17%, respectively, of transduced cells compared with means of 9% (range, 0.4% to 18.6% of TIM3) and 11.7% (range, 7.7% to 22.5% of PD1) in nontransduced (NT-LSTs) T-cell products (Fig 1D). DNRII-LSTs displayed LMP specificity as evaluated by IFN-γ ELISPOT assay (Fig 1E) and [Cr51] release cytotoxicity assay (Figs 1F and 1G). To confirm the protective function of DNRII, we added recombinant TGF-β1 to DNRII-LSTs and NT-LSTs and compared their thymidine uptake 72 hours later (Fig 1H). Recombinant TGF-β1 inhibited the thymidine uptake of NT-LSTs by a mean of 57.74% compared with 14.64% in DNRII-LSTs (P = .01563, Wilcoxon signed rank test).
Fig 1.

Characteristics of infused dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein (LMP)–specific T cells (DNRII-LSTs). (A) Gating strategy for detecting DNRII-transduced cells by flow cytometry. T cells that were transduced with SFG:DNRII (truncated TGFβRII) were stained with anti-CD3 and TGFβRII monoclonal antibody. Expression of the transgenic TGFβRII receptor was determined on the basis of comparison with nontransduced LSTs. (B) DNRII expression is shown as both percentage of TGFβDNRII expression on T cells by flow cytometry (blue bar represents nontransduced cells and gold bar represents transduced cells; n = 5) and the number of copies on quantitative PCR (qPCR; gray bar represents transduced cells; n = 8). A mean of 62.44% (range, 23.27% to 89.03%) of CD3+ T cells transduced with the clinical-grade SFG:DNRII vector expressed the mutant receptor construct (DNRII) as determined by flow cytometry. Transgene levels ranged from 35,841 to 168,408 transgene copies/100 ng DNA. (C) Phenotypic characteristics of DNRII-LSTs as determined by flow cytometry. Percentages were based on the lymphocyte gate and are expressed as means ± standard deviation (n = 8). DNRII-LSTs displayed a predominantly effector memory phenotype (CD45RAneg/CCR7neg/CD62Lneg) (mean, 61%; range, 40% to 76%) with populations demonstrating a CD45RAneg/CCR7neg/CD62Lpos phenotype that was intermediate between central memory (CCR7+CD45RA−) and effector memory (mean, 31.03%; range, 12.17% to 56.56%). (D) Expression of the exhaustion markers, TIM3, PD1, and LAG3, as determined by flow cytometry. Percentages were based on the lymphocyte gate and are expressed as means ± standard deviation (n = 4). The exhaustion markers, TIM3 and PD1, were expressed on a mean of 20% (range, 2.4% to 33.3%) and 17% (range, 9.7% to 22.9%), respectively, of transduced cells compared with means of 9% (range, 0.4% to 18.6% TIM3) and 11.7% (range, 7.7% to 22.5% programmed death-1 [PD1]) in nontransduced T-cell products. However, LAG3 was negative in all products. (E) Ex vivo expanded DNRII-LSTs are specific for Epstein-Barr virus (EBV) antigens, LMP-1 and LMP-2, and recognize EBV antigen–expressing autologous lymphoblastoid cell lines (EBV-LCL), as measured by interferon-gamma (IFN-γ) enzyme-linked immunospot (ELISPOT). Black lines indicate mean values (n = 7). Transduced T cells demonstrated LMP specificity as evaluated by IFNγ ELISPOT assay (median, 4.5 spot-forming cells/1 × 105cells; range, 0 to 186.5 for LMP-1; median, 36.5 spot-forming cells/1 × 105cells; range, 6 to 146 for LMP-2). (F) DNRII-LSTs lyse EBV antigen–expressing autologous targets as measured by chromium release assay. Specific lysis at 20:1 effector:target ratio (E:T; except for patient 7, denoted as a star, at 40:1) is shown. Black lines indicate mean values (n = 8). (G) Representative plot of cytotoxicity as measured by chromium release assay at different E:T ratios (patient 8). (H) Proliferation assay that compare thymidine uptake in nontransduced (gold bars) and transduced cells (red bars) in the presence and absence (blue and gray bars, respectively) of transforming growth factor-β. Bars show means ± standard deviation of thymidine uptake counts per minute (CPMA; n = 7). FSC, forward scatter; SFC, spot-forming cell; SSC, side scatter.
DNRII-LSTs Expand In Vivo Without Prior Lymphodepletion and Without Immediate or Delayed Toxicity
Eight patients were infused with DNRII-LSTs and their characteristics are summarized in Table 1. Seven patients had relapsed disease and one patient was in remission at the time of DNRII-LST infusion. All infusions were well tolerated, and autologous (patients 1 to 4 and 6 to 8) DNRII-LSTs did not cause autoimmunity and donor-derived (patient 5) DNRII-LSTs did not induce graft-versus-host disease. Furthermore, there were no toxicities as a result of cytokine release syndrome. DNRII-LSTs were detected in the peripheral blood (PB) of all patients, with transgene copy numbers that ranged from 0.7 to 1476.9 copies/1,000 ng DNA (median, 37.75) and peaked from 3 hours to 3 weeks after infusion (Fig 2). By using flow cytometry, up to 1.3% of circulating CD3+ T cells were TGFβRII positive (Appendix Fig A1, online only). Overall, the persistence of DNRII-LSTs in PB ranged from 2 to 51 months (median, 17 months; Table 2). The rise in DNRII+ T cells in the PB was associated with a concomitant 2- to 64-fold increase in the frequency of LMP-specific T cells as evaluated by IFN-γ ELISPOT assay (Figs 3A-3D).
Fig 2.

In vivo expansion and persistence of infused dominant-negative transforming growth factor-β type 2 receptor (DNRII)–transduced T cells. In vivo expansion and persistence of infused DNRII-transduced T cells were assessed by quantitative PCR of peripheral blood from patients with Epstein-Barr virus–positive Hodgkin lymphoma. DNRII levels are expressed as transgene copies per 1,000 ng DNA (blue line). Days of T-cell infusions are indicated by gray arrows. (A-H) Patients 1 to 8.
Table 2.
Clinical Outcomes
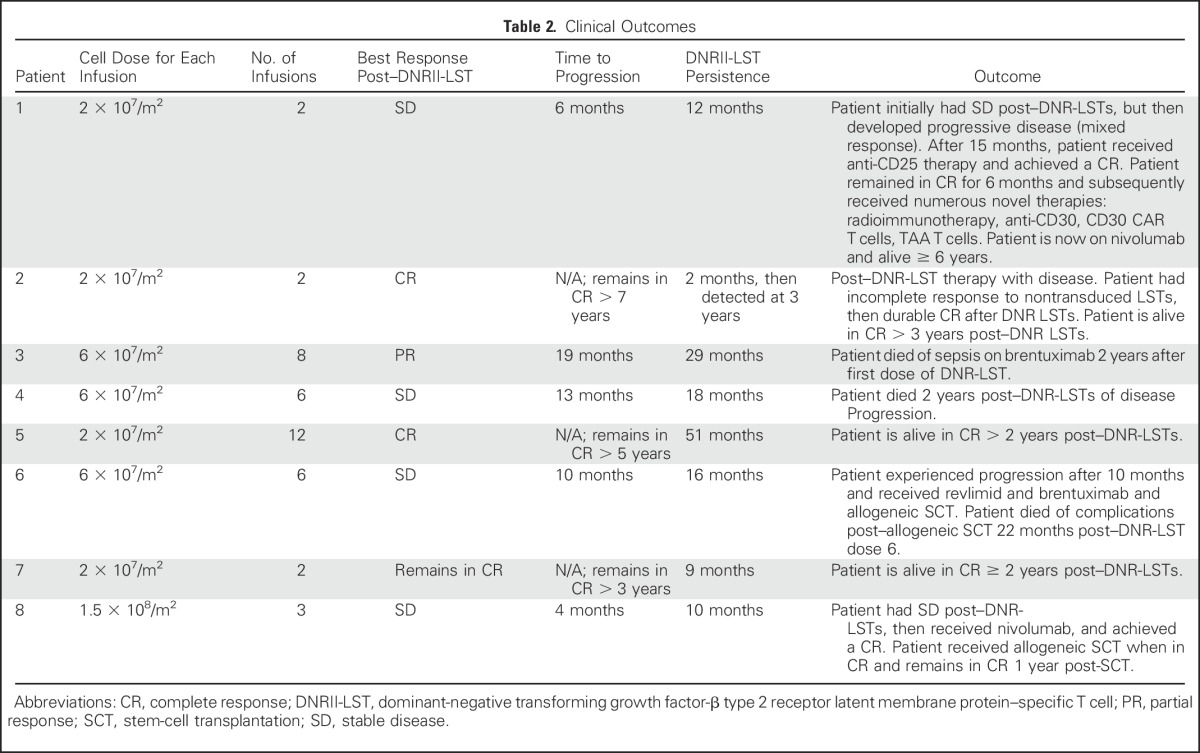
Fig 3.

Clinical responses and immune reconstitution. (A) In patient 2, whose axillary lymph nodes responded to an infusion of dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein (LMP)–specific T cells (DNRII-LSTs; as shown by positron emission tomography [PET]/computed tomography scan), there is a rise in both DNRII-transduced T cells as detected by quantitative PCR and LMP-2–specific T cells as detected by interferon-gamma (IFN-γ) enzyme-linked immunospot (ELISPOT) assay. At the time the patient achieved a complete remission, 2 months postinfusion, DNRII-transduced T cells were no longer detectable in the peripheral blood. (B) A PET scan demonstrating increased uptake in the ilium is observed in patient 3. The follow-up scan 8 weeks post–T-cell infusion 1 shows a partial response that coincided with an increase in DNRII-transduced T cells by quantitative PCR and detectable Epstein-Barr virus (EBV)–specific T cells (against lymphoblastoid cell line targets) and, to a lesser extent, LMP-2–specific T cells (against LMP-2 pepmix) by IFNγ ELISPOT assay. (C) A PET scan demonstrating increased uptake in the lumbar spine and sacrum is observed in patient 5 after receiving both radiotherapy and donor lymphocyte infusion post–allogeneic stem-cell transplatation. The follow-up scan 8 weeks post–donor-derived DNRII-LST infusion 1 shows a good partial response that coincided with an increase in DNRII-transduced T cells by quantitative PCR and EBV-, LMP-1–, and LMP-2–specific T cells as determined by IFNγ ELISPOT assay. (D) A PET scan demonstrating increased uptake in the axilla is observed in patient 8. The follow-up scan 8 weeks post–DNRII-LST infusion 1 shows overall stable disease and an increase in DNRII-transduced T cells by quantitative PCR. Blue lines denote quantitative PCR copies of the DNRII transgene, gold lines denote spot-forming cells as measured by IFNγ ELISPOT assay, and red arrows denote days of T-cell infusions. SFC, spot-forming cell.
Clinical Outcomes
Of the seven patients with relapsed disease who were refractory to standard treatment (Tables 1 and 2), two patients (patients 2 and 5) achieved complete clinical responses (CR), one (patient 3) achieved a partial response for 19 months, and four patients (patients 1, 4, 6, and 8) had stable disease for 4 to 13 months (Figs 3A-3D and Appendix Fig A2, online only). Reponses were observed in patients who received multiple (range, 2 to 12) doses of DNRII-LSTs at 2 × 107 cells/m2 per dose, 6 × 107/m2 or 1.5 × 108/m2 per dose (total dose range, 4 × 107/m2 to 4.5 × 108/m2 LSTs).
Patient 1 had stable disease after infusion of DNRII-LSTs, then developed progressive disease, characterized as mixed response, which became a CR after the patient received anti-CD25 radioimmunotherapy on another protocol.20 She remained in CR for approximately 6 months, then experienced relapse. A biopsy of an involved lymph node at the time of relapse showed an influx of CD3+ T cells but also residual disease; however, staining demonstrated that the Hodgkin Reed-Sternberg cells that had been LMP-1 negative, but Epstein Barr encoded region–positive (data not shown) and LMP-2 positive pre–DNRII-LST infusion were now LMP-2 negative, which was consistent with tumor immune escape (Figs 4A-4C).
Fig 4.

Tumor immune escape in patient 1. Patient 1 achieved a mixed clinical response after dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein (LMP)–specific T cells (DNRII-LSTs), which became a complete response (CR) after receiving anti-CD25 radioimmunotherapy. She remained in CR for approximately 6 months, then experienced relapse. Biopsies were obtained preinfusion and at the time of disease progression post–DNRII-LSTs and anti-CD25–directed therapy, when immunohistochemistry (IHC) was performed. (A) A biopsy performed preinfusion shows the presence of LMP-2–positive H-RS cells by IHC. (B and C) Biopsy of an involved lymph node performed at the time of disease progression post–DNRII-LSTs and anti-CD25–directed therapy shows H-RS cells that are negative for LMP-2, despite an influx of (C) CD3+ T cells.
Patient 2 had experienced relapse after autologous transplant and received nine doses of NT-LSTs over a 2-year period. The patient had an incomplete response to LSTs4 (Deauville score range, 2 to 4), but achieved a CR (Deauville score, 2) within 8 weeks of receiving the first dose of DNRII-LSTs (Fig 3A) and remains in CR > 7 years post–DNRII-LST treatment.
Patient 3 (Fig 3B) had a PR after two doses of DNRII-LSTs. She received an additional infusion, and a repeat scan showed continued improvement; however, 6 weeks after infusion 4 (8 months from infusion 1), a PET scan demonstrated increased standardized uptake value in axillary lymph nodes, which suggests interval worsening of malignancy. Needle biopsy of an axillary lymph node displayed a heavy infiltrate of CD3+ T cells (Fig 5A) as well as a small residual LMP-positive tumor cell population (Fig 5B). T cells that were obtained from this lymph node biopsy were expanded ex vivo. ELISPOT assay demonstrated that the majority of T cells were specific for LMP-1 or LMP-2 in a pattern that was comparable to the DNRII-LST product (Fig 5C). Epitope mapping of these ex vivo expanded T cells demonstrated that the tumor-infiltrating LSTs were specific for an LMP-1 epitope that was strongly represented in the infused LST product (Fig 5D); however, infiltrating LSTs were negative for the DNRII transgene by quantitative PCR. The patient received four additional doses of DNRII-LSTs over the next 12 months; however, 8 weeks after her eighth and final dose of DNRII-LST, there was evidence of disease progression in the axilla as well as new osseous lesions, predominantly in the pelvis, that were concerning for the development of osseous involvement by lymphoma. The patient refused all chemotherapy throughout, but did receive two doses of brentuximab over 2 years after her first dose of DNRII-LSTs. She ultimately died of sepsis before restaging could be performed.
Fig 5.

Latent membrane protein (LMP)-1 T-cell accumulation at tumor site after dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) LMP-specific T-cell (DNRII-LST) infusion in patient 3. Patient 3 achieved a partial response after DNRII-LST therapy; however, 8 months postinfusion 1, increasing standardized uptake values (SUVs) were evident on positron emission tomography scanning in the axilla. (A) Using immunohistochemistry, CD3+ infiltrating T cells were observed in an axillary lymph node biopsy. (B) However, small residual numbers of LMP-2–positive tumor cells remained. (C) T cells obtained from this lymph node biopsy were expanded ex vivo in response to LMP-1 and LMP-2 pepmixes. Enzyme-linked immunospot (ELISPOT) assay demonstrated that the majority of tumor-infiltrating LSTs were specific for LMP-1, but LMP-2–specific T cells were also detected. (D) Epitope mapping showed that tumor-infiltrating LSTs were specific for an LMP-1 epitope that was strongly represented in the infused LST product as determined by IFNγ ELISPOT assay. CTL, cytotoxic T lymphocyte; EBV, Epstein-Barr virus; LCL, lymphoblastoid cell line; SFC, spot-forming cell.
Patient 4 was ultimately resistant to all therapies and died of disease progression 2 years post–DNRII-LST treatment.
Patient 5 experienced relapse in the bone after allogeneic HSCT with tumor that was refractory to radiotherapy, followed by three doses of donor lymphocyte infusion that were administered over a 5-month period (Deauville score, 4). He received his first dose of DNRII-LST 3 months after his last dose of donor lymphocyte infusion and achieved a PR after the first two DNRII-LST doses (Deauville score, 3). The patient ultimately achieved a CR after a total of 12 doses of donor-derived DNRII-LSTs (Fig 3C). He remains in CR > 5 years after the last dose of DNRII-LSTs (Appendix Fig A3, online only).
Patient 6 experienced disease progression within 3 months of receiving NT-LST therapy alone, but achieved disease stabilization for 10 months after six DNRII-LST infusions. DNRII-LSTs were detectable in the PB at the time of last blood draw, which was 6 months after the sixth infusion of DNRII-LST. The patient went on to receive revlimid and brentuximab and eventually achieved a CR and proceeded to allogeneic HSCT, but died of gram-negative sepsis after HSCT (22 months after her last dose of DNRII-LSTs).
Patient 7 received two doses of DNRII-LSTs because of a high risk for relapse (Table 1) and remains in complete remission for > 2 years (Table 2).
Patient 8 received three doses of DNRII-LSTs after experiencing relapse post–high-dose chemotherapy, followed by autologous stem-cell rescue and mantle radiation. The patient had evidence of increasing DNRII-LSTs in the PB as determined by flow cytometry (Appendix Fig A1) and quantitative PCR that corresponded with decreasing standardized uptake values on PET scan (Appendix Fig A2); however, overall, imaging did not meet the criteria for PR. The patient went on to receive the PD1 inhibitor, nivolumab, and achieved a CR. He then proceeded to allogeneic stem-cell transplantation and remains in CR for > 1 year post-HSCT.
None of the patients who received autologous DNRII-LSTs experienced cytokine release syndrome or had any elevation of IL-6, TNF-α, or IFN-γ levels in plasma samples that were obtained before or during the first year after infusion (Fig 6). TGF-β was detected in the plasma of all patients, and sCD137 levels were only detectable in the plasma of patients who were treated with DL 2 and 3. In contrast, the recipient of donor-derived DNRII-LSTs—patient 5, who achieved a CR—had elevated TNF-α and IL-6 within 2 weeks of infusion that persisted for 2 weeks (Fig 6). Nonetheless, he remained asymptomatic throughout his treatment course.
Fig 6.

Cytokine levels postinfusion. Plasma samples obtained from patients pre– versus post–dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein (LMP)–specific T cell (DNRII-LST) infusion were evaluated using the luminex assay for concentrations of the following cytokines: (A) TGF-β1. (B) IL-10. (C) TNF-α. (D) IL-6. (E) sCD137. Data from all patients are shown and divided according to dose levels.
DISCUSSION
To counter the production of TGF-β, a ubiquitous tumor immune evasion strategy that markedly inhibits the function of tumor-specific T cells, we expressed a DNRII in tumor-specific T cells. As previously demonstrated in vitro and in murine models, DNRII was expressed at high frequency and provided resistance to the inhibitory levels of TGF-β without impairing antigen specificity.9,21 In these preclinical studies, cytolytic activity and the ability to secrete Th1 cytokines in response to antigen in the presence of TGF-β was preserved in DNRII-modified T cells. In contrast, the presence of TGF-β had devastating effects on the growth and function of unmodified EBV-specific T cells in vitro and in vivo.9,21 After infusion in patients with relapsed/refractory EBV-positive HL, DNRII-LSTs expanded and persisted for up to ≥ 4 years after infusion. No patient received lymphodepleting chemotherapy before infusion, but DNRII-LSTs alone produced therapeutic effects that included tumor CR in two of seven patients with active disease, one subsequent CR after CD25 targeted radioimmunotherapy, one PR that lasted 19 months, and stable disease in three patients for 4, 10, and 13 months. Detection of the DNRII transgene in patient blood after infusion correlated with an increase in the frequency of LMP-specific T cells. There was no toxicity associated with DNRII, which allayed fears that the loss of a homeostatic receptor might produce uncontrolled T-cell proliferation.
Approximately 40% of HLs are EBV positive and express EBV antigens that can be exploited for immunotherapy with EBV-directed T cells; however, HL uses numerous tumor immune escape strategies to evade attack by EBV-specific cytotoxic T lymphocytes, including downregulating the majority of approximately 90 EBV antigens and recruiting immunosuppressive cell types that secrete inhibitory cytokines, such as TGF-β. We previously overcame the limited expression of viral antigens by reactivating and expanding T cells that were specific for the subdominant tumor-associated EBV antigens, LMP-1 and/or LMP-2, and infused these LSTs into patients with relapsed EBV-positive HL and non-HL. Objective clinical responses—CR or PR—with LST therapy alone were observed in 13 of 21 patients, including four of eight patients with HL4; however, in contrast to the current study in which all patients were immune competent, of the four patients with HL who achieved a response in the previous study,4 three had underlying immune deficiency so that the immune evasion strategies that were used by their tumors might have been weaker than those observed in the majority of patients with this disease.
TGF-β is abundant in many tumor environments and is secreted not only by tumors, but also by infiltrating stromal cell, such as T regulatory cells, myeloid-derived suppressor cells, and tumor-associated macrophages.22 Mutations in the TGF-β receptor signaling pathway are commonly found in solid tumors and prevent TGF-β–mediated tumor cell differentiation and apoptosis.23 A dominant-negative TGF-β receptor (DNRII) that lacks its intracellular signaling domains was first described by Yin et al.24 Our group later demonstrated that the expression of the same DNRII in T cells that were cultured in vitro protected not only DNRII T cells, but also NT T cells from TGF-β, so that both fractions proliferated similarly in the presence of inhibitory concentrations of TGF-β.12 This conferred protection suggests that DNRII-expressing cells may be able to sequester TGF-β from the tumor environment, thereby inhibiting the development of myeloid-derived suppressor cells and cancer-associated fibroblasts, restraining angiogenesis and tumor migration, depleting T regulatory cells, and enabling the function of natural killer cells. Hence, the benefits of dominant-negative TGF-β receptor expression may go beyond simply protecting DNRII T cells from TGF-β.
Infusion of DNRII-LSTs was followed by substantial in vivo expansion as measured by quantitative PCR and immunoassays to measure the frequency of LSTs in PB. Although these levels of T-cell expansion as measured by the DNRII transgene are lower than those observed after infusion of CD19-CAR T cells,5-7,25 patients who were treated in this study had no prior lymphodepletion. The lower levels of LST expansion may also explain the lack of cytokine release syndrome associated with the infusion of DNRII-LSTs.
In this study, as few as 4 × 107 DNRII-LSTs/m2 produced tumor PRs or CRs in three of seven patients. We cannot assess the relative potency of DNRII-modified versus unmodified T cells in this small, phase I study, but all patients who were enrolled in this trial were immunocompetent—that is, patients who did not have a known primary immune deficiency or post-transplant lymphoproliferative disorder before HL diagnosis—and had multiply relapsed chemorefractory disease; thus, their prognosis was worse than that of patients who were treated in our earlier study of unmodified LSTs.4 Furthermore, observations in patient 2, who achieved PR post–unmodified LSTs and durable CR post–DNR-LSTs, and patient 6, who achieved stabilized disease for 10 months after DNR-LST versus 3 months post-LST alone, are consistent with greater activity of DNRII-LSTs versus LSTs alone. Therefore, it is tempting to speculate that DNRII-LSTs compare favorably with unmodified LSTs,4 as immunocompetent patients with HL who received DNRII-LSTs achieved an overall response rate (CR/PR) of 43% (three of seven patients) versus 20% (one of five patients) in the immunocompetent HL group who received unmodified LSTs.4 Moreover, as discussed above, two patients achieved greater responses to DNRII-LSTs than to unmodified LSTs.
Although HL is a curable disease, this outcome requires prolonged intensive and highly toxic treatment regimens with severe bystander organ toxicity, immune suppression, and late effects, including second cancers and cardiac disease.26 Such problems are compounded in patients who experience relapse and receive additional cytotoxic therapies. These limitations have spurred the development of targeted immunotherapies, such as tumor-specific monoclonal antibody–toxin conjugates and adoptive transfer of tumor-directed T cells, including those that express CARs.1,3,5,27 Antibody–toxin conjugates have demonstrated success in the treatment of HL, and brentuximab vedotin has produced CRs in approximately 30% of patients with a 1-year progression-free survival of approximately 50%27; however, antibodies have a limited in vivo half-life. T cells that were modified with CARs targeting CD30 have been evaluated in clinical trials for HL, which is CD19 negative,28 but targeting a single epitope with a transgenic receptor may result in immune escape by antigen editing, as reported for CD19-CAR treatment of leukemia and B-cell non-HLs.29 In contrast, T cells that target viral antigens via their native TCRs persist long term, do not require prior chemotherapy to potentiate their action, have minimal toxicity, and do not damage healthy tissues4; however, immune escape may remain a problem even when targeting multiple antigens, as demonstrated by patient 1, who experienced relapse with EBV-negative disease. More recently, antibodies to the PD1/programmed death-ligand 1 checkpoint axis have been reported to produce an overall response rate of 87%, with CR in 17% of patients with otherwise resistant HL.1 Of note, approximately 30% of infused DNRII-LSTs expressed PD1. In future, it will be of interest to assess whether PD1 or related checkpoint inhibitors can be safely combined with this and other T-cell immunotherapies to produce additive or even synergistic benefits.
Even in the absence of checkpoint blockade, DNRII-LSTs persisted for > 4years in PB, which may be ascribed to the presence in the infused population of early differentiated memory T cells with long survival potential.7 These cells receive potent stimulation from professional antigen-presenting cells in the form of EBV-infected, nonmalignant B cells in lymph nodes and at tumor sites.30 Despite the long-term persistence, the initial expansion of the transferred DNRII-LSTs was at least an order of magnitude less than that observed when virus-specific T cells and CD19-CAR T cells are administered after HSCT or lymphodepletion.6,31-35 Now that we have demonstrated safety and activity with our initial approach, it will be possible to discover whether the addition of lymphodepletion is safe and enhances initial in vivo expansion.
Overall, our findings emphasize the importance of strategies to overcome tumor immune evasion by TGF-β production and underline the feasibility and therapeutic potential of counteracting a single, but common, tumor immune editing mechanism. This strategy may be exploited to enhance the immunotherapy of the many other tumors that exploit TGF-β for survival.
Appendix
Patients and Latent Membrane Protein Status of the Tumors
The protocol for the use of dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein (LMP)–specific T cells (DNRII-LSTs) as therapy for Epstein Barr virus (EBV)–positive Hodgkin lymphoma (HL) was approved by the US Food and Drug Administration (FDA), the Recombinant DNA Advisory Committee, and the Baylor College of Medicine Institutional Review Board and Institutional Biosafety Committee. The study included patients with EBV-associated HL as detected by immunohistochemistry for LMP1 and/or in situ hybridization for Epstein Barr encoded region. Patients were eligible for this institutional review board, institutional biosafety committee, FDA-approved protocol if they had EBV-positive HL and experienced relapse after receiving standard therapy, including allogeneic hematopoietic stem-cell transplantation, or, as in one case, were considered to be at high risk for relapse as defined by first or subsequent relapse that required high-dose chemotherapy and autologous stem-cell rescue. Patients received two infusions of DNRII-transduced T cells 2 weeks apart. On dose levels 1, 2, and 3, patients received two infusions of 2 × 107cells/m2 per dose, 6 × 107cells/m2 per dose, and 1.5 × 108cells/m2per dose, respectively. If patients achieved a partial response or stable disease 8 weeks after DNRII-LSTs, they were eligible to receive up to 10 additional T-cell infusions at the same dose level as the initial infusion (Table 1). Blood was monitored at regular intervals for toxicities and for DNRII-LSTs by quantitative PCR and flow cytometry.
The dominant-negative receptor for TGF-β (TGFβRIIdcyt) was a gift of Joan Massague (Memorial Sloan Kettering Cancer Center, New York, NY).11,12 Clinical-grade DNRII retroviral vector (SFG-TGFβRIIdcyt) was generated from PG13 producer lines as previously described.15 PG13 cells were transduced with retroviral supernatant from transfected Phoenix-Eco cells, and transduced PG13 cells were single-cell cloned. Clones with the highest biologic titer were expanded in T-75 flasks to 80% confluence. A master vector producer bank and clinical grade lots of viral supernatant were made and tested according to FDA guidelines.16
Generation and Transduction of DNRII-LSTs
At least 5 × 106 peripheral blood mononuclear cells (PBMCs) that were derived from each patient—or their healthy sibling in the one allograft recipient—were used to establish an EBV-transformed B-lymphoblastoid cell line (LCL) for use as antigen-presenting cells (Smith CY, et al: J Hematother 4:73-79, 1995). Dendritic cells (DCs) that were transduced with the Ad5f35dLMP1-I-LMP2 vector were used as antigen-presenting cells to stimulate and expand LSTs from nonadherent PBMCs. To generate DCs, PBMCs were adhered to tissue culture plates for 2 hours, then nonadherent cells were cryopreserved and adherent cells were cultured with 800 U/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; Sargramostim Leukine; Immunex, Seattle, WA) and 1,000 U/mL IL-4 (R&D Systems) for 5 days. IL-4 and GM-CSF were replenished on day 3. On day 5, immature DCs were harvested by vigorous pipetting, transduced with the Ad5f35dLMP1-I-LMP2 vector,4 and matured with IL-4, GM-CSF, TNF-α (R&D systems), and PGE1 (Merck, Darmstadt, Germany). Before coculture with nonadherent PBMCs—with or without IL-15—DCs were γ-irradiated (30 Gy). From day 10, responder T cells were restimulated weekly with irradiated LCLs were transduced with the same LMP vector. After two stimulations with transduced LCLs, LSTs were transduced with retroviral supernatant and expanded in the presence of rhIL-2. LMP specificity was determined using HLA-peptide multimers and interferon-gamma (IFN-γ) enzyme-linked immunospot (ELISPOT) assays, LST phenotype by flow cytometry, transduction efficiency by real-time PCR, DNRII function by using [3H]thymidine assays in the presence and absence of TGF-β, and cytotoxic function using chromium release assays.
Immunophenotyping
DNRII-LSTs were stained for CD3, CD4, CD8, CD16, CD56, TCRαβ, TCRγδ, CD19, CD28, CD62L, CCR7, CD45RA, and CD45RO (Becton Dickinson). To assess the expression of DNRII, DNRII-LSTs (1 × 106) were incubated with 10 μl TGFβRII antibody (R&D Systems). For each sample, 10,000 cells were analyzed by FACSCalibur using Cell Quest software (Becton Dickinson).
[3H]Thymidine Uptake Assay
Effects of TGF-β on the proliferation of nontransduced and DNRII-transduced LSTs were evaluated using [3H]thymidine uptake assays.12 In brief, LSTs were seeded at 5 × 105 cells per well and maintained in the presence or absence of human TGF-β (R&D Systems; 2.5 ng/5 × 105 cells) and stimulated with IL-2 and LCLs for 72 hours at 37°C. Eighteen hours after pulsing with 5 μC [3H]thymidine (Amersham Pharmacia Biotech, Little Chalfont, United Kingdom), cells were harvested onto glass fiber filter strips and the incorporated [3H]thymidine was measured using a TriCarb 2500 TR β-counter (Packard Bioscience, Downers Grove, IL).
LMP Multimers and Peptides
To identify LSTs in the LST products in PBMCs and lymph node tissue, soluble HLA-peptide tetramers that were prepared by the Baylor College of Medicine Tetramer Core Facility or pentamers (Proimmune, Springfield, VA; together termed multimers) were used as previously described.4 For each sample, 5 × 105 cells were stained and 100,000 were analyzed. Panels of 15mer peptides that overlapped by 11 amino acids (pepmixes) covering the entire amino acid sequences of LMP-1 and LMP-2 from the white prototype EBV strain, B95-8, were synthesized by JPT Technologies (Berlin, Germany) as previously described.17 For LMP-2, 23 peptide pools composed of two to 12 15mer peptides were prepared so that each 15mer peptide was represented in two pools.18 For LMP-1, 10 peptide pools were prepared with each 15mer peptide represented in one pool. These LMP peptide libraries were designed to identify all possible HLA class I–restricted epitopes, which have lengths of eight to 10 amino acids.16
ELISPOT Assay
ELISPOT analysis was used to determine the frequency and function of T cells that secreted IFNγ in response to LMP-1 and LMP-2 pepmixes.19 ELISPOT assays were performed to determine the frequency of LMP-1– and LMP-2–specific T cells within LSTs and in patient PBMCs pre- and post-LST infusion. To reduce interassay variability, patient PBMC samples were cryopreserved and batched for ELISPOT analysis after > 6 weeks of follow-up. PBMCs that were stimulated with cytomegalovirus pepmix and staphylococcal enterotoxin B (1 μg/mL; Sigma-Aldrich, St Louis, MO) served as controls. IFNγ spot-forming cells (SFCs) were quantified by Zellnet Consulting (New York, NY), and the frequency of SFCs was calculated on the basis of the input cell numbers.
Quantification of the Transduction Rate by Real-Time PCR
DNA was extracted from transduced and nontransduced LSTs and PBMCs using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) according to manufacturer instructions. Real-time quantitative PCR was performed to quantify the transduction of DNR-transduced LSTs. A custom TaqMan Gene Expression Assay (×20) that was specific for the SFG.DNRIIdcyt sequence was developed. The forward primer (5′-GGCAGCAGAAGCTGAGTTCATA-3′) binds 23 base pairs (bp) upstream of XhoI in the 3′-SFG.MCS vector polylinker site used for TGFβRIIdcyt cloning (NcoI, MluI, SphI, XhoI, ClaI). The reverse primer (5′-CCACTGATATCCTGTCTTTAACAAATTG-3′) is 51 bp downstream of the cloning site, and the probe (5′-FAM-CTCGAGATCGATCCGG-MGB-NFQ-3′) spans the linker. For real-time quantitative PCR amplification, we used the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) and the manufacturer’s TaqMAN program settings, with a reaction volume of 50 μL that contained 2× TaqMan Universal PCR Master Mix (Applied Biosystems), 900 nM primers, 250 nM probe (the final concentration in the reaction when using 20× TaqMan Assay), the DNA template (1,000 ng), and nuclease-free water. We established the standard curve using serial dilutions of the plasmid that encodes the transgene (from 300,000 to three copies per reaction), and each sample was analyzed in triplicate. A TaqMan β-actin real-time quantitative PCR amplification was run in parallel using TaqMan β-actin Detection Reagents kit (Applied Biosystems) to confirm the integrity of the DNA sample. A correlation coefficient of > 0.99 was found > 6 orders of magnitude after the amplification of the SFG.DNRIIdcyt 74-bp sequence.
Statistical Analysis
Descriptive statistics, such as mean, standard deviation, and median, were used to summarize the data, including SFCs for LSTs at pre- and postinfusion time points as well as changes in SFCs from preinfusion. Continuous variables were assessed for the normality assumption, and data were log-transformed if needed. Differences between pre- and postinfusion LST SFCs were compared using paired t tests on log-transformed data or using the nonparametric Wilcoxon signed rank test. P values < .05 were considered statistically significant.
Fig A1.

Detection of circulating infused T cells by flow cytometry in patient 8. Flow cytometry analysis was used to compare the frequency of dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII)–transduced T cells pre- and postinfusion in patient 8 using an antibody that was specific for TGF-β receptor II (TGFβRII). Percentages of TGFβRII-positive T cells in the peripheral blood are shown pre- and 1, 2 (pre-CTL 2), and 3 weeks postinfusion. CTL: cytotoxic T lymphocyte.
Fig A2.

Radiographic changes after infusion of dominant-negative transforming growth factor-β (TGF-β) type 2 receptor (DNRII) latent membrane protein–specific T cells (DNRII-LSTs) in patient 8. Whereas it was documented that patient 8 had stable disease overall, decreases in standardized uptake values (SUVs) were demonstrated on positron emission tomography scan within 2 months of DNRII-LST infusion, corresponding to a rise in DNRII-transduced T cells as determined by quantitative PCR.
Fig A3.

Complete response post–dominant-negative transforming growth factor-β type 2 receptor latent membrane protein–specific T cells (DNRII-LSTs) in patient 5. Serial positron emission tomography scans evaluated by an independent radiologist confirming complete remission in patient 5 after 12 DNRII-LST infusions. (A) Left common iliac. (B) Left presacral/internal iliac. (C) Left internal iliac.
Footnotes
Supported by National Institutes of Health Grant No. P01-CA094237, SPORE in Lymphoma Grant No. P50-CA126752, and a Leukemia and Lymphoma Society SCOR Grant No. 7018-04, as well as by the shared resources of the Dan L. Duncan Cancer Center support Grant No. P30-CA125123
Clinical trial information: NCT00368082.
See accompanying article on page 1140
AUTHOR CONTRIBUTIONS
Conception and design: Catherine M. Bollard, Hao Liu, Malcolm K. Brenner, Helen E. Heslop, Cliona M. Rooney
Provision of study materials or patients: Adrian P. Gee
Collection and assembly of data: Catherine M. Bollard, Tamara Tripic, Conrad Russell Cruz, Stephen Gottschalk, Vicky Torrano, Olga Dakhova, George Carrum, Carlos A. Ramos, Hao Liu, Meng-Fen Wu, Andrea N. Marcogliese, Cecilia Barese, Youli Zu, Daniel Y. Lee, Owen O'Connor, Adrian P. Gee, Malcolm K. Brenner, Helen E. Heslop, Cliona M. Rooney
Data analysis and interpretation: Catherine M. Bollard, Tamara Tripic, Conrad Russell Cruz, Gianpietro Dotti, Stephen Gottschalk, Hao Liu, Meng-Fen Wu, Malcolm K. Brenner, Helen E. Heslop, Cliona M. Rooney
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Catherine M. Bollard
Consulting or Advisory Role: Cellectis, NexImmune
Stock or Other Ownership: Mana Therapeutics
Tamara Tripic
No relationship to disclose
Conrad Russell Cruz
Stock or Other Ownership: Mana Therapeutics
Travel, Accommodations, Expenses: Torque
Gianpietro Dotti
Research Funding: Bluebird Bio, Cell Medica, Bellicum Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Patents in the field of T-cell immunotherapy
Stephen Gottschalk
Consulting or Advisory Role: AbbVie, Celgene, Merrimack Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Patent applications: Eight focused on cell therapy for cancer, one focused on oncolytic viruses, one focused on cancer gene therapy
Vicky Torrano
No relationship to disclose
Olga Dakhova
No relationship to disclose
George Carrum
No relationship to disclose
Carlos A. Ramos
Consulting or Advisory Role: Novartis
Research Funding: Tessa Therapeutics (Inst)
Hao Liu
No relationship to disclose
Meng-Fen Wu
No relationship to disclose
Andrea N. Marcogliese
No relationship to disclose
Cecilia Barese
No relationship to disclose
Youli Zu
No relationship to disclose
Daniel Y. Lee
Employment: Cell Medica
Consulting or Advisory Role: Cell Medica
Owen O'Connor
Consulting or Advisory Role: Celgene
Mundipharma
Research Funding: Spectrum Pharmaceuticals, Celgene, Mundipharma, Trillium Therapeutics, ADCT, Epizyme, Affimed Therapeutics
Travel, Accommodations, Expenses: TG Therapeutics
Adrian P. Gee
No relationship to disclose
Malcolm K. Brenner
Stock or Other Ownership: Viracyte
Consulting or Advisory Role: Marker Therapeutics, Neon, Memgen, Torque, Unum Therapeutics, Nankwest, Poseida, Cellmedica (I), Formula Pharmaceuticals, Bellicum Pharmaceuticals (Inst)
Research Funding: Bluebird Bio
Helen E. Heslop
Stock or Other Ownership: Viracyte, Marker Therapeutics
Consulting or Advisory Role: Chimerix, Novartis
Research Funding: Celgene, Cell Medica
Patents, Royalties, Other Intellectual Property: Cell Medica
Cliona M. Rooney
Stock or Other Ownership: Viracyte, Marker Therapeutics
Consulting or Advisory Role: Cell Medica, CellGenix
Research Funding: Tessa Therapeutics
Patents, Royalties, Other Intellectual Property: Patent applications in the field of cellular immunotherapy
REFERENCES
- 1.Ansell SM, Lesokhin AM, Borrello I, et al. : PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372:311-319, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armand P: Immune checkpoint blockade in hematologic malignancies. Blood 125:3393-3400, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Batlevi CL, Matsuki E, Brentjens RJ, et al. : Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol 13:25-40, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollard CM, Gottschalk S, Torrano V, et al. : Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol 32:798-808, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramos CA, Heslop HE, Brenner MK: CAR-T cell therapy for lymphoma. Annu Rev Med 67:165-183, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Dudley ME, Kassim SH, et al. : Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33:540-549, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riddell SR, Sommermeyer D, Berger C, et al. : Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J 20:141-144, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R, Zinzani PL, Fanale MA, et al. : Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 35:2125-2132, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falchi L, Sawas A, Deng C, et al. : High rate of complete responses to immune checkpoint inhibitors in patients with relapsed or refractory Hodgkin lymphoma previously exposed to epigenetic therapy. J Hematol Oncol 9:132, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Younes A, Santoro A, Shipp M, et al. : Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol 17:1283-1294, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massagué J: TGF-beta signal transduction. Annu Rev Biochem 67:753-791, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Bollard CM, Rössig C, Calonge MJ, et al. : Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 99:3179-3187, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Taylor GS, Long HM, Brooks JM, et al. : The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 33:787-821, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Cheson BD, Fisher RI, Barrington SF, et al. : Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol 32:3059-3068, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pule M. A., Savoldo B, Myers GD, et al. : Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14:1264-1270, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang ST, Ghosh D, Kirschner DE, et al. : Peptide length-based prediction of peptide-MHC class II binding. Bioinformatics 22:2761-2767, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Meij P, Leen A, Rickinson AB, et al. : Identification and prevalence of CD8+ T-cell responses directed against Epstein-Barr virus-encoded latent membrane protein 1 and latent membrane protein 2. Int J Cancer 99:93-99, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Kern F, Faulhaber N, Frömmel C, et al. : Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur J Immunol 30:1676-1682, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Bollard CM, Aguilar L, Straathof KC, et al. : Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin’s disease. J Exp Med 200:1623-1633, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreitman RJ: Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr Pharm Des 15:2652-2664, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster AE, Dotti G, Lu A, et al. : Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother 31:500-505, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas D. A., Massagué J: TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8:369-380, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Massagué J: TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616-630, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin JJ, Selander K, Chirgwin JM, et al. : TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 103:197-206, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gill S, June CH: Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263:68-89, 2015 [DOI] [PubMed] [Google Scholar]
- 26.Castellino SM, Geiger AM, Mertens AC, et al. : Morbidity and mortality in long-term survivors of Hodgkin lymphoma: A report from the Childhood Cancer Survivor Study. Blood 117:1806-1816, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Younes A: Brentuximab vedotin for the treatment of patients with Hodgkin lymphoma. Hematol Oncol Clin North Am 28:27-32, 2014 [DOI] [PubMed] [Google Scholar]
- 28.Ramos CA, Ballard B, Zhang H, et al. : Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest 127:3462-3471, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner R, Wu D, Cherian S, et al. : Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127:2406-2410, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Küppers R, Engert A, Hansmann ML: Hodgkin lymphoma. J Clin Invest 122:3439-3447, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heslop HE, Slobod KS, Pule MA, et al. : Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 115:925-935, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadopoulou A, Gerdemann U, Katari UL, et al. : Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med 6:242ra83, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Restifo NP: Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62-68, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turtle CJ, Hanafi LA, Berger C, et al. : CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126:2123-2138, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turtle CJ, Hanafi LA, Berger C, et al. : Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8:355ra116, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]