

Abstract
Germline mutations in breast cancer susceptibility gene 1 or 2 (BRCA1 or BRCA2) significantly increase cancer risk in hereditary breast and ovarian cancer syndrome (HBOC). Both genes function in the homologous recombination (HR) pathway of the DNA double‐strand break (DSB) repair process. Therefore, the DNA‐repair defect characteristic of cancer cells brings about a therapeutic advantage for poly(ADP‐ribose) polymerase (PARP) inhibitor‐induced synthetic lethality. PARP inhibitor‐based therapeutics initially cause cancer lethality but acquired resistance mechanisms have been found and need to be elucidated. In particular, it is essential to understand in detail the mechanism of DNA damage and repair to PARP inhibitor treatment. Further investigations have shown the roles of BRCA1/2 and its associations to other molecules in the DSB repair system. Notably, the repair pathway chosen in BRCA1‐deficient cells could be entirely different from that in BRCA2‐deficient cells after PARP inhibitor treatment. The present review describes synthetic lethality and acquired resistance mechanisms to PARP inhibitor through the DSB repair pathway and subsequent repair process. In addition, recent knowledge of resistance mechanisms is discussed. Our model should contribute to the development of novel therapeutic strategies.
Keywords: DNA replication, HBOC syndrome, homologous recombination, non‐homologous DNA end‐joining, PARP inhibitor
Abbreviations
- 53BP1
p53‐binding protein 1
- APE
AP‐endonuclease
- BER
base excision repair
- BRCA1/2
breast cancer susceptibility gene 1/2
- CtIP
C‐terminal‐binding protein interacting protein
- DSB
double‐strand break
- HBOC
hereditary breast and ovarian cancer syndrome
- HR
homologous recombination
- MRE11
meiotic recombination 11
- NHEJ
non‐homologous end joining
- PARP
poly(ADP‐ribose) polymerase
- PARylation
poly ADP‐ribosylation
- RIF1
Rap1‐interacting factor 1
- seDSB
single‐ended DSB
- SSB
single‐strand break
1. INTRODUCTION
Hereditary breast and ovarian cancer syndrome is caused by germline mutations in BRCA1 or BRCA2 genes.1, 2 Approximately 10% of all breast cancer cases are inherited, and half of them are HBOC.3 These patients have elevated risks of developing ovarian, breast and other cancers.
Breast cancer susceptibility gene 1 and 2 proteins function in a DNA repair pathway for DSB by a process called HR.4, 5 It uses homologous DNA sequences of sister chromatids to ensure genomic stability. In cancer cells, however, DNA repair function is often modulated. Many anticancer agents induce cell death by damaging DNA and accumulating mutations. Therefore, cell death is rarely stimulated in cancer cells with enhanced functions of DNA repair (chemotherapy resistance).
Advances in BRCA1 and BRCA2 research have led to the development of novel therapeutic regimens based on synthetic lethality such as PARP inhibitors. Synthetic lethality is frequently induced in cells with reduced repair function (chemotherapy sensitivity). Consequently, HBOC patients are highly sensitive to DNA‐damaging agents such as PARP inhibitor, platinum‐based agents, and topoisomerase inhibitors.6 In synthetic lethality theory, cells survive even if one of two specific genes involved in cell survival is inhibited. However, cell death is induced when two genes are simultaneously repressed. Poly(ADP‐ribose) polymerase inhibitor treatment is a novel therapy for HBOC patients that targets BRCA1/2 mutations as well as PARP.7, 8 This polymerase plays a role in DNA single‐strand break (SSB) repair. The inhibitor suppresses PARP's SSB repair function during the S‐phase of the cell cycle resulting in unrepaired DNA and DSB formation. Notably, this cell cycle‐specific DSB requires HR for correct repair. Therefore, although HR‐proficient cells can repair DNA lesions, HBOC patients have BRCA1 and BRCA2 mutations whose cancer cells cannot repair DSB as a result of dysregulation of the HR repair pathway and are sensitive to PARP inhibitors. Indeed, recent studies show that complicated molecular mechanisms affect DSB repair.
Normal cells mainly repair DSB by two mechanisms during the cell cycle: HR and NHEJ.9 Optimum pathway selection is necessary for DSB repair under specific conditions. Although NHEJ repairs DSB throughout the cell cycle, HR only functions in the S/G2 phase following DNA replication.10, 11, 12, 13 Many reports have investigated the mechanism of “DSB repair pathway choice” in the S/G2 phase where NHEJ overlaps with HR. Understanding of this pathway choice may best explain how PARP inhibitor‐induced DSB in S‐phase is repaired in several situations.
In the present review, we summarize our current knowledge on the synthetic lethality between BRCA1/2 dysfunction and PARP inhibitors focusing on the molecular mechanisms that regulate the two major DSB repair pathways, molecular defects, and pathway choice. We also discuss the synthetic lethal effect and acquired resistance to PARP inhibitors.
2. PARP INHIBITOR‐INDUCED DNA DAMAGE
Poly(ADP‐ribose) polymerase‐1 (PARP1) is a member of the PARP family that plays a vital role in the repair process of SSB in base excision repair (BER).14, 15 Cells receive constitutive attacks by endogenous and exogenous factors that lead to DNA damage. Base lesions are mainly repaired by BER. At first, damaged sites cleaved by glycosylase and APE create a single‐strand DNA nick. Then, PARP‐1 recognizes it as a SSB and synthesizes PAR polymers covalently at the site as PARylation.16, 17 As a result, PARP1 interacts with proteins such as DNA polymerase β (pol β), DNA ligase III, and X‐ray repair cross‐complementing protein 1 (XRCC1), which are recruited at the SSB site in the BER process. However, in the presence of a PARP inhibitor, PARylation is inhibited by PARP‐1 activity trapping.18 The unrepaired damaged DNA encounters the replication fork during replication in S‐phase. The collision causes the fork to stall and makes a DSB.19 In general, DSB induced by DNA‐damaging agents has two DNA ends; however, DSB generated through replication fork stalling has only one DNA end, called seDSB (Figure 1). Double‐strand break could be a severe threat to genomic stability and must be corrected. Moreover, unlike two‐ended DSB, seDSB needs to be repaired by a more limited pathway to avoid genomic instability and cell lethality.
Figure 1.
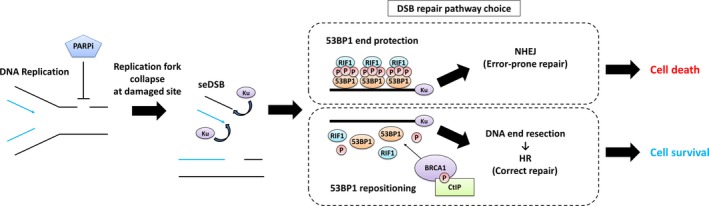
Poly(ADP‐ribose) polymerase (PARP) inhibitor‐induced cell fate through double‐strand break (DSB) repair pathway choice. Single‐ended DSB (seDSB) is generated after PARP inhibitor‐induced DNA replication fork collapse during S phase. The DSB end protected by the p53‐binding protein 1/Rap1‐interacting factor 1 (53BP1‐RIF1) complex is repaired by error‐prone non‐homologous end joining (NHEJ) pathway causing cell death. In contrast, the DSB end released by breast cancer susceptibility gene 1/C‐terminal‐binding protein interacting protein (BRCA1‐CtIP) interaction is resected, leading to the homologous recombination (HR) pathway, resulting in cell survival
3. DSB REPAIR PATHWAY CHOICE
Two principal pathways repair DSB: HR or NHEJ. Homologous recombination error‐free repair uses sister chromosomes as a homologous template. NHEJ error‐prone repair directly ligates damaged DNA ends. DSB repair is regulated in a cell cycle‐dependent method where HR functions in S/G2 and NHEJ in all phases in a competitive way. When DSBs are generated, abundant Ku heterodimers (Ku70 and Ku80 subunits) bind to DSB ends with high affinity.20 Then, either the NHEJ process, recruitment of DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), X‐ray cross‐complementation group 4 (XRCC4), and DNA ligase IV (Lig4),21 starts, or DNA ends are resected (DNA end resection) as an initiating step for HR. Molecular mechanisms regulate each DSB repair pathway (Figure 1).
53BP1, RIF1, CtIP, and BRCA1 play key roles in pathway choice. 53BP1 rapidly participates in repair by surrounding DSB sites after its generation and protects damaged ends from excessive end resection.22, 23 Then, ataxia telangiectasia mutated kinase (ATM)‐dependent phosphorylation of 53BP1 recruits the 53BP1‐binding factor RIF124 and blocks CtIP‐dependent DNA end resection.25 These steps lead to the NHEJ pathway. By contrast, BRCA1 modulates DSB repair pathway with its antagonistic relationship to 53BP1 and RIF1. Several studies reported that cyclin‐dependent kinase (CDK)‐dependent CtIP interaction with BRCA1 is important for promoting end resection and suppression of 53BP1‐RIF1 signaling.26, 27, 28 It was also shown that BRCA1‐induced dephosphorylation of 53BP1 causes RIF1 release from the damaged site and repositioning of 53BP1.29 In contrast, the mechanism of DNA end resection suppression by 53BP1‐RIF1 activity is consistent with a report that loss of BRCA1 decelerates CtIP‐dependent DNA end resection.30 Therefore, dysfunction of this process would allow MRE11‐induced endonuclease activity to be an initiation step31 such that CtIP‐BRCA1 signaling directs the repair pathway from NHEJ to HR.32, 33 Also, the number of replication protein A (RPA) foci resections is reduced in the absence of BRCA1 but is still moderate compared to when CtIP is depleted.29 These findings indicate that CtIP‐dependent end resection is available even when BRCA1 is not present.34 Loss of BRCA1 directs repair to the NHEJ pathway but may not induce a strong inhibition of end resection. In summary, CtIP‐BRCA1 and 53BP1‐RIF1 regulate each pathway during S/G2 phases (Figure 2A) but the detailed mechanisms need to be further investigated.
Figure 2.

Mechanisms of synthetic lethality to poly(ADP‐ribose) polymerase (PARP) inhibitor in breast cancer susceptibility gene 1/2 (BRCA1/2)‐deficient cells through double‐strand break (DSB) repair and ensuing pathways during S/G2 phase. PARP inhibitor‐induced cell fate analyzed in (A) wild‐type (WT), (B) BRCA1‐deficient, and (C) BRCA2‐deficient cells. 53BP1, p53‐binding protein 1; CDK, cyclin‐dependent kinase; CtIP, C‐terminal‐binding protein interacting protein; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; HR, homologous recombination; Lig4, DNA ligase IV; NHEJ, non‐homologous end joining; RIF1, Rap1‐interacting factor 1
4. PARP INHIBITOR‐INDUCED SYNTHETIC LETHALITY
Breast cancer susceptibility gene 1/2‐mutated cancer cells respond well to Poly(ADP‐ribose) polymerase (PARP) inhibitors through synthetic lethality.7, 8 Poly(ADP‐ribose) polymerase inhibitor‐induced cell cycle‐dependent seDSB could be repaired by HR through the same pathway selection mechanism in the S/G2 phase. Although cells require NHEJ for two‐ended DSB repair to survive,35 it causes chromosomal aberration, genomic instability in PARP inhibitor‐treated cells, and cell death because seDSB has no other DNA end that can be correctly ligated.36 Therefore, stalled replication fork‐induced seDSB needs to be repaired by HR (Figures 1, 2A). Both BRCA1 and BRCA2 are known to play critical roles in the HR pathway. Their roles in synthetic lethality will be discussed in the context of other molecules. In addition, the mechanism of synthetic lethality in BRCA1/2‐mutated cells based on the DSB repair pathway models will be explained.
Breast cancer susceptibility gene 1, a key player in DSB repair, directs HR with CtIP by promoting DNA end resection by suppression of the 53BP1‐RIF1 pathway.29, 32 The presence of BRCA1 leads to dysfunctional NHEJ and functional HR in DSB repair. It could also help maintain genomic stability by preventing error‐prone repair of accidentally generated seDSB.37 Conversely, loss of BRCA1‐induced 53BP1‐RIF1 signaling restoration directs the repair pathway to NHEJ and causes RIF1‐dependent partial suppression of end resection. BRCA1 also recruits Rad51 onto the resected DNA strand by replacing RPA and colocalizing with Rad51 and BRCA2 to activate HR38, 39 such that it cannot function in BRCA1‐deficient cells. Therefore, impaired BRCA1 facilitates NHEJ and suppresses HR leading to genome instability. The dysfunctional HR‐induced persistently unrepaired DSB leads to senescence or apoptosis in cells40, 41 and promotion of NHEJ results in chromosome aberrations and cell death. Thus, acceleration of NHEJ and suppression of HR could be a promising target for PARP‐inhibitor cancer treatment (Figure 2B).
Different situations arise in BRCA2‐ and BRCA1‐mutated cells because BRCA2 does not affect DSB repair selection but functions downstream of the HR pathway. Functional BRCA1 suppresses the 53BP1‐RIF1 signaling in the S/G2 cell cycle phase so that the HR pathway is chosen for PARP inhibitor‐induced seDSB repair in both normal and BRCA2‐mutated cells. Then, seDSB undergoes HR after end resection. However, DSB repair will be stalled and BRCA2 loss of function results in cell death (Figure 2C). Also, NHEJ inhibition reduces error‐prone repair, chromosome aberrations, and rescued PARP inhibitor‐induced cell lethality.42, 43 This model shows that cells harboring the BRCA1 mutation contain more gene variability and response to PARP inhibitor than those with the BRCA2 mutation because of NHEJ pathway predominance. Hence, BRCA1 and BRCA2 have different mechanisms of cellular lethality. Furthermore, Fanconi anemia‐related tumor suppressors, including BRCA1 and BRCA2, protect nascent DNA from MRE11‐dependent nucleolytic degradation in the replication fork and maintain genome stability.44 Consequently, impairment of replication fork protection as a result of BRCA1 or BRCA2 dysfunction is considered to be an important mechanism for synthetic lethality by PARP inhibition.45
Some investigations indicated that other molecules besides BRCA1/2 could be potential targets of synthetic lethality. Loss of CtIP sensitized tumors both in vitro and in vivo to PARP inhibitor.46, 47 This response may be a result of CtIP‐dependent end resection suppression that promotes the NHEJ pathway. Interestingly, dysfunction of Rad51 and other molecules involved in the HR pathway also triggered a sensitive response, blocking HR as BRCA2 defective.48, 49 Taken together, end resection and HR‐related factors (CtIP, BRCA1, MRE11, Rad51, BRCA2 etc.) are potential PARP inhibitor targets for cancer treatment.
5. PARP INHIBITOR RESISTANCE MECHANISMS
Poly(ADP‐ribose) polymerase inhibitor‐induced seDSB requires HR to be correctly repaired, and NHEJ is toxic to cells with seDSB. Consequently, resistance to PARP inhibitor might be acquired when the pathway choice shifts from NHEJ to HR. Here we present examples of resistance mechanisms that include restoration of HR function and avoidance of the NHEJ pathway.
5.1. Secondary mutations in BRCA1/2
Cancer recurrence frequently occurs in HBOC patients despite the targeted treatment of tumors with BRCA1/2 mutations with several DNA damaging agents. Sakai et al50, 51 (2008 and 2009) first elucidated the in vitro mechanism by which the secondary mutations that restored BRCA2 function recovered DNA repair capacity, and became resistant to DNA‐damaging agents such as cisplatin and PARP inhibitor. Functional restoration in BRCA2 caused by additional mutations canceled the original mutation‐induced frameshift by reverting to an unimpaired C‐terminal DNA‐binding domain, nuclear localization signal, and Rad51‐binding domain.51, 52 This type of resistance mechanism was also found in BRCA1‐mutated cancer cells.53 Furthermore, clinical data showed that secondary mutations restored not only BRCA1/2 but also Rad51 in recurrent and metastatic tumors from several cancers.54, 55, 56, 57 These findings indicate that replication stress‐induced DNA damage would mainly depend on the HR pathway. Functional recovery of BRCA1/2‐mediated HR would induce a therapeutic window collapse in the clinical strategy that uses DNA repair capacity differences between normal and cancer cells. Consequently, functional restoration may be a major resistance mechanism in PARP inhibitor‐based therapy.
5.2. Loss of 53BP1 in BRCA1‐mutated cells
In BRCA1‐mutated cells, PARP inhibitor effectively induces cell death by promoting NHEJ by activation of the 53BP1‐RIF1 pathway and reducing HR efficiency through incomplete end resection and Rad51 recruitment. Several groups discovered that the loss of 53BP1 function caused PARP inhibitor resistance in BRCA1‐defective cells.25, 58 Dysfunctional 53BP1 by frameshift mutation was also identified in PARP inhibitor‐resistant BRCA1‐mutated tumors in mice.59 Considering the preference for the DSB repair pathway, loss of 53BP1 inactivates RIF1‐dependent regulation of end resection in the absence of BRCA1, resulting in the promotion of HR. Moreover, loss of 53BP1 causes ring finger protein 8 (RNF8)‐induced Rad51 recruitment even in the absence of BRCA1.60 This combined HR pathway restoration of end resection and Rad51 recruitment functions as a cell survival backup mechanism in BRCA1‐mutated cells treated with PARP inhibitor (Figure 3A). Markedly, 53BP1 rescues proliferation defects in BRCA1 but not in BRCA2‐deficient mouse embryonic fibroblasts (MEF).58 Both BRCA1 and BRCA2 defects in cells tend to induce spontaneous replication stress because of lower HR activity. However, BRCA2 is an indispensable factor in the HR process after end resection. Therefore, the repair pathway shift through 53BP1 loss may not affect cell survival in BRCA2‐deficient cells (Figure 3B).
Figure 3.
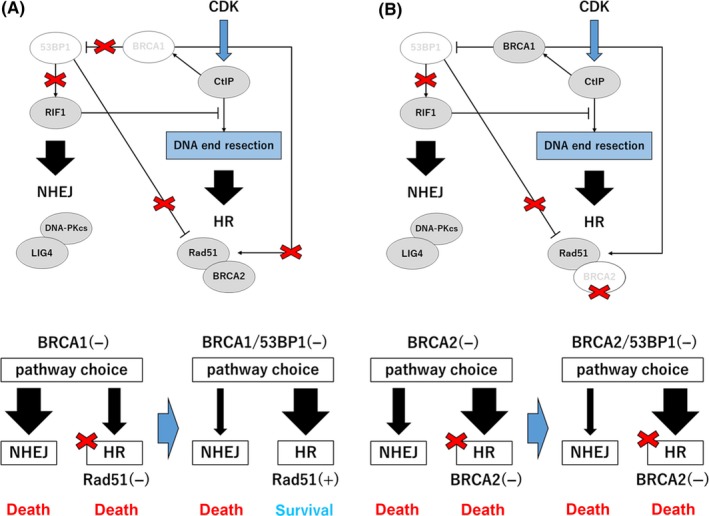
Loss of p53‐binding protein 1 (53BP1)‐induced resistance mechanism to poly(ADP‐ribose) polymerase (PARP) inhibitor in breast cancer susceptibility gene 1 (BRCA1)‐ and breast cancer susceptibility gene 2 (BRCA2)‐deficient cells through double‐strand break (DSB) repair and ensuing pathways during the S/G2 cell cycle phase were compared. Pathway differences in PARP inhibitor sensitivity with additional loss of 53BP1 were analyzed in (A) BRCA1‐deficient and (B) BRCA2‐deficient cells. CDK, cyclin‐dependent kinase; CtIP, C‐terminal‐binding protein interacting protein; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; HR, homologous recombination; Lig4, DNA ligase IV; NHEJ, non‐homologous end joining; RIF1, Rap1‐interacting factor 1
5.3. Additional resistance mechanisms to PARP inhibitor therapy
To date, in addition to secondary mutation and loss of 53BP1 function, different mechanisms underlying PARP inhibitor resistance have been described and resistance mechanisms can be divided into three groups (Table 1). The first group, “restoration of homologous recombination” includes demethylation of the BRCA1 promoter,61 the aforementioned secondary mutation of BRCA1/2,50, 51, 52, 53, 54, 55, 56, 57 and loss of HR suppression factors such as 53BP1.58, 59, 60, 62, 63, 64 The second contains acquisition of replication fork protection.65, 66, 67, 68, 69 The third has the P‐glycoprotein (also known as multidrug resistance protein 1 [MDR 1] or ATP‐binding cassette sub‐family B member 1 [ABCB 1]). Indeed, increased PARP inhibitor efflux by overexpression of this transmembrane transporter has recently been reported.70, 71
Table 1.
PARP inhibitor resistance mechanisms in BRCA1/2‐associated cancers
| Resistant type | HR defect | PARP inhibitor resistance mechanism | References |
|---|---|---|---|
| Restoration of homologous recombination | BRCA1/2 mutation | BRCA1/2 second mutation | 50, 51, 52, 53, 54, 55, 56, 57 |
| Hypermethylation of BRCA1 promoter | Demethylation of BRCA1 promoter | 61 | |
| BRCA1 mutation |
Dysfunction of HR suppression factors 53BP1, REV7, JMJD1C, RIF1 |
25, 58, 59, 60, 62, 63, 64 | |
| Protection of replication forks | BRCA2 mutation | Restoration of fork protection: Inhibition of molecules related with degradation of stalled replication forks | 65, 66, 67, 68, 69 |
| Increased efflux of PARP inhibitor | BRCA1/2 mutation | Increased expression of P‐glycoprotein (MDR1) | 70, 71 |
53BP1, p53‐binding protein 1; BRCA1/2, breast cancer susceptibility gene 1/2; HR, homologous recombination; PARP, poly(ADP‐ribose) polymerase; RIF1, Rap1‐interacting factor 1.
6. CONCLUSIONS/FUTURE DIRECTIONS
Studies on PARP inhibitor‐based clinical investigations are subject to heated discussions not only for HBOC but also for other types of cancer with DNA repair defects. The practical knowledge gained from clinical data preceded detailed elucidation of the PARP inhibitor‐induced DNA damage mechanism and subsequent complicated repair process. Here, we have discussed synthetic lethality and potential resistance mechanisms to PARP inhibitor mainly in connection with DSB repair pathways. In particular, BRCA1, together with several other molecules, has several roles as a mediator of the HR pathway to sustain genome stability. Also, the factor‐like loss of 53BP1 recovers the HR pathway even in the absence of BRCA1. Therefore, the clinical strategies to overcome the acquired resistance to PARP inhibitor treatment for BRCA1‐ and BRCA2‐mutated tumors should be different. In addition, other DSB repair pathways (microhomology‐mediated end joining [MMEJ] and single‐strand annealing [SSA]) could be sensitized to PARP inhibitor, but this hypothesis requires further investigation. The present review will contribute to the future development of both fundamental and clinical studies.
CONFLICT OF INTEREST
YM reports receiving patent royalties from the University of Utah, USA, and honoraria from AstraZeneca K.K. The other authors have no conflicts of interest to declare.
ACKNOWLEDGMENT
This work was supported by JSPS KAKENHI Grant Number JP16H04693 (YM).
Sunada S, Nakanishi A, Miki Y. Crosstalk of DNA double‐strand break repair pathways in poly(ADP‐ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2‐mutated cancer. Cancer Sci. 2018;109:893–899. https://doi.org/10.1111/cas.13530
Funding information
Japan Society for the Promotion of Science (Grant/Award Number: ‘JP16H04693’).
REFERENCES
- 1. Miki Y, Swensen J, Shattuck‐Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66‐71. [DOI] [PubMed] [Google Scholar]
- 2. Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789‐792. [DOI] [PubMed] [Google Scholar]
- 3. Buys SS, Sandbach JF, Gammon A, et al. A study of over 35,000 women with breast cancer tested with a 25‐gene panel of hereditary cancer genes. Cancer. 2017;123:1721‐1730. [DOI] [PubMed] [Google Scholar]
- 4. Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014;343:1470‐1475. [DOI] [PubMed] [Google Scholar]
- 5. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vollebergh MA, Lips EH, Nederlof PM, et al. Genomic patterns resembling BRCA1‐ and BRCA2‐mutated breast cancers predict benefit of intensified carboplatin‐based chemotherapy. Breast Cancer Res. 2014;16:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917‐921. [DOI] [PubMed] [Google Scholar]
- 8. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434:913‐917. [DOI] [PubMed] [Google Scholar]
- 9. Shibata A, Jeggo PA. DNA double‐strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 2014;26:243‐249. [DOI] [PubMed] [Google Scholar]
- 10. Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double‐strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706‐5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Orthwein A, Fradet‐Turcotte A, Noordermeer SM, et al. Mitosis inhibits DNA double‐strand break repair to guard against telomere fusions. Science. 2014;344:189‐193. [DOI] [PubMed] [Google Scholar]
- 12. Terasawa M, Shinohara A, Shinohara M. Canonical non‐homologous end joining in mitosis induces genome instability and is suppressed by M‐phase‐specific phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP‐ribose) polymerase 1 accelerates single‐strand break repair in concert with poly(ADP‐ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597‐5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single‐strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP‐ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249‐268. [PMC free article] [PubMed] [Google Scholar]
- 18. Satoh MS, Lindahl T. Role of poly(ADP‐ribose) formation in DNA repair. Nature. 1992;356:356‐358. [DOI] [PubMed] [Google Scholar]
- 19. Heacock ML, Stefanick DF, Horton JK, Wilson SH. Alkylation DNA damage in combination with PARP inhibition results in formation of S‐phase‐dependent double‐strand breaks. DNA Repair (Amst). 2010;9:929‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double‐strand break repair. Nature. 2001;412:607‐614. [DOI] [PubMed] [Google Scholar]
- 21. Jeggo PA. DNA breakage and repair. Adv Genet. 1998;38:185‐218. [DOI] [PubMed] [Google Scholar]
- 22. Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double‐strand breaks. J Cell Biol. 2000;151:1381‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Virgilio M, Callen E, Yamane A, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang H, Shi LZ, Wong CC, et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR‐mediated double‐strand break repair. PLoS Genet. 2013;9:e1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1‐associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci. 2012;125:3529‐3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19:1‐9. [DOI] [PubMed] [Google Scholar]
- 29. Isono M, Niimi A, Oike T, et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017;18:520‐532. [DOI] [PubMed] [Google Scholar]
- 30. Cruz‐Garcia A, Lopez‐Saavedra A, Huertas P. BRCA1 accelerates CtIP‐mediated DNA‐end resection. Cell Rep. 2014;9:451‐459. [DOI] [PubMed] [Google Scholar]
- 31. Shibata A, Moiani D, Arvai AS, et al. DNA double‐strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yun MH, Hiom K. CtIP‐BRCA1 modulates the choice of DNA double‐strand‐break repair pathway throughout the cell cycle. Nature. 2009;459:460‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Escribano‐Diaz C, Orthwein A, Fradet‐Turcotte A, et al. A cell cycle‐dependent regulatory circuit composed of 53BP1‐RIF1 and BRCA1‐CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872‐883. [DOI] [PubMed] [Google Scholar]
- 34. Polato F, Callen E, Wong N, et al. CtIP‐mediated resection is essential for viability and can operate independently of BRCA1. J Exp Med. 2014;211:1027‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sunada S, Kanai H, Lee Y, et al. Nontoxic concentration of DNA‐PK inhibitor NU7441 radio‐sensitizes lung tumor cells with little effect on double strand break repair. Cancer Sci. 2016;107:1250‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gelot C, Magdalou I, Lopez BS. Replication stress in mammalian cells and its consequences for mitosis. Genes (Basel). 2015;6:267‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savage KI, Harkin DP. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015;282:630‐646. [DOI] [PubMed] [Google Scholar]
- 38. Scully R, Chen J, Plug A, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265‐275. [DOI] [PubMed] [Google Scholar]
- 39. Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999;59(7 suppl):1752s‐1756s. [PubMed] [Google Scholar]
- 40. Galbiati A, Beausejour C, d'Adda di Fagagna F. A novel single‐cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017;16:422‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. White RR, Vijg J. Do DNA double‐strand breaks drive aging? Mol Cell. 2016;63:729‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP‐ribose) polymerase (PARP) inhibitor lethality in homologous recombination‐deficient cells. Proc Natl Acad Sci USA. 2011;108:3406‐3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Somyajit K, Mishra A, Jameei A, Nagaraju G. Enhanced non‐homologous end joining contributes toward synthetic lethality of pathological RAD51C mutants with poly (ADP‐ribose) polymerase. Carcinogenesis. 2015;36:13‐24. [DOI] [PubMed] [Google Scholar]
- 44. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ying S, Hamdy FC, Helleday T. Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72:2814‐2821. [DOI] [PubMed] [Google Scholar]
- 46. Gaymes TJ, Mohamedali AM, Patterson M, et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP‐ribose) polymerase inhibitors in myeloid malignancies. Haematologica. 2013;98:1397‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J, Ding Q, Fujimori H, Motegi A, Miki Y, Masutani M. Loss of CtIP disturbs homologous recombination repair and sensitizes breast cancer cells to PARP inhibitors. Oncotarget. 2016;7:7701‐7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res. 2006;66:8109‐8115. [DOI] [PubMed] [Google Scholar]
- 49. Min A, Im SA, Yoon YK, et al. RAD51C‐deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther. 2013;12:865‐877. [DOI] [PubMed] [Google Scholar]
- 50. Sakai W, Swisher EM, Jacquemont C, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2‐mutated ovarian carcinoma. Cancer Res. 2009;69:6381‐6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2‐mutated cancers. Nature. 2008;451:1116‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111‐1115. [DOI] [PubMed] [Google Scholar]
- 53. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1‐mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581‐2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229:422‐429. [DOI] [PubMed] [Google Scholar]
- 55. Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008‐3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pishvaian MJ, Biankin AV, Bailey P, et al. BRCA2 secondary mutation‐mediated resistance to platinum and PARP inhibitor‐based therapy in pancreatic cancer. Br J Cancer. 2017;116:1021‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kondrashova O, Nguyen M, Shield‐Artin K, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high‐grade ovarian carcinoma. Cancer Discov. 2017;7:984‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple‐negative and BRCA‐mutated breast cancers. Nat Struct Mol Biol. 2010;17:688‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1‐mutated mouse mammary tumors. Cancer Discov. 2013;3:68‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakada S, Yonamine RM, Matsuo K. RNF8 regulates assembly of RAD51 at DNA double‐strand breaks in the absence of BRCA1 and 53BP1. Cancer Res. 2012;72:4974‐4983. [DOI] [PubMed] [Google Scholar]
- 61. Ter Brugge P, Kristel P, van der Burg E, et al. Mechanisms of therapy resistance in patient‐derived xenograft models of BRCA1‐deficient breast cancer. J Natl Cancer Inst. 2016;108:djw148 https://doi.org/10.1093/jnci/djw148. [DOI] [PubMed] [Google Scholar]
- 62. Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature. 2015;521:541‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watanabe S, Watanabe K, Akimov V, et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1‐mediated chromatin response to DNA breaks. Nat Struct Mol Biol. 2013;20:1425‐1433. [DOI] [PubMed] [Google Scholar]
- 64. Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1‐dependent nonhomologous end joining and suppression of DNA double‐strand break resection. Mol Cell. 2013;49:858‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ray Chaudhuri A, Callen E, Ding X, et al. Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature. 2016;535:382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rondinelli B, Gogola E, Yucel H, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371‐1378. [DOI] [PubMed] [Google Scholar]
- 67. Lemacon D, Jackson J, Quinet A, et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81‐dependent fork rescue in BRCA2‐deficient cells. Nat Commun. 2017;8:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dungrawala H, Bhat KP, Le Meur R, et al. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol Cell. 2017;67:374‐386. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kolinjivadi AM, Sannino V, De Antoni A, et al. Smarcal1‐mediated fork reversal triggers Mre11‐dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell. 2017;67:867‐881. e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jaspers JE, Sol W, Kersbergen A, et al. BRCA2‐deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015;75:732‐741. [DOI] [PubMed] [Google Scholar]