

Abstract
In recent years, it has become clear that members of the signal transducer and activator of transcription (STAT) family of genes play an important role in cancer. The STAT family consists of seven genes, STAT1‐4,STAT5A, STAT5B and STAT6, that are involved in regulating cellular proliferation, apoptosis, angiogenesis and the immune system response. Constitutive activation of STAT3, via mutational changes, is important in oncogenesis in both solid and hematopoietic cancers. In the case of hematopoietic neoplasms, STAT3 driver mutations have been described in T‐cell large granular lymphocytic (T‐LGL) leukemia and chronic natural killer lymphoproliferative disorders (CLPD‐NK) and are seen in 30%‐40% of T‐LGL leukemia patients. STAT5B is also mutated in T‐LGL leukemia and CLPD‐NK, but in a much smaller proportion. Here we review past and current research on STAT genes in hematopoietic and solid cancers with emphasis on STAT3 and STAT5B and their roles in the pathogenesis of hematopoietic malignancies, particularly T‐LGL leukemia and CLPD‐NK.
Keywords: CLPD‐NK, JAK‐STAT pathway, STAT3, STAT5B, T‐LGL leukemia
1. INTRODUCTION
The STAT family, encompassing 7 members (STAT1‐4, STAT5A, STAT5B and STAT6), has been studied widely and has many different biological functions, including roles in cell differentiation, proliferation, inflammation and apoptosis. These proteins participate in the pathogenesis of many inflammatory, autoimmune and neoplastic diseases due to their involvement in diverse signaling events downstream of cytokines, interleukins (IL‐6, which is part of the IL‐6 signaling pathway, is the most important activator of STAT3) and growth factor receptors.1 They operate both as cytoplasmic signaling proteins and as nuclear transcription factors with a diverse set of target genes, including oncogenes and tumor suppressors.2, 3, 4
Of the 7 members, STAT3 and STAT5, particularly STAT3, are the most significant in cancer development; meanwhile STAT1, STAT2, STAT4 and STAT6 appear to have more limited roles in oncogenesis.
Signal transducer and activator of transcription (STAT) are part of the JAK (Janus kinase)‐STAT pathway, which is a signaling pathway that allows direct communication between transmembrane receptors and the nucleus. Ligands in the form of cytokines, interleukins and growth factors bind to their respective transmembrane receptors and upon ligand binding, receptor‐associated JAK become activated and phosphorylate each other and the intracellular portion of their receptors. This phosphorylation then recruits STAT monomers to the phosphorylated receptors, which then undergo tyrosine phosphorylation themselves and dimerize. The STAT‐STAT dimer then translocates from the cytoplasm to the nucleus, where it can bind to the DNA promotor sequences of target genes, and thereby directly regulates gene expression by either enhancing or inhibiting gene transcription. It is now clear that STAT bind to tens of thousands of different sites in the genome and thereby regulate transcription of thousands of protein‐coding genes.2, 3, 4, 5, 6 The JAK‐STAT pathway is intricately regulated to create a fine balance between enhancement and inhibition because persistent stimulation of STAT can result in both malignancy, autoimmune inflammatory diseases.3, 5, 6
In addition, recent studies have shown that STAT3 not only regulates gene expression by acting as a transcription factor but regulates gene expression ultimately through epigenetic mechanisms such as DNA methylation and chromatin modification.1
The JAK part of JAK‐STAT pathway is well researched and JAK mutations and their importance in pathogenesis of hematological disorders are well studied, with JAK2 V617F being the most well‐known mutation, which is detected in the majority of myeloproliferative neoplasms, such as polycythemia vera, primary myelofibrosis and essential thrombocytosis. Numerous JAK‐STAT pathway inhibitors have been developed; for instance, the JAK1/2 inhibitor ruxolitinib is approved for treatment of polycythemia vera and primary myelofibrosis, and the pan‐JAK inhibitor tofacitinib has also been used in treatment of rheumatologic diseases.2, 5 Studies have also demonstrated that STAT3 may be the ultimate downstream target of JAK inhibitors and in a study by Chen et al,7 it was demonstrated that JAK inhibitors may have a place in treatment of anaplastic lymphoma kinase negative anaplastic large‐cell lymphoma (p‐STAT3(+) ALK(–) ALCL) even without occurrence of JAK or STAT3 mutations.
In this review, we focus on the STAT portion of the JAK‐STAT pathway with emphasis on STAT3 and STAT5B and their roles in the pathogenesis of diverse diseases with main emphasis on hematopoietic malignancies where we focus on STAT3 and STAT5B in T‐cell large granular lymphocytic (T‐LGL) leukemia, CLPD‐NK (Chronic NK lymphoproliferative disorders), B and T cell lymphomas, in addition to melanoma and other tumors.
2. OVERVIEW AND INCIDENCE OF SOMATIC STAT GENE MUTATIONS IN CANCER
The STAT proteins all consist of the same 6 domains (Figure 1): an N‐terminal domain followed by a coiled‐coil domain, a DNA‐binding domain (DBD), a linker, an Src‐homology2 (SH2) domain and a C‐terminal transactivation domain (TAD) (Figure 1). The SH2 domain is highly conserved and is the target of most STAT inhibitors that have been tested to this date. The SH2 domain together with the N‐terminal domain mediate homo and heterodimer formation of STAT monomers. The coiled‐coil domain functions as a nuclear localization signal, while TAD undergoes serine phosphorylation and thereby recruits additional transcriptional activators and enhances the transcriptional activity of STAT. DBD, as the name implicates, determines DNA association.3, 4, 5, 6, 8
Figure 1.
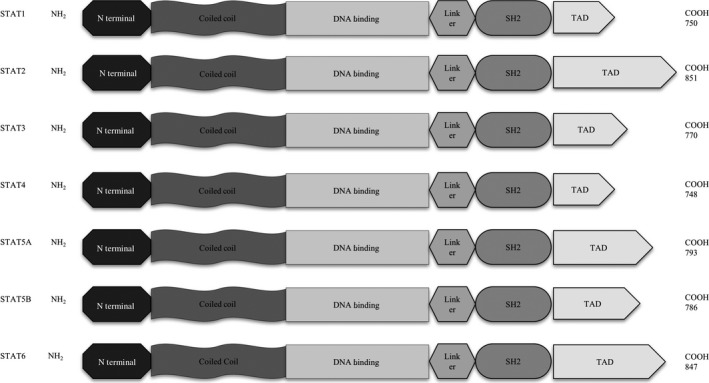
Signal transducer and activator of transcription (STAT) family members: 7 proteins that all consist of the same 6 domains: amino acid length is indicated on the right for each member. SH2, Scr homology domain 2; TAD, transactivation domain
STAT3 and STAT5B are the 2 members of the STAT family that are constitutively activated in multiple cancers. Constitutive activation of STAT3 can be caused by 4 mechanisms: increased upstream stimulation of kinases, lack of negative regulation, positive feedback loops and somatic mutations that cause hyperactivity to STAT3. STAT3 is predominantly mutated in hematopoietic neoplasms, especially in T‐LGL leukemia where a high mutation rate (30%‐40%) is observed and in CLPD‐NK (Figure 2) .
Figure 2.
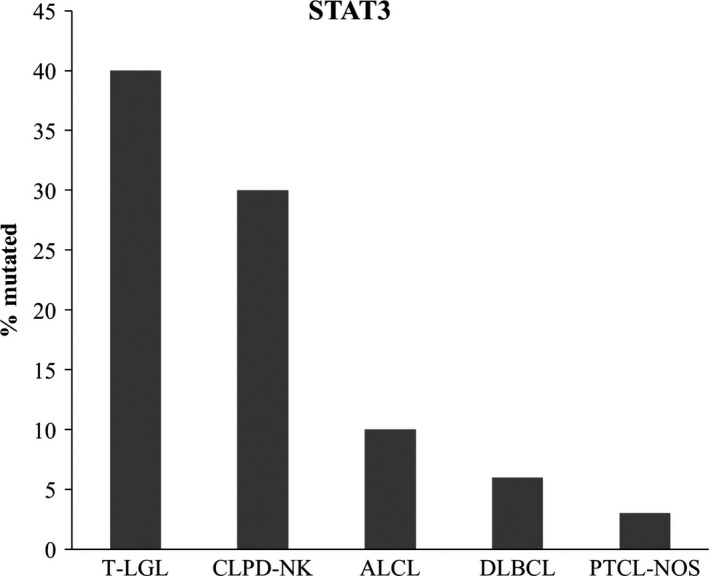
Frequency of STAT3 mutations in T‐LGL (T‐cell large granular lymphocytic) leukemia, CLPD‐NK (chronic NK lymphoproliferative disorder),18,19,21‐23,26 PTCL‐NOS (peripheral T‐cell lymphomas not otherwise specified),52 ALCL (anaplastic large T‐cell lymphoma)34,36 and DLBCL (Diffuse large B cell lymphoma).31,32 STAT3 plays a major role in pathogenesis of CLPD‐NK and T‐LGL leukemia where it is highly mutated
In addition to its role in oncogenesis, STAT3 is also involved in the pathogenesis of autoimmune diseases. De novo germline activating STAT3 mutations can result in early onset autoimmune disorders, while inactivating germline mutations are known to result in Hyper IgE syndrome (HIES).9, 10 In a study by Flanagan et al a cohort of 65 patients with early‐onset autoimmunity (all patients were under 5 years of age at the time of the genetic testing) were tested for de novo STAT3 mutations by exome sequencing, and 4 different de novo heterozygous missense mutations were identified (K392R, N646K, K658N and T716M). Flanagan et al11 conclude that these mutations were disease‐causing in these unrelated individuals. Inactivating germline mutations in STAT3 are, in contrast, known to cause HIES, which is characterized by eosinophilia, eczema and recurrent bacterial infections, among other symptoms.9, 10
It is now evident that STAT3 plays a critical role in cancer. Based on data collected from the Catalogue of Somatic Mutations In Cancer (COSMIC), the frequency of somatic STAT3 and other STAT family member mutations in different cancer subtypes (ie, skin cancers, melanoma, gastrointestinal neoplasms, neural tumors and hematopoietic neoplasms) are shown in Figure 3.12 As expected, STAT3 is the most frequently mutated gene from the STAT family in hematopoietic cancers, followed by STAT5B. STAT1 is unexpectedly the most frequently mutated in melanoma, followed by STAT5B and STAT4, even though the overall occurrence of STAT mutations is low in all entities other than T‐LGL and CLPD‐NK (Figures 2 and 3).12
Figure 3.
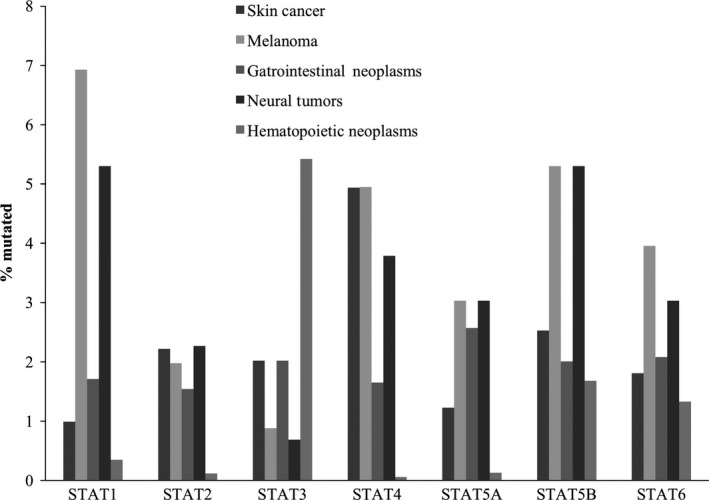
Mutational frequencies of Signal transducer and activator of transcription (STAT) genes in different entities. Data was obtained from the Catalogue of Somatic Mutations In Cancer (COSMIC). STAT3 is, as expected, the most frequent gene mutated in hematopoietic entities
3. GENETICS AND PATHOLOGIC FEATURES OF CANCERS WITH STAT GENE MUTATIONS
Here we review the mutational status of STAT3 and STAT5B in T‐LGL, CLPD‐NK, aggressive B and T cell lymphomas, melanoma and other tumors, with emphasis on T‐LGL leukemia and CLPD‐NK, where STAT3 mutational status is useful as a diagnostic marker.
3.1. T‐cell large granular lymphocytic leukemia and chronic natural killer lymphoproliferative disorders
T‐cell large granular lymphocytic leukemia and CLPD‐NK are rare lymphoproliferative disorders ranging from 2% to 5% of all chronic lymphoproliferative disorders and are characterized by expansions of CD3+CD8+ cytotoxic T‐cells or CD16+CD56+ NK‐cells, respectively. No significant differences in clinical course or therapy exist between T‐LGL leukemia and CLPD‐NK. The median age for those diagnosed with T‐LGL leukemia is 55‐60 years old, and only 20%‐25% of patients are younger than 50 years old: diagnoses are equally distributed among men and women.13, 14
Patients affected by this disease typically show cytopenias (neutropenia and/or anemia) and may have splenomegaly with recurrent infections; some cases are associated with autoimmune diseases, such as rheumatoid arthritis (RA) and even systemic lupus erythematosus (SLE).13, 14 Interestingly, the clinical phenotype presents itself differently in various ethnic groups. In Asian cohorts, as studied by Ishida et al and Qui et al, pure red blood cell aplasia (PRCA) is more frequently associated with a T‐LGL leukemia diagnosis; patients are typically not neutropenic, unlike patient cohorts from Western countries where rheumatoid arthritis (RA) and infections caused by neutropenia are seen more frequently.15, 16, 17
STAT3 is the first molecular marker that is highly specific for T‐LGL leukemia, and several mutational hotspots have been detected in 30%‐40% of T‐LGL leukemia patients and in one‐third of the patients with CLPD‐NK; a smaller subset of T‐LGL leukemia patients (2%) have STAT5B mutations.18, 19, 20, 21, 22, 23 In a study by Fasan et al,18 a cohort of 55 patients with unselected TCR rearrangements were studied and STAT3 mutations were detected in 72.7% of the patients (40 out of 55).
In most T‐LGL leukemia cases, a clonal expansion of CD8+ large granular lymphocytes exists, with a smaller subset originating from CD4+ LGLs. In a study by Andersson et al,24 STAT5B mutations were detected in 55% (6 of 11 patients) of the patients with CD4+T‐LGL leukemia and no STAT5B mutation was detected in CD8+ T‐LGL leukemia. The authors of this study concluded that STAT5B mutations are more unique for the CD4+ phenotype than for the CD8+ phenotype. Clinically, these patients had an indolent disease, which argues against previous assumptions about a more clinically aggressive disease in T‐LGL leukemia patients with STAT5B mutations.24 Larger cohorts need to be studied to confirm a potential correlation between the CD4+ phenotype and existence of STAT5B mutations, and to determine the clinical course in this group of patients.24
As described above, STAT3 is mutated in 30%‐40% of T‐LGL cases and STAT3 mutational hotspots are mainly located in the SH2 domain, as shown in Figure 4. However, more recently, in a study by Andersson et al, activating STAT3 mutations were also detected in the DBD and the coiled‐coil domain in a cohort of patients without known SH2 hotspot mutations in STAT3 or STAT5B. Phenotypically, there was no difference between patients with somatic mutations in the SH2 domain compared to those with mutations in the DBD and in the coiled‐coil domain Figure 4.25
Figure 4.
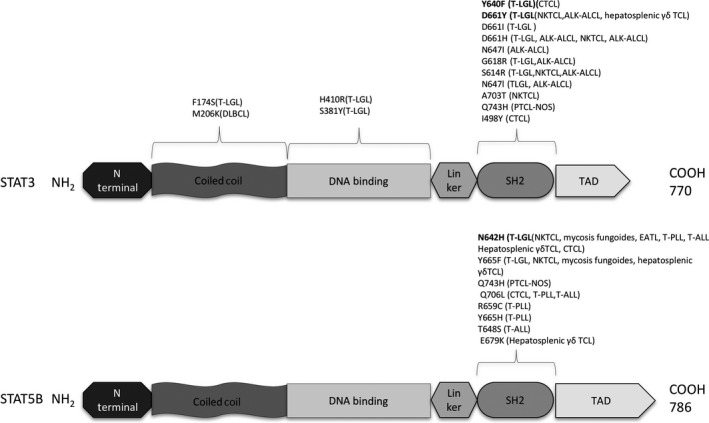
Mutations in STAT3 and STAT5B are predominantly localized in the SH2 domain; however, recent studies have identified additional mutations in the DNA binding domain and coiled‐coil domain of STAT3, which are shown here: mutational hotspot changes are in bold
Studies have, furthermore, shown that almost all T‐LGL leukemia patients have activation of the STAT3 pathway, even without somatic mutations in the STAT3 gene. Andersson et al23 performed exome sequencing on 3 STAT3‐mutation negative T‐LGL leukemia patients and found an activated STAT3 pathway by RNA expression and pSTAT analysis, indicating that T‐LGL leukemia patients have activation of STAT3 responsive genes even without the actual STAT3 somatic mutations. More research involving patient cohorts diagnosed with T‐LGL leukemia who lack STAT3/STAT5B mutations needs to be conducted to characterize the mutational landscape in this heterogeneous subgroup of T‐LGL leukemia patients. Wild‐type and STAT3 mutant patients share many clinical features phenotypically and are believed to respond similarly to potential STAT inhibitors.23, 26
Despite the high mutational frequency of STAT3 mutations in T‐LGL leukemia and CLPD‐NK, there are some studies that question the role of the STAT3 mutations as the driver mutations in this entity. In a study by Dutta et al, for instance, it was demonstrated that expression of the STAT3 mutations were insufficient to induce LGL leukemia in mice models, and it was suggested that STAT3 mutations do not play a causal role in development of T‐LGL leukemia and that additional gene mutations and deregulation of other signaling pathways in association with STAT3 mutations may cause T‐LGL leukemia. However, it is well known that mouse models may not always recapitulate human cancer biology due to tissue architectural differences, different susceptibilities to malignant transformation, and engagement of different oncoprotein signaling pathways between mice and humans.27
Cases of benign large granular lymphocytosis with clonal expansion of T cells in reactive processes, such as in viral infections and autoimmune diseases, complicate the diagnosis of large granular lymphocytic leukemia. Somatic STAT3 mutations can provide a diagnostic tool to distinguish true malignant lymphoproliferation involving T and NK cells from reactive expansions of LGL, and also provide a novel therapeutic target for this entity.6, 28
3.2. Aggressive B‐cell lymphomas
B cell lymphomas are more common than T cell lymphomas and account for approximately 85% of all non‐Hodgkin lymphomas (NHL). Diffuse large B cell lymphoma (DLBCL) is the most common form of NHL, accounting for 80% of aggressive lymphomas. Multiple genetic sequencing studies with DLBCL cohorts have been performed and demonstrate that STAT3 mutations are not frequent in this entity. In a study by Guangzhen Hu et al,29 all exons of STAT3 gene from 40 DLBCL tumors were sequenced. Two point mutations were identified in all cases: 1 missense mutation M206K and 1 synonymous mutation 2552G > A. M206K was located in the coiled‐coil domain of STAT3 and shown to be an activating mutation that increased STAT3 signaling and thereby caused cell proliferation.30
STAT3 somatic mutations have also been identified in DLBCL not otherwise specified (DLBCL‐NOS) and in B‐cell lymphoma, unclassifiable, with features intermediate between diffuse large B‐cell lymphoma and Burkitt lymphoma (BCLU, DLBCL/BL). In addition, in DLBCL a higher mutation rate in STAT3 is seen in cases with CD30+ overexpression compared to those without CD30+ overexpression.31
Even though the occurrence of somatic STAT3 mutations are low in DLBCL, the prevalence of pSTAT3 expression in DLBCL is high and varies in different studies from 11% to 67%. In a study by Ok et al, which included 443 patients with DLBCL, 16% of the patients showed expression of pSTAT3. Expression of pSTAT3 seemed to correlate with advanced stage, multiple sites of extra nodal involvements, activated B‐cell like (ABC) subtype, MYC expression and MYC/BCL2 double expression. DLBCL patients with pSTAT3 expression demonstrated a more unfavorable prognosis with poorer overall survival and progression free survival.32 It has even been suggested that these patients are molecularly distinct from pSTAT3 (−) DLBCL patients and may benefit from molecularly targeted therapy against STAT3.32
Immunohistochemistry can be helpful to detect nuclear expression of STAT3 in DLBCL patients to identify a subgroup of patients with poorer clinical outcome who can be given a more aggressive treatment from the beginning. However, more research needs to be done to determine the significance of pSTAT3 expression in DLBCL patients and whether the presence of pSTAT3 worsens these patients' prognosis.33
3.3. Aggressive T‐cell lymphomas
T‐cell malignancies other than T‐LGL leukemia and CLPD‐NK demonstrate STAT3 mutations, as shown in Figure 2, although the occurrence is lower than in T‐LGL leukemia and CLPD‐NK. STAT3 mutations are more frequently detected in CD30+ T‐cell lymphomas than in CD30− T‐cell lymphomas and have been identified in anaplastic lymphoma kinase negative anaplastic large cell lymphoma, ALK(−) ALCL, ALK(+) ALCL, in peripheral T‐cell lymphomas not otherwise specified (PTCL‐NOS) and in cutaneous T cell lymphoma (CTCL).34, 35, 36
Genetic mutational sequencing studies of STAT5B have detected mutations in a subset of patients with mycosis fungoides, in enteropathy associated T‐cell lymphoma (EATL) and in T‐cell acute lymphoblastic leukemia (T‐ALL).2 In a study by Kiel et al, a patient cohort of 50 patients with T‐cell prolymphocytic leukemia (T‐PLL) was studied by whole exome sequencing (WES) and whole genome sequencing (WGS), and a high percentage of the patients, 18 out of 50 (36%), had somatic mutations in STAT5B. In another study of T‐PLL by Lopez et al, STAT5B mutations were detected in 8 out of 39 patients (21%), and in both studies the mutations were located in the SH2 domain of the STAT5B; no STAT3 mutations were found in these studies. In addition to STAT5B, JAK3 was also frequently mutated in T‐PLL in both studies, even though STAT5B and JAK3 were mutually exclusive. These studies confirmed the significance of JAK‐STAT pathway in T‐PLL and suggested that therapeutically targeting the JAK‐STAT pathway may potentially improve treatment of T‐PLL.37, 38
3.4. Melanoma
Melanoma is a genomic unstable disease and has the highest basal mutation rate of any cancer sequenced to date. Different driver mutations have been discovered for this disease, with discovery of BRAF V600E mutation in over 50% of melanomas in 2002 being perhaps most important.39 While most STAT genes are not mutated in any significant frequency in melanoma, surprisingly, based on COSMIC data, several STAT genes show relevant mutational frequencies: STAT1 (6.93%) followed by STAT5B (5.3%), STAT4 (4.95%) and STAT3 (0.88%), as shown in Figure 3. Although the mutation rate of STAT genes, in particular STAT3, is not high in melanoma, studies have shown that STAT3 activation plays a role in tumor growth and angiogenesis of melanoma in tumor xenograft studies.
In a study by Xie et al, it was demonstrated that STAT3 activation is higher in brain metastasis than in primary melanoma, suggesting a possible association between STAT3 activation and development of brain metastasis in melanoma patients. STAT3 activity is believed to drive overexpression of the metastatic molecules and thereby contribute to brain metastasis by increasing tumor invasion and angiogenesis.40 More studies involving melanoma patients need to be conducted to address whether increased levels of STAT3 protein in primary melanoma could indicate development of brain metastasis later in these patients: if this is proven, STAT3 might have potential as a targeted therapy for brain metastasis in melanoma and may improve the prognosis in this patient cohort that usually has a very dismal prognosis.
3.5. Other tumors
The STAT family of genes are not significantly mutated in other cancers beyond those listed above; however, constitutive activation of STAT3 has been identified in head and neck cancers, multiple myeloma as well as breast cancer, prostate cancer, ovarian cancer, GI cancer, and neural tumors such as glioblastoma multiforme (GBM).41, 42, 43
In recent studies, it has been demonstrated that STAT3 is activated in significantly higher levels in GBM tumor cells than in normal brain cells. The high STAT3 activation seems to be a negative prognostic indicator in GBM, which is the most common primary brain tumor and patients have a dismal prognosis.43 More research is obviously needed to determine if STAT3 activation plays any role in GBM patients and if it has a negative prognostic effect.
4. THERAPIES TARGETING THE STAT PATHWAY
The development of STAT inhibitors has become an area of major focus as such inhibitors may help in treatment of STAT3 and STAT5B mutated T‐LGL leukemia, CLPD‐NK and other types of solid and hematopoietic malignancies, as well as potential therapeutics in autoimmune and inflammatory disorders. To this date, no clinically effective inhibitor specifically of STAT proteins has been identified.44, 45, 46 However, mechanistically, STAT, in particular STAT3, can be targeted in multiple ways directly or indirectly: by inhibiting upstream kinases, such as JAK2 inhibitors, inhibiting dimerization of STAT monomers by targeting SH2 domain, developing compounds that cause dephosphorylating of STAT, inhibiting STAT DNA binding, and neutralizing antibody against upstream receptors and ligands and TAD inhibitors. As mentioned above, currently there is no STAT inhibitor available in the clinic in part due to numerous challenges facing development of these inhibitors: issues with bioavailability, in vivo efficacy, adverse side effects and selectivity.28 Significant homology, for instance, exists between STAT1 and STAT3, STAT1 facilitates functions such as apoptosis and pathogen defense, and an off‐target STAT1 blockade by a STAT3 inhibitor can result in unwanted adverse effects such as increased survival of tumor cells.28, 46
To this date, targeted therapies focused on inhibiting the SH2 domain in STAT3 have been investigated in several phase I/II clinical trials in both solid tumors and hematopoietic cancers, including OPB‐31121 which was studied in a phase I/II trial for hepatocellular carcinoma but had unacceptable adverse effects, such as peripheral neuropathy, and had limited efficacy.47, 48 OPB‐51602 was another inhibitor which appeared safe and effective in the treatment of solid tumors such as lung cancer but was also associated with a high risk of peripheral neuropathy and had poor bioavailability.45 Inhibitors of DBD, TAD and N‐terminal domains of STAT3 have also been investigated as potential targets for therapy, and research is ongoing to develop not only STAT3 inhibitors but also inhibitors of other members of the STAT family of genes.28, 45, 46, 47
STAT6, for instance, plays an important role in allergic pathways acting downstream of IL‐4 and IL‐13 and phosphopeptides blocking the SH2 domain of STAT6 are under investigation to inhibit phosphorylation and further downstream signaling of this pathway, which may be useful in allergic diseases such as bronchial asthma.
5. CONCLUSIONS
It is now evident that STAT proteins, in particular STAT3 and STAT5B, play an essential role in oncogenesis. Multiple signaling pathways, beyond the canonical JAK‐STAT pathway are involved in particularly activating STAT3: for instance, the IL‐6 signaling pathway, which encompasses multiple cytokines, such as IL‐6 and IL‐11, activates STAT3.
Recent studies have revealed that known oncogenes such as BCR‐ABL and RAS can upregulate expression of IL‐6, and thereby indirectly activate the JAK‐STAT3 pathway. STAT3 inhibitors may, therefore, have the potential to block the effects of these oncogenes.1
As described elsewhere, STAT3 activation is usually tightly regulated through several mechanisms, such as protein inhibitor of activated STAT (PIAS), the suppressors of cytokine signaling (SOCS) and several tyrosine protein phosphatases (PTP), which dephosphorylate activated STAT. Hyperactive growth factor receptors and overexpression of stimulatory ligands in the form of IL‐6 and IL‐10 can, however, lead to aberrant activation of STAT3. Activated STAT3 is then translocated to the nucleus where it enhances transcription of downstream target genes. STAT3‐regulated genes encode cytokines, growth receptors and angiogenic factors such as IL‐6, IL‐10 and vascular endothelial growth factor (VEGF), which, in turn, activate STAT3 signaling and hereby a feed‐forward loop is created between activated STAT3 and cancer cells. In addition, STAT3 enhances gene expression of specific genes epigenetically, such as by DNA methylation, to maintain its own activation in tumor cells and tumor‐associated immune cells.29, 49, 50
The majority of mutations occurring in the SH2 domain of STAT3, such as Y640F and D661V, are gain‐of‐function mutations leading to increased phosphorylation at tyrosine residue Tyr705 of STAT3, which is needed for protein dimerization and activation of STAT3. Once STAT3 is activated it can evade anti‐tumor immune responses by enhancing expression of IL‐6, IL‐10 and VEGF in the tumor microenvironment and by activating transcription of oncogenes which are involved in immune suppression.29
In recent studies, G‐protein‐coupled receptors and Toll‐like receptors have also been identified as new receptor groups, which are involved in activating JAK‐STAT3 pathway. Additional studies need to be conducted to investigate these new receptors and their potential as targets for blocking this pathway.1, 51
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Shahmarvand N, Nagy A, Shahryari J, Ohgami RS. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018;109:926–933. https://doi.org/10.1111/cas.13525
Contributor Information
Nahid Shahmarvand, Email: nshahmar@stanford.edu.
Robert S. Ohgami, Email: rohgami@stanford.edu
REFERENCES
- 1. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736‐746. [DOI] [PubMed] [Google Scholar]
- 2. Yu H, Jove R. The STATs of cancer–new molecular targets come of age. Nat Rev Cancer. 2004;4:97‐105. [DOI] [PubMed] [Google Scholar]
- 3. Gotthardt D, Sexl V. STATs in NK‐cells: the good, the bad, and the ugly. Front Immunol. 2017;7:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19:2496‐2504. [DOI] [PubMed] [Google Scholar]
- 5. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F. STATs: an old story, yet mesmerizing. Cell J. 2015;17:395‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen J, Zhang Y, Petrus MN, et al. Cytokine receptor signaling is required for the survival of ALK− anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc Natl Acad Sci U S A. 2017;114:3975‐3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Teramo A, Gattazzo C, Passeri F, et al. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T‐type large granular lymphocyte leukemia. Blood. 2013;121:54, S1. [DOI] [PubMed] [Google Scholar]
- 9. Heimall J, Davis J, Shaw PA, et al. Paucity of genotype‐phenotype correlations in STAT3 mutation positive hyper IgE syndrome (HIES). Clin Immunol. 2011;139:75‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saikia B, Goel S, Suri D, Minz RW, Rawat A, Singh S. Novel mutation in SH2 domain of STAT3 (p. M660T) in hyper‐IgE syndrome with sterno‐clavicular and paravertebral abscesses. Indian J Pediatr. 2017;84:494‐495. [DOI] [PubMed] [Google Scholar]
- 11. Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early‐onset multi‐organ autoimmune disease. Nat Genet. 2014;46:812‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forbes SA, Beare D, Bindal N, et al. COSMIC: high‐resolution cancer genetics using the catalogue of somatic mutations in cancer. Curr Protoc Hum Genet. 2016;91:10. 11.1‐10.11.37. [DOI] [PubMed] [Google Scholar]
- 13. Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129:1082‐1094. [DOI] [PubMed] [Google Scholar]
- 14. Matutes E. Large granular lymphocytic leukemia. Current diagnostic and therapeutic approaches and novel treatment options. Expert Rev Hematol. 2017;10:251‐258. [DOI] [PubMed] [Google Scholar]
- 15. Qiu ZY, Fan L, Wang L, et al. STAT3 mutations are frequent in T‐cell large granular lymphocytic leukemia with pure red cell aplasia. J Hematol Oncol. 2013;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin X, Yu Y, Yan S, Wang R, Liu X, Chen C. Pure red cell aplasia and autoimmune hemolytic anemia sequentially occurring in a patient with large granular T‐lymphocytic leukemia. Intern Med. 2016;55:1491‐1496. [DOI] [PubMed] [Google Scholar]
- 17. Ishida F, Matsuda K, Sekiguchi N, et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer Sci. 2014;105:342‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fasan A, Kern W, Grossmann V, Haferlach C, Haferlach T, Schnittger S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia. 2013;27:1598‐1600. [DOI] [PubMed] [Google Scholar]
- 19. Rajala HL, Porkka K, Maciejewski JP, Loughran TP Jr, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia‐novel STAT3 and STAT5b mutations. Ann Med. 2014;46:114‐122. [DOI] [PubMed] [Google Scholar]
- 20. Rajala HL, Olson T, Clemente MJ, et al. The analysis of clonal diversity and therapy responses using STAT3 mutations as a molecular marker in large granular lymphocytic leukemia. Haematologica. 2015;100:91‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajala HL, Eldfors S, Kuusanmaki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541‐4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905‐1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andersson EI, Rajala HL, Eldfors S, et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT‐pathway and T‐cell activation. Blood Cancer J. 2013;3:e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andersson EI, Tanahashi T, Sekiguchi N, et al. High incidence of activating STAT5B mutations in CD4‐positive T‐cell large granular lymphocyte leukemia. Blood. 2016;128:2465‐2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersson E, Kuusanmaki H, Bortoluzzi S, et al. Activating somatic mutations outside the SH2‐domain of STAT3 in LGL leukemia. Leukemia. 2016;30:1204‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T‐cell large granular lymphocyte leukemia. Blood. 2012;120:3048‐3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dutta A, Yan D, Robert EH, Golam M. STAT3 mutations are not sufficient to induce large granular lymphocytic leukaemia in mice. Br J Haematol. 2016. https://doi.org/10.1111/bjh.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Munoz J, Dhillon N, Janku F, Watowich SS, Hong DS. STAT3 inhibitors: finding a home in lymphoma and leukemia. Oncologist. 2014;19:536‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2017;415:117‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu G, Witzig TE, Gupta M. A novel missense (M206K) STAT3 mutation in diffuse large B cell lymphoma deregulates STAT3 signaling. PLoS ONE. 2013;8:e67851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohgami RS, Ma L, Monabati A, Zehnder JL, Arber DA. STAT3 mutations are present in aggressive B‐cell lymphomas including a subset of diffuse large B‐cell lymphomas with CD30 expression. Haematologica. 2014;99:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ok CY, Chen J, Xu‐Monette ZY, et al. Clinical implications of phosphorylated STAT3 expression in De Novo diffuse large B‐cell lymphoma. Clin Cancer Res. 2014;20:5113‐5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu ZL, Song YQ, Shi YF, Zhu J. High nuclear expression of STAT3 is associated with unfavorable prognosis in diffuse large B‐cell lymphoma. J Hematol Oncol. 2011;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ohgami RS, Ohgami JK, Pereira IT, Gitana G, Zehnder JL, Arber DA. Refining the diagnosis of T‐cell large granular lymphocytic leukemia by combining distinct patterns of antigen expression with T‐cell clonality studies. Leukemia. 2011;25:1439‐1443. [DOI] [PubMed] [Google Scholar]
- 36. Khoury JD, Medeiros LJ, Rassidakis GZ, et al. Differential expression and clinical significance of tyrosine‐phosphorylated STAT3 in ALK+ and ALK− anaplastic large cell lymphoma. Clin Cancer Res. 2003;9:3692‐3699. [PubMed] [Google Scholar]
- 37. Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T‐cell prolymphocytic leukemia. Blood. 2014;124:1460‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lopez C, Bergmann AK, Paul U, et al. Genes encoding members of the JAK‐STAT pathway or epigenetic regulators are recurrently mutated in T‐cell prolymphocytic leukaemia. Br J Haematol. 2016;173:265‐273. [DOI] [PubMed] [Google Scholar]
- 39. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949‐954. [DOI] [PubMed] [Google Scholar]
- 40. Xie TX, Huang FJ, Aldape KD, et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188‐3196. [DOI] [PubMed] [Google Scholar]
- 41. Wong AL, Soo RA, Tan DS, et al. Phase I and biomarker study of OPB‐51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26:998‐21005. [DOI] [PubMed] [Google Scholar]
- 42. Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113:365‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang N, Ahn SH, Kong DS, Lee HW, Nam DH. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol Cell Endocrinol. 2017;451:53‐65. [DOI] [PubMed] [Google Scholar]
- 44. Sen M, Thomas SM, Kim S, et al. First‐in‐human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ogura M, Uchida T, Terui Y, et al. Phase I study of OPB‐51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106:896‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bendell JC, Hong DS, Burris HA 3rd, et al. Phase 1, open‐label, dose‐escalation, and pharmacokinetic study of STAT3 inhibitor OPB‐31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74:125‐130. [DOI] [PubMed] [Google Scholar]
- 47. Hayakawa F, Sugimoto K, Harada Y, et al. A novel STAT inhibitor, OPB‐31121, has a significant antitumor effect on leukemia with STAT‐addictive oncokinases. Blood Cancer J. 2013;3:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim MJ, Nam HJ, Kim HP, et al. OPB‐31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013;335:145‐152. [DOI] [PubMed] [Google Scholar]
- 49. Pilati C, Letouze E, Nault JC, et al. Genomic profiling of hepatocellular adenomas reveals recurrent FRK‐activating mutations and the mechanisms of malignant transformation. Cancer Cell. 2014;25:428‐441. [DOI] [PubMed] [Google Scholar]
- 50. Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK‐STAT3 signaling. JAKSTAT. 2013;2:e23828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nairismagi ML, Tan J, Lim JQ, et al. JAK‐STAT and G‐protein‐coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T‐cell lymphoma. Leukemia. 2016;30:1311‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ohgami RS, Ma L, Merker JD, Martinez B, Zehnder JL, Arber DA. STAT3 mutations are frequent in CD30+ T‐cell lymphomas and T‐cell large granular lymphocytic leukemia. Leukemia. 2013;27:2244‐2247. [DOI] [PubMed] [Google Scholar]