

Abstract
Molecularly targeted therapy has enabled outstanding advances in cancer treatment. Whereas various anti‐human epidermal growth factor receptor 2 (HER2) drugs have been developed, trastuzumab is still the only anti‐HER2 drug presently available for gastric cancer. In this study, we propose novel treatment options for patients with HER2‐positive gastric cancer. First, we determined the molecular profiles of 12 gastric cancer cell lines, and examined the antitumor effect of the pan‐HER inhibitors afatinib and neratinib in those cell lines. Additionally, we analyzed HER2 alteration in 123 primary gastric cancers resected from Japanese patients to clarify possible candidates with the potential to respond to these drugs. In the drug sensitivity analysis, both afatinib and neratinib produced an antitumor effect in most of the HER2‐amplified cell lines. However, some cells were not sensitive to the drugs. When the molecular profiles of the cells were compared based on the drug sensitivities, we found that cancer cells with lower mRNA expression levels of IGFBP7, a tumor suppressor gene that inhibits the activation of insulin‐like growth factor‐1 receptor (IGF‐1R), were less sensitive to pan‐HER inhibitors. A combination therapy consisting of pan‐HER inhibitors and an IGF‐1R inhibitor, picropodophyllin, showed a notable synergistic effect. Among 123 clinical samples, we found 19 cases of HER2 amplification and three cases of oncogenic mutations. In conclusion, afatinib and neratinib are promising therapeutic options for the treatment of HER2‐amplified gastric cancer. In addition to HER2 amplification, IGFBP7 might be a biomarker of sensitivity to these drugs, and IGF‐1R‐targeting therapy can overcome drug insensitiveness in HER2‐amplified gastric cancer.
Keywords: Afatinib, gastric cancer, HER2, IGFBP7, neratinib
Abbreviations
- CCLE
Broad‐Novartis Cancer Cell Line Encyclopedia
- CI
combination index
- EGFR
epidermal growth factor receptor
- HER
human epidermal growth factor receptor
- IGF‐1R
insulin‐like growth factor 1 receptor
- IGFBP7
insulin‐like growth factor‐binding protein 7
- IGF
insulin‐like growth factor
- IHC
immunohistochemistry
- MTS
3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium
- PARP
poly(ADP‐ribose) polymer
- p‐HER2
phoshorylated HER2
- PPP
picropodophyllin
- qPCR
quantitative real‐time PCR
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
Significant advancements have been achieved in the development of novel anti‐HER2 drugs, resulting in increased therapeutic opportunities for patients with HER2‐positive malignant tumors, especially breast cancer. However, trastuzumab remains the only anti‐HER2 drug with established clinical evidence for the treatment of HER2‐positive gastric cancer.1 Regarding HER2‐targeting small molecular drugs, phase III clinical trials have not been carried out, other than those for lapatinib, and the superiority of lapatinib against HER2‐positive gastric cancer was not established in either the Tytan trial or the LoGic trial. Moreover, little is known about HER2 mutation in gastric cancer.2, 3
Afatinib and neratinib are irreversible human EGFR TKIs that also bind to the kinase domains of HER2 and HER4. Both drugs are called pan‐HER inhibitors. Afatinib has already been approved and clinically used as a treatment option for EGFR‐mutant non‐small‐cell lung cancers, and it has also been reported to be effective against HER2‐mutant or HER2‐amplified non‐small‐cell lung cancers.4, 5 Neratinib has been approved for the treatment of breast cancer by the US FDA, and an improvement in the overall survival of post‐operative HER2‐positive breast cancer patients after trastuzumab treatment has been reported.6 Because these two drugs are promising candidates as anti‐HER2 small molecular drugs for HER2‐positive gastric cancer, we examined the antitumor effects of afatinib and neratinib against gastric cancer. We also analyzed HER2 alterations, including the mutation status, in primary gastric cancer from 123 Japanese patients who underwent a gastrectomy at our institution.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
Twelve gastric cancer cell lines (ECC10, GCIY, KATO‐III, MKN7, MKN74, NCI‐N87, NUGC3, NUGC4, OCUM‐1, SH‐10‐TC, SNU‐16, and SNU‐216) were used in this study. ECC10, GCIY, and MKN7 were provided by Riken BRC through the National Bio‐Resource Project of Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. NUGC3, MKN74, and OCUM‐1 were provided by Japanese Collection of Research Bioresources/Health Science Research Resources Bank (JCRB/HSRRB), Osaka, Japan. KATO‐III, NCI‐N87, NUGC4, SNU‐16, SH‐10‐TC were purchased from ATCC (Manassas, VA, USA), and SNU‐216 was obtained from the Korean Cell Line Bank. All the cells, other than GCIY and OCUM‐1, were cultured in RPMI‐1640 media supplemented with 10% FBS. GCIY and OCUM‐1 were cultured in minimum essential media (Sigma‐Aldrich) with 15% FBS and DMEM with 0.5 mmol/L sodium pyruvate and 10% FBS, respectively. Afatinib, neratinib, and PPP were purchased from Selleckchem and MedChem Express. Gefitinib was purchased from Sykkinase. Trastuzumab and pertuzumab were obtained from Chugai Pharmaceutical Co.
2.2. Clinical tumor samples
Primary gastric cancer tumor samples were obtained from 123 patients who underwent a gastrectomy at Okayama University Hospital (Okayama, Japan). The median patient age was 68 years (range, 36‐90 years), and all the patients were Japanese. Institutional Review Board permission and informed consent were obtained at our institution.
2.3. DNA and RNA extraction
The genomic DNA and RNA of 12 cell lines were extracted using the DNeasy Blood and Tissue Kit and the RNeasy mini Kit (Qiagen), respectively, according to the manufacturer's instructions. RNA was reversed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Genomic DNA was extracted from clinical samples using overnight digestion with SDS and proteinase K (Life Technologies) and then obtained using standard phenol‐chloroform extraction and ethanol precipitation.
2.4. Copy number, gene expression assay, and FISH
Copy number variations and the gene expression of HER2 were determined by the ΔΔCT method of qPCR (StepOnePlus real‐time PCR system; Applied Biosystems). All the samples were analyzed in triplicate. Based on the results of our previous study, we defined the copy number of control human genomic DNA (Thermo Fisher Scientific) as 2 and amplification as values >4 in cell lines and those >5 in clinical samples.7, 8, 9, 10 The expression level of HER2 in NCI‐N87, which showed the highest level of HER2 expression among the 12 cell lines, was defined as 1. A dual‐color FISH assay was carried out using the LSI HER2 SpectrumOrange/CEP17 SpectrumGreen probe (Abbott Molecular).
2.5. Western blot analysis and IHC
The detailed protocols of the total cell lysate extraction, Western blot analysis, and IHC have been described previously.11, 12 The primary antibodies were as follows: p‐HER2 (Tyr1221/1222), HER2, p‐EGFR (Tyr1068), EGFR, p‐MAPK (Erk1/2) (Thr202/Tyr204), MAPK (Erk1/2), p‐AKT (Ser473), AKT, cleaved PARP (Asp214), IGF‐I receptor β (IGF‐1R), p‐IGF‐I receptor β (phospho‐IGF‐1R) (Tyr1135/1136) (Cell Signaling Technology), and actin (Santa Cruz Biotechnology). The following secondary antibodies were used in the Western blotting: anti‐rabbit, anti‐mouse (Santa Cruz Biotechnology). To detect specific proteins, the membranes were examined using the ECL Prime Western Blotting Detection System (GE Healthcare) and LAS‐3000 (Fuji‐film). As for IHC staining, the clinical tumors were stained using anti‐HER2 primary antibody purchased from Roche Diagnostics.
2.6. Cell growth inhibition assay
The cell growth inhibition rate was determined using a modified MTS assay with CellTiter 96 AQueous bromide One Solution Reagent (Promega) or an MTT assay with Thiazolyl Blue Terazolium (Sigma‐Aldrich). In the MTS and MTT assays, cells were seeded in 96‐well tissue culture plates (2000 cells/well) and a 10‐cm dish (2.0 × 105 cells/dish), respectively, and drug dilutions were added to each well at 12 hours after seeding. Cell growth was measured at 3 days after drug treatment. The inhibitory effects against cell growth are shown as the IC50. In the MTT assay, after the drug‐containing medium was completely removed after 3 days of treatment, the cells were incubated with MTT solution (600 μL MTT and 2400 μL RPMI‐1640) for 2 hours; DMSO was then added for purple formazan solubilization. In the MTT assay, the concentrations of drugs were fixed at 500 nmol/L. For the combination treatments, the concentration of each drug was 500 nmol/L. The combination effect of the two drugs was evaluated by CI. The CI was calculated from the results of MTS assays performed for both single agent and dual medication, using Calcusyn software (Biosoft). The combination effect with CI < 1 was defined as synergistic.
2.7. Direct sequencing assay
The direct sequencing of exon 8, the transmembrane domain (exon 17), and the kinase domain (exons 18‐24) of HER2 was carried out for each of the 12 cell lines and 123 primary gastric cancer samples. The sequences of the primers were the same as previously reported.13, 14 The detailed PCR protocol has been previously reported, and all the PCR products were incubated using exonuclease I and shrimp alkaline phosphatase (Amersham Biosciences). DNA sequence alterations were evaluated using MutationTaster2.15
2.8. Xenograft mouse model
All the experimental mice were provided with food and water and were handled in accordance with the Policy on the Care and Use of Laboratory Animals, Okayama University. BALB/c‐nu/nu female mice were purchased from Charles River Laboratories. Approximately 1.0 × 106 cells of the HER2‐amplified cell lines (NCI‐N87, NUGC4, and SNU‐216) suspended in 100 μL RPMI‐1640 media and Matrigel Basement Membrane Matrix (Corning, Corning, NY, USA) were s.c. transplanted into two places on the back side of each mouse. Because the tumor growth of SNU‐216 was extremely slow, we carried out the in vivo assay using NCI‐N87 and NUGC4 only. Mice were randomly divided into three groups that received a placebo, 20 mg/kg afatinib, or 40 mg/kg neratinib. Drugs were diluted in 0.5 w/v (%) methyl cellulose (Wako Pure Chemical Industries) and were given orally five times per week for 3 weeks. The size of the tumors was measured three times a week, and tumor volume was calculated using the following formula: volume (mm3) = length (longest diameter) × width (shortest diameter)2 × 0.5. Drug treatment was generally started once the tumor volume exceeded 50 mm3.
2.9. Statistical analysis
In this study, the statistical analysis was mainly carried out using R, and numerical data were expressed as the mean ± SD. The difference between two groups was considered statistically significant at P < 0.05.
2.10. Bioinformatics analysis
We used the microarray data (Affymetrix U133+2 arrays), genetic alterations, and drug sensitivities of the cell lines shown in the CCLE. In the clinical bioinformatics analysis, we investigated the HER2 alterations listed in the TCGA, MSK‐IMPACT, and METABRIC databases, in addition to our original data.16, 17, 18 Bioinformatics data on HER2 alterations from these datasets are available through cBioportal for the Cancer Genomics website and are available to the public.19, 20
3. RESULTS
3.1. Gene alterations and activation of HER2 in gastric cancer cell lines
We examined the copy number and gene expression of HER2 in 12 gastric cancer cell lines (Figure 1A). Five of the 12 cell lines (NCI‐N87, MKN7, SNU‐216, GCIY, and NUGC4) showed the amplification of HER2, and NCI‐N87 showed a remarkably high copy number of HER2. Among these cell lines, only ECC10 showed a HER2 mutation (L755S) (Figure 1B). Regarding other HER family members, the amplifications of EGFR in MKN74 and NUGC4 and of a HER3 mutation in NUGC3 (G1273R) were reported in the CCLE database (Table 1). The activation of downstream pathway molecules of HER2 and phosphorylated HER2 were analyzed using Western blotting (Figure 1C). Three (MKN7, NCI‐N87, and SNU‐216) of the five HER2‐amplified gastric cancer cell lines showed overexpression of p‐HER2. In the HER2 mutant ECC10 cell line, p‐HER2 was not detected. Because total‐HER2 and phospho‐HER2 in GCIY and NUGC4, which had been diagnosed as HER2‐amplified from the copy number variation of HER2 examined by qPCR, were barely detectable in Western blot analyses, we analyzed these cell lines using FISH (Figure 1D). Based on the results, GCIY and NUGC4 were confirmed as HER2‐amplified cell lines.
Figure 1.
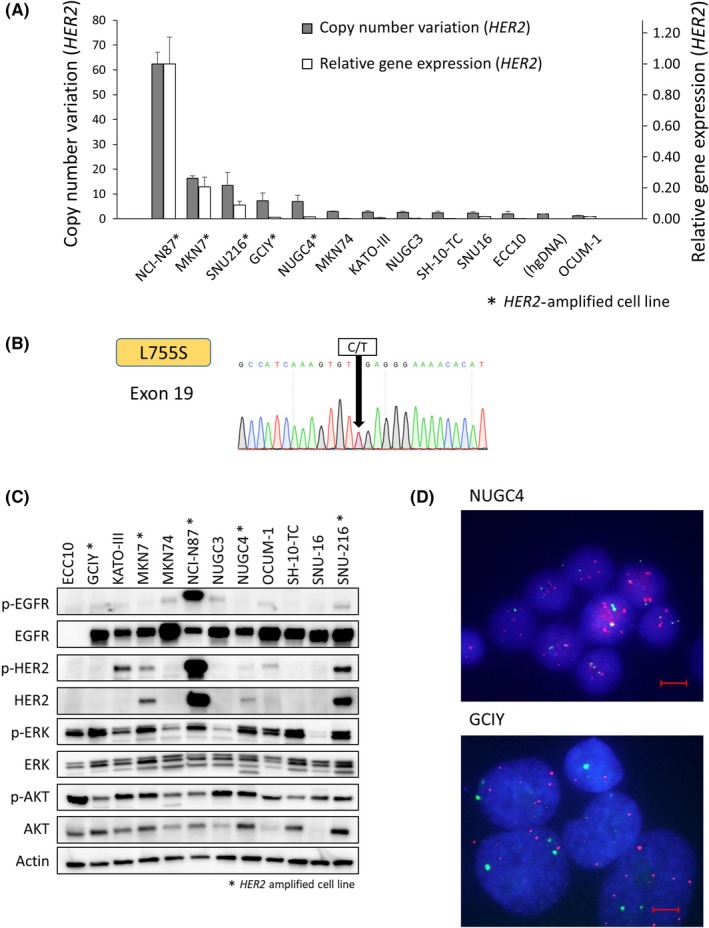
Genetic alteration of HER2 and activation of human epidermal growth factor receptor 2 (HER2) and downsignal pathway proteins in gastric cancer cell lines. A, Copy number variation and relative gene expression of HER2 were analyzed using quantitative real‐time PCR. Five cell lines (NCI‐N87, MKN7, SNU‐216, GCIY, and NUGC4) showed HER2 amplification. B, Direct Sanger sequencing of ECC10 shows a HER2 mutation, L755S, in a kinase domain. C, Activation of HER2 and downsignal pathway proteins are shown. Total‐HER2 was upregulated in the HER2‐amplified MKN7, NCI‐N87, NUGC4, and SNU‐216 cell lines. *HER2‐amplified cell line. D, FISH analysis shows that HER2 is amplified in NUGC4 and GCIY. Blue, nucleus; green, CEP17; red, HER2. p‐, phosphorylated. Scale bar, 10 μm
Table 1.
Genomic alterations of HER family members in gastric cancer cell lines
| Cell line | Sex | HER family status | Afatinib | Neratinib | Gefitinib | Pertuzumab | Trastuzumab | ||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (nmol/L) | Sensitivity | IC50 (nmol/L) | Sensitivity | IC50 (nmol/L) | IC50 (μg/mL) | IC50 (μg/mL) | |||
| NUGC4 | F |
HER2 amp (CNV, 6.95) EGFR amp |
0.7 | High | 0.8 | High | 168.2 | >103 | >103 |
| NCI‐N87 | M | HER2 amp (CNV, 62.41) | 3.1 | High | <0.16 | High | 1110.3 | >103 | >103 |
| NUGC3 | M | HER3 mut (G1273R) | 21.3 | High | 30.7 | High | 447.2 | >103 | >103 |
| SNU‐216 | F | HER2 amp (CNV, 13.40) | 29.0 | High | 47.7 | High | 6391.9 | >103 | >103 |
| GCIY | F | HER2 amp (CNV, 7.28) | 39.5 | High | 63.8 | High | 449.4 | >103 | >103 |
| MKN7 | M | HER2 amp (CNV, 16.30) | 798.4 | Moderate | 757.3 | Moderate | >104 | >103 | >103 |
| KATO‐III | M | N/A | 1398.6 | Insensitive | 320.1 | Moderate | 7640.9 | >103 | >103 |
| SNU‐16 | F | N/A | 1754.2 | Insensitive | 1055.7 | Insensitive | >104 | >103 | >103 |
| SH‐10‐TC | N/A | N/A | 2524.1 | Insensitive | 3417.1 | Insensitive | >104 | >103 | >103 |
| ECC10 | M | HER2 mut (L755S) | 4461.7 | Insensitive | 519.2 | Moderate | >104 | >103 | >103 |
| MKN74 | M | N/A | 6758.1 | Insensitive | >104 | Insensitive | >104 | >103 | >103 |
| OCUM‐1 | F | N/A | >104 | Insensitive | 5000 | Insensitive | >104 | >103 | >103 |
3.2. Effects of afatinib and neratinib in HER2‐amplified cell lines
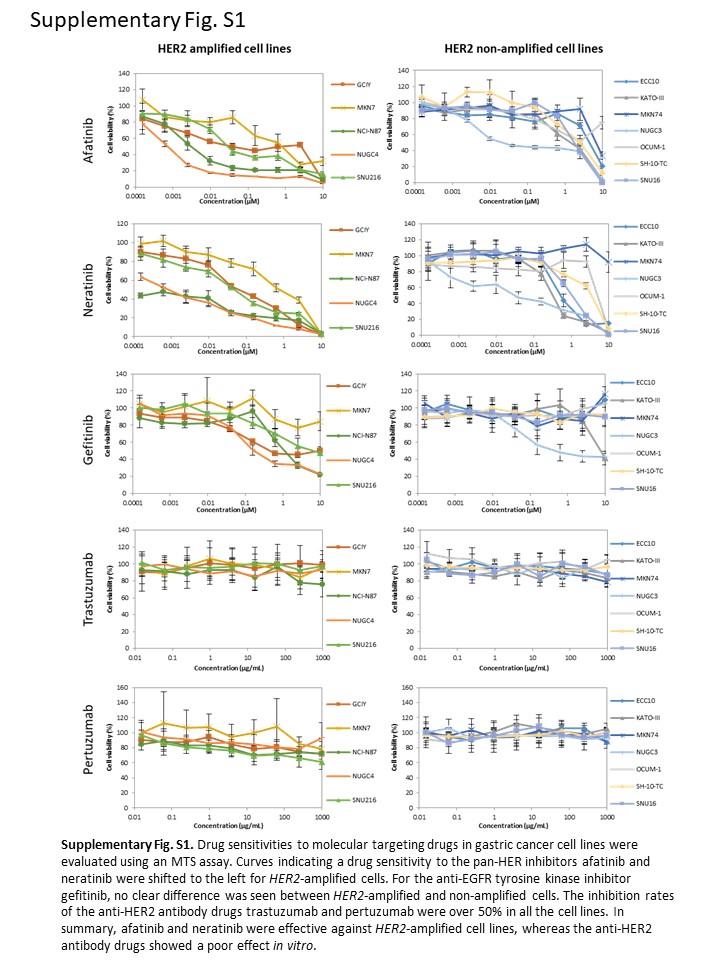
We examined the effects of pan‐HER inhibitors (afatinib and neratinib), anti‐HER2 antibody (trastuzumab and pertuzumab), and EGFR inhibitor (gefitinib) in 12 gastric cancer cell lines (Table 1, Figure S1). We classified drug sensitivity into three groups based on the IC50 values determined using an MTS assay. The IC50 values <100 nmol/L, 100‐1000 nmol/L, and >1000 nmol/L were defined as highly sensitive, moderately sensitive, and insensitive to TKI, respectively. Four (GCIY, NCI‐N87, NUGC4, and SNU‐216) of the five HER2‐amplified cell lines and NUGC3 were highly sensitive to both pan‐HER inhibitors. The IC50 value of afatinib and neratinib in these cells ranged from 0.7 to 39.5 nmol/L and from <0.15 to 63.8 nmol/L, respectively. NUGC4, the most afatinib‐sensitive cell line, also showed EGFR amplification. Interestingly, the HER2‐amplified MKN7 cell line was not highly sensitive to either of the pan‐HER inhibitors, compared with other HER2‐amplified cell lines; nevertheless, the activation of HER2 was revealed using Western blotting. In addition, MKN7 cells were also insensitive to lapatinib, which inhibits both EGFR and HER2 according to the CCLE database (IC50 >8 μM). In most cell lines, the sensitivity of afatinib and neratinib showed the same tendency, except in the ECC10 and KATO‐III cell lines, which were insensitive to afatinib and moderately sensitive to neratinib. All 12 cell lines were insensitive to trastuzumab and pertuzumab at a concentration of 1000 μg/mL.
Next, we evaluated the effect of afatinib and neratinib on the HER2 pathway in TKI highly sensitive HER2‐amplified cell lines (NCI‐N87, NUGC4, and SNU‐216), a moderately sensitive HER2‐amplified cell line (MKN7), and an insensitive cell line (MKN74). To analyze HER2 and downstream pathway proteins, cell lysate was collected after 12 hours of treatment. The phosphorylation of HER2 was inhibited by both afatinib and neratinib in all four HER2‐amplified cell lines, including MKN7 (Figure 2). In addition to p‐HER2, total HER2 was also downregulated by the pan‐HER inhibitors in all four HER2‐amplified cell lines. Regarding downstream pathway molecules, the phosphorylation of ERK and AKT was inhibited in all four HER2‐amplified cell lines. By contrast, p‐ERK and p‐AKT were not downregulated in an insensitive cell line (MKN74). Phosphorylation of EGFR was suppressed in all five cell lines.
Figure 2.
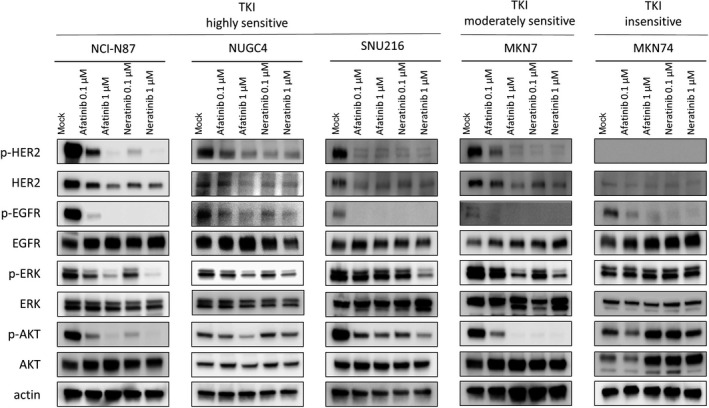
Influence of afatinib or neratinib on human epidermal growth factor receptor 2 (HER2) and the downsignal pathway in gastric cancer cell lines. Both afatinib and neratinib downregulate the phosphorylation (p‐) of HER2 and epidermal growth factor receptor (EGFR) in tyrosine kinase inhibitor (TKI) highly and moderately sensitive cells. In addition to p‐HER2, total‐HER2 was also downregulated by pan‐HER inhibitors in HER2‐amplified cells. In NCI‐N87, NUGC4, SNU‐216, and MKN7 cells, p‐ERK and p‐AKT were downregulated in a dose‐dependent manner
3.3. Antitumor effect of afatinib or neratinib on a xenograft mouse model
Based on the in vitro experiment results, we undertook an in vivo experiment to test the effect of afatinib or neratinib against HER2‐amplified gastric cancer cell lines (Figure 3A,B). HER2‐amplified NCI‐N87 and NUGC4 cells were used in this experiment, and the doses of both drugs were decided based on the results of previous studies.4, 21 Both afatinib and neratinib showed significant antitumor effects against NCI‐N87 and NUGC4. In particular, both drugs showed remarkable effects against NCI‐N87 (P < 0.0001).
Figure 3.
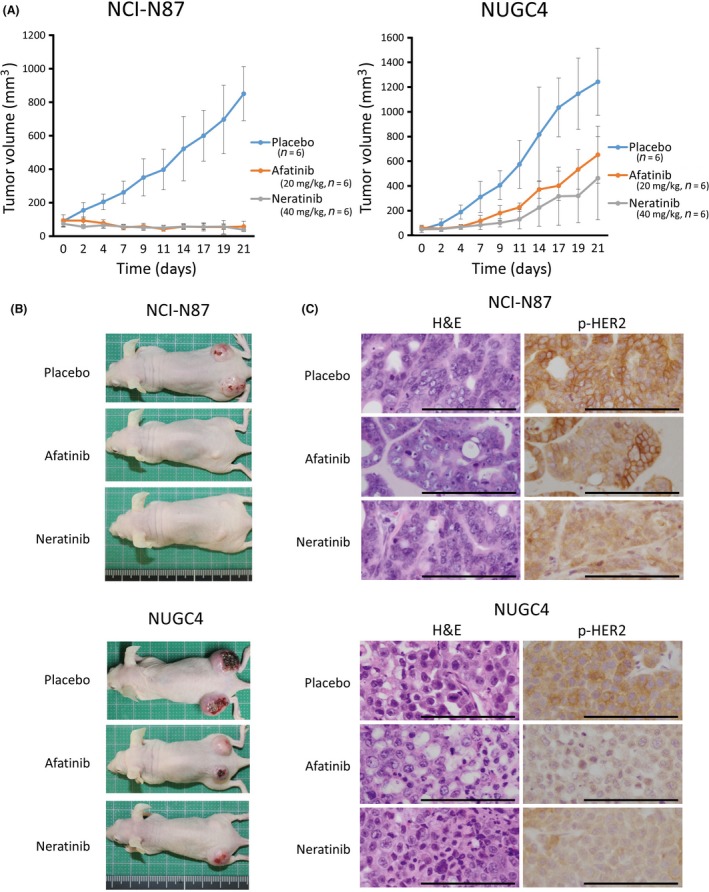
Both afatinib and neratinib exerted an antitumor effect against HER2‐amplified gastric cancer cells in vivo. A, In HER2‐amplified NCI‐N87 and NUGC4 xenografts, afatinib and neratinib inhibited cell growth in a nude mouse xenograft model. In particular, both drugs showed remarkable effects in NCI‐N87 cells (P < 0.0001). The effects of afatinib and neratinib did not differ significantly in either cell line (NCI‐N87, P = 0.6095; NUGC4, P = 0.2780). B, Photographs of the xenograft model taken on day 21. C, Representative images of immunohistochemical staining in tumors resected from xenograft model mice. In the pan‐human epidermal growth factor receptor (HER) inhibitor‐treated mice, the expression level of phosphorylated (p‐)HER2 in tumors were not completely suppressed (positive weak p‐HER2 expression), compared with the level in placebo‐treated mice. Scale bar, 100 mm
The expression level of p‐HER2 in tumors obtained from mice xenografts after treatment was examined using IHC. In both NCI‐N87 and NUGC4 cells treated with afatinib and neratinib, the expression of p‐HER2 was suppressed, compared with the expression level in tumors treated with the placebo (Figure 3C). However, p‐HER2 in mice xenografts was not completely suppressed, despite strong antitumor effects with pan‐HER inhibitors. The reason for this phenomenon may be that the cells maintaining weak p‐HER2 expression even with pan‐HER inhibitor treatment could barely survive.
3.4. IGFBP7 is a possible novel biomarker of afatinib or neratinib sensitivity in HER2‐amplified gastric cancer
As HER2‐amplified gastric cancer did not always show a high sensitivity to afatinib or neratinib, we further searched for a novel gene associated with afatinib or neratinib sensitivity. We divided the 12 cell lines into two groups: a TKI highly sensitive group (GCIY, NCI‐N87, NUGC3, NUGC4, and SNU‐216), in which the IC50 values of afatinib and neratinib were both <100 nmol/L; and a TKI moderately sensitive or insensitive group (ECC10, KATO‐III, MKN7, MKN74, OCUM‐1, and SH‐10‐TC), in which the IC50 value of at least one drug was >100 nmol/L. Then we compared microarray data obtained from the CCLE database between the two groups. The ratios of the mean expression values (approximately 54 000 mRNA) in the TKI highly sensitive group to the values in the moderately sensitive or insensitive group were then calculated. Thirty probes with the highest ratios and 30 probes with the lowest ratios were selected as candidate gene markers for sensitivity to pan‐HER inhibitors (Tables S1 and S2). Among these probes, 201163_s_at corresponded to IGFBP7, and we focused on IGFBP7 and IGF‐1R to determine their abilities to predict and overcome resistance to pan‐HER inhibitors in HER2‐amplified gastric cancer.
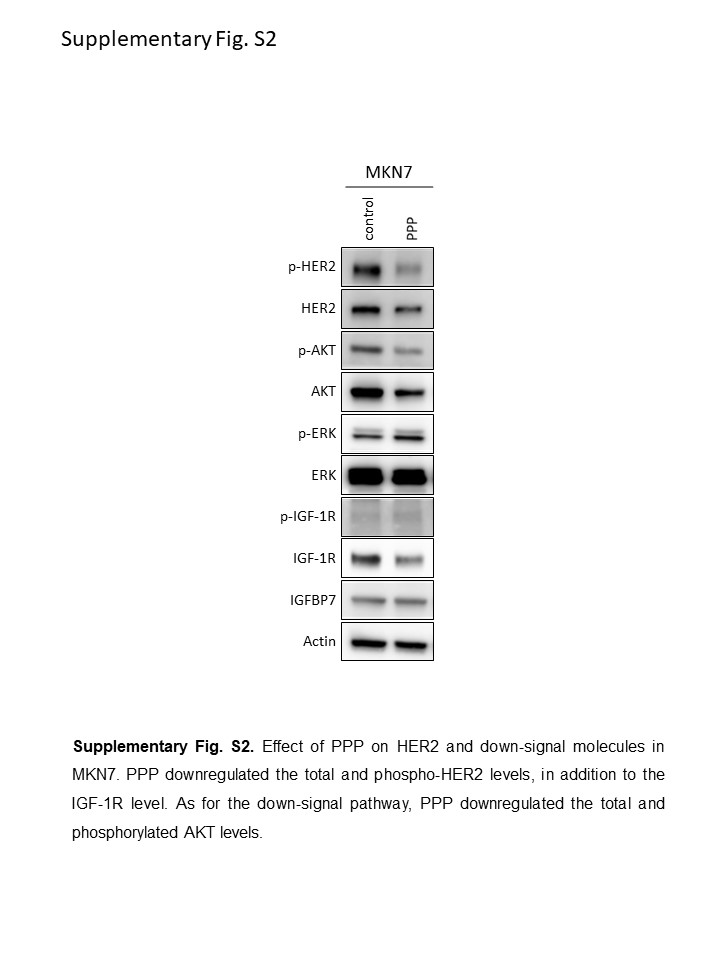
Western blot analysis was used to determine the expression levels of IGFBP7 and IGF‐1R in the TKI highly sensitive group and the moderately sensitive or insensitive group using the NCI‐N87, NUGC4, and SNU‐216 and the MKN7 and MKN74 cell lines, respectively (Figure 4A). The results showed that IGFBP7 was upregulated in the TKI highly sensitive group only, and the expression of total IGF‐1R itself was less strongly correlated with TKI sensitivity. Subsequently, to determine whether IGF‐1R had any impact on sensitivity to afatinib or neratinib, we undertook an MTT assay using the HER2‐amplified MKN7 cell line, which had high levels of HER2 expression in both a gene expression assay and Western blot analysis (Figure 4B). To downregulate the activation of IGF‐1R, we used PPP, an IGF‐1R‐targeting small molecule drug. Picropodophyllin inhibits phosphorylation of IGF‐1R without interfering with insulin receptor activity and is reported to decrease IGF‐1‐stimulated phosphorylation of AKT.22, 23 The cell proliferation ratios of the cells treated with afatinib, neratinib, or PPP compared with a control were 62.3%, 45.9%, and 54.1%, respectively. In contrast, the cell proliferation ratios when combinations of PPP plus afatinib or PPP plus neratinib were used were 26.9% and 12.9%, respectively, which were significantly lower than the single‐use results (P < 0.0001). To reveal the synergistic effect of pan‐HER inhibitors and PPP, we also undertook MTS assays and calculated the CI (Figure 4C). The effect of afatinib, neratinib, or PPP alone were compared to the effect of combination therapy (afatinib and PPP, or neratinib and PPP). The combination therapy was effective in MKN7 cells, in which the expression of IGFBP7 was downregulated, and the effect was proved to be synergistic by the CI. In a Western blot analysis, PPP downregulated IGF‐1R as well as the total and phospho‐HER2 levels in the MKN7 cell line (Figure S2).
Figure 4.
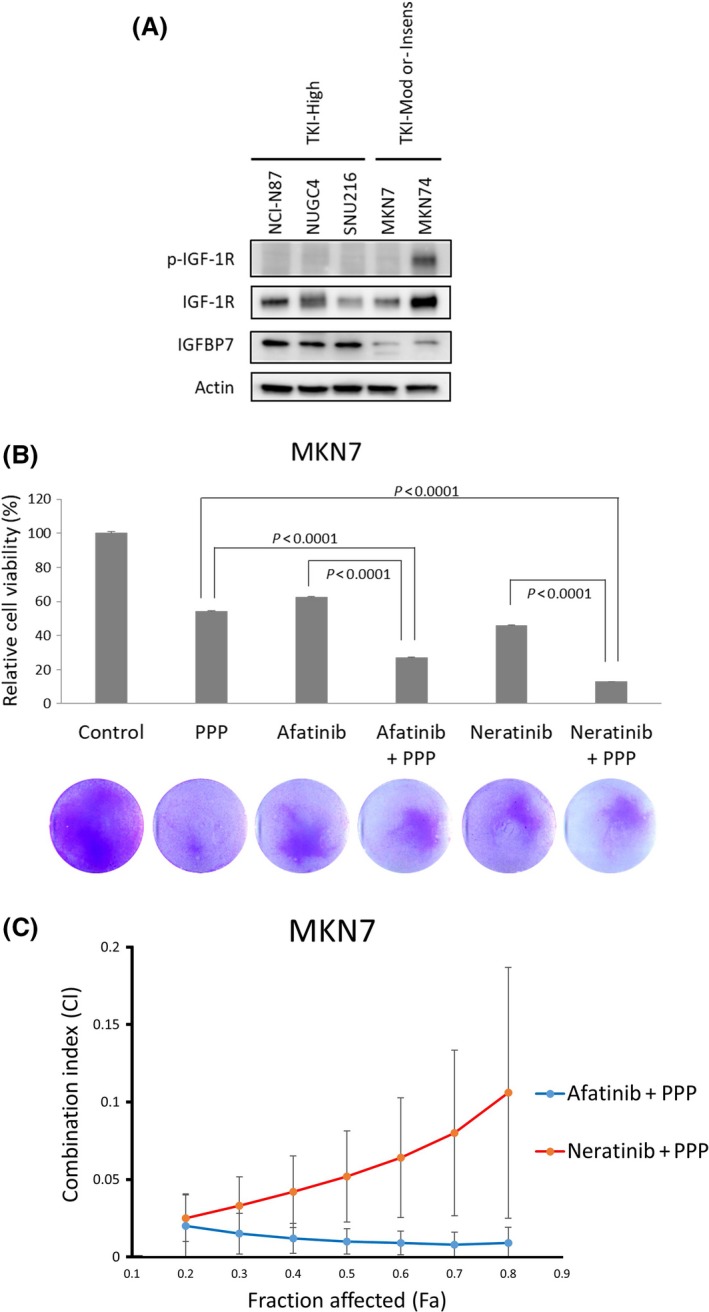
Relation between the effect of pan‐human epidermal growth factor receptor (HER) inhibitors and insulin‐like growth factor‐binding protein 7 (IGFBP7) and insulin‐like growth factor 1 receptor (IGF‐1R) in gastric cancer cell lines. A, IGFBP7 was upregulated in tyrosine kinase inhibitor (TKI) highly sensitive (TKI‐High) cells. Phosphorylated (p‐)IGF‐1R was upregulated in MKN74 cells, which were insensitive to both afatinib and neratinib. TKI‐ Insens, TKI insensitive; TKI‐Mod, TKI moderately sensitive. B, In an MTT assay, a combined treatment co‐targeting HER2 and IGF‐1R produced a synergistic effect in MKN7 cells. In this assay, the concentrations of afatinib, neratinib, and picropodophyllin (PPP) were all 500 nmol/L, and the cells were exposed to the drugs for 3 days. MKN7 cells, cultured under the same conditions as in the MTT assay, were stained using crystal violet (photographs). C, Combination index (CI) and fraction affected (Fa) were calculated from the results of 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assay. The calculated CI was <1 at all concentrations of combinations of afatinib and PPP, and neratinib and PPP), and the effects were shown to be synergistic
3.5. HER2 alteration in primary gastric cancers
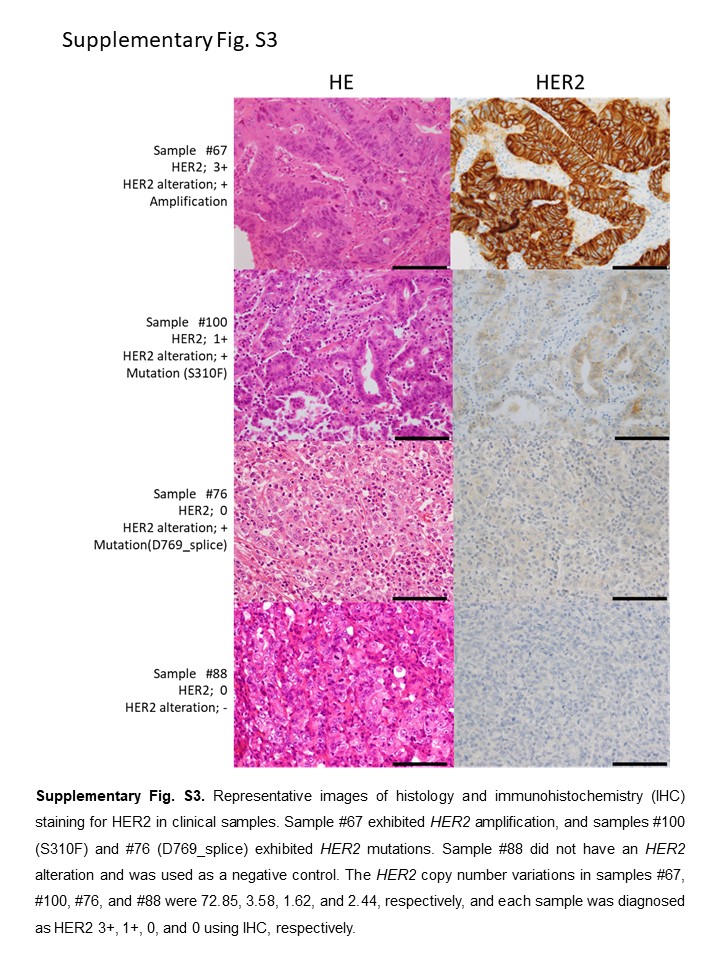
To clarify possible candidates likely to benefit from pan‐HER inhibitors, we analyzed 123 DNA samples obtained from primary gastric cancer patients. All the patients were Japanese, and 82 (66.7%) were men. The pathological stage and histology of the specimens are shown in Figure 5A,B. We analyzed the HER2 copy number variation and mutations in exons 8 and 17‐24. The HER2 copy number ranged from 1.27 to 192.04, and HER2 was amplified in 19 cases (15.4%) (Figure 5C). HER2 amplification was detected in both early and advanced cancer and in both diffuse and intestinal type (Table S3). Three mutations, S310F, D769_splice, and T862A, were detected in this study (Figure 5D); all of these mutations were evaluated as “disease causing” by MutationTaster2. Duplications of the HER2 mutation were not detected in this study, and HER2 amplification and mutation were mutually exclusive. S310F and T862A have already been reported as oncogenic mutations,24 and D769_splice was detected as a novel mutation in HER2. Approximately half of the HER2‐amplified tumors were HER2‐positive when examined using IHC. The tumor with the S310F mutation was HER2‐negative (1+) when examined using IHC (Figure S3), although S310F is known to be an oncogenic change.
Figure 5.
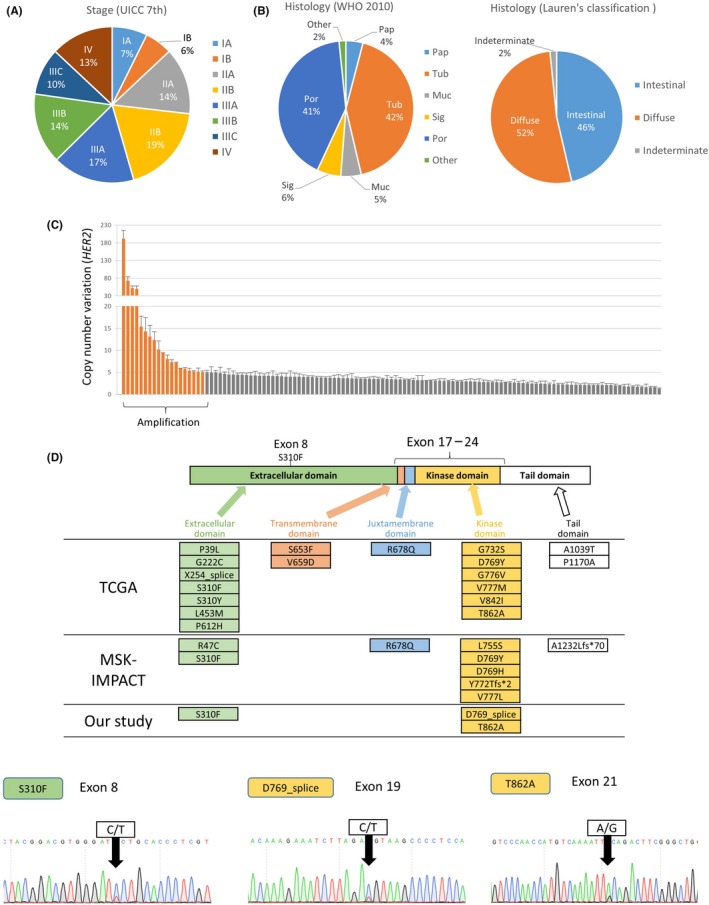
Analysis of HER2 alterations in clinical samples of gastric cancer. A, Samples were collected from all cancer stages. B, Distribution of histological classifications. Muc, mucinous adenocarcinoma; Pap, papillary adenocarcinoma; Por, poorly differentiated adenocarcinoma; Sig, signet‐ring cell carcinoma; Tub, tubular adenocarcinoma. C, Copy number variation in HER2 as analyzed using quantitative real‐time PCR. D, The HER2 structure is composed of an extracellular domain, transmembrane domain, juxtamembrane domain, kinase domain, and tail. We undertook Sanger direct sequencing for exon 8 and exons 17‐24 and compared the results with the TCGA and MSK‐IMPACT databases.16, 17 Chromatograms showing the three mutation sites that we identified are shown below
4. DISCUSSION
The molecularly targeted treatment of gastric cancer has lagged behind that of lung cancer and breast cancer, considering the dramatic developments in genome analysis techniques and personalized medicine. Until now, the effect of pan‐HER inhibitors, afatinib and neratinib, has been reported in some articles, using a limited number of cell lines.25 In the present study, we determined the molecular profiles of 12 gastric cancer cell lines and showed the potent antitumor effects of afatinib and neratinib against HER2‐amplified gastric cancer cells in vitro and in vivo. Because the mechanism of pan‐HER inhibitors is completely different from that of anti‐HER2 antibody drugs, we believe that afatinib and neratinib could be promising therapeutic options against HER2‐positive gastric cancer.
Although the amplification of HER2 can be used as a biomarker of drug sensitivity to pan‐HER inhibitors, a few exceptions have been noted. For instance, MKN7, which has a high copy number and high gene expression of HER2, did not have a high sensitivity to either drug, and NUGC3, which shows neither amplification nor a mutation of EGFR or HER2, showed a high sensitivity. To find novel biomarkers, we carried out a comprehensive gene expression analysis of gastric cancer cell lines in silico. Among the candidates for a novel biomarker gene, we focused on IGFBP7, as this gene reportedly inhibits the activation of IGF‐1R, which is known to mediate anti‐apoptotic signals and cell proliferation, by binding to unoccupied IGF‐1R and suppressing downstream signaling.26, 27 Actually, IGFBP7 reportedly plays a role as a tumor suppressor gene in breast cancer, colorectal cancer, and hepatocellular carcinoma.28, 29, 30 The association of HER family protein and IGF‐1R has also been well‐documented,31, 32 and low expression of IGFBP7 is correlated with poor prognosis in high‐grade serous ovarian carcinoma.33 The activation of IGF‐1R might influence the sensitivity of pan‐HER inhibitors indirectly, as IGF‐1R reportedly forms a heterodimer with EGFR.32 The effect of co‐targeting HER2 and IGF‐1R therapy has already been reported in HER2 non‐overexpressing breast cancer.34 Based on these facts, we decided to undertake experiments to overcome the resistance against afatinib or neratinib in HER2‐amplified, but not highly sensitive, cells using a combination therapy consisting of a pan‐HER inhibitor and an IGF‐1R inhibitor. As a result, the combination of afatinib or neratinib with PPP produced a sufficient effect. Although the detailed mechanisms remain unclear, PPP downregulated total and phosphorylated HER2 in addition to IGF‐1R, suggesting that PPP might potentiate the anti‐HER2 effect of pan‐HER inhibitors.
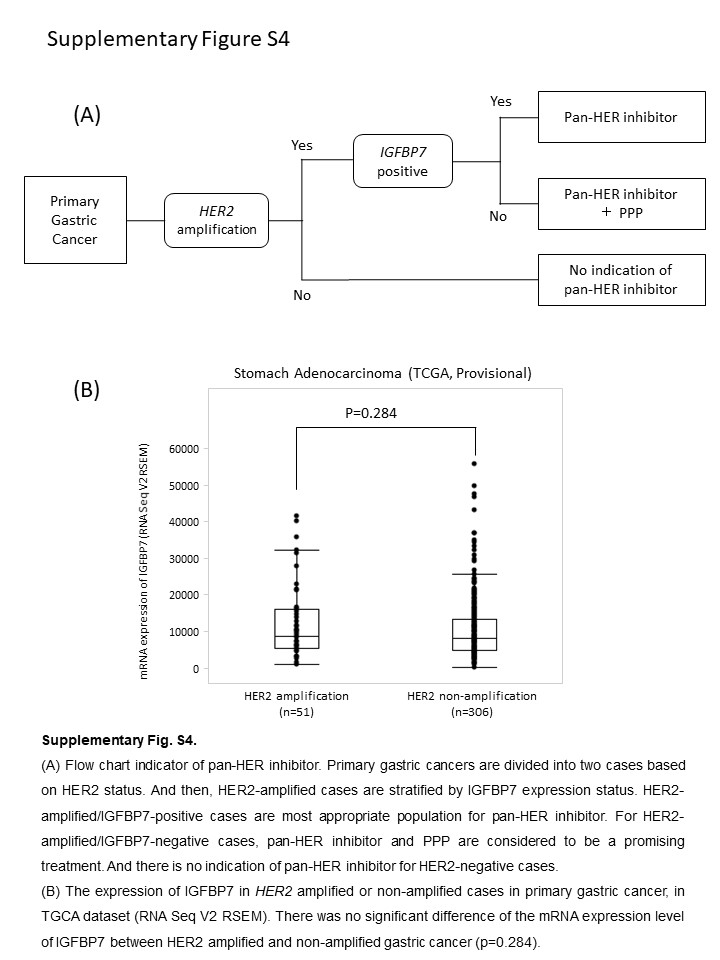
From these findings, we propose a novel indication criteria of pan‐HER inhibitors against primary gastric cancer, as shown in Figure S4A. Primary gastric cancers are divided into HER2‐amplified or non‐amplified cases. HER2‐amplified cases are stratified by IGFBP7 expression status. HER2‐amplified/IGFBP7‐positive cases are the most appropriate cohort for treatment with pan‐HER inhibitors. For HER2‐amplified/IGFBP7‐negative cases, pan‐HER inhibitor plus PPP is considered to be a promising treatment. There is no indication for pan‐HER inhibitors among the HER2‐non‐amplified group. Because there was no significant difference between mRNA expression level of IGFBP7 in HER2‐amplified gastric cancer and that of HER2‐non‐amplified cases (Figure S4B), IGFBP7 would be an additional factor that determines the effect of pan‐HER inhibitors on HER2‐amplified gastric cancer.
The in vitro effects of trastuzumab and pertuzumab, which is another anti‐HER2 antibody drug, were poor in the present study, and their effects in clinical medicine might mainly be the result of antibody‐dependent cell‐mediated cytotoxicity.35
Heterogeneity in gastric cancer is well‐documented and is one of the most important causes of insufficient efficacy of molecularly targeted therapy in gastric cancer patients.36, 37, 38 To overcome the resistance caused by heterogeneity, we tried to narrow the gastric cancer patient candidates who would receive benefit from treatment with pan‐HER inhibitors. In this study, we defined HER2 amplification by qPCR and FISH analysis, and most of the HER2‐amplified cell lines showed high sensitivity to pan‐HER inhibitors, including GCIY and NUGC4, in which total‐HER2 was barely detected by Western blot analysis. However, HER2 is mainly evaluated by IHC, which reflects the protein level of HER2 in tumor samples, in clinical medicine. From the result of the present study, we recognize that HER2 amplification as a more appropriate biomarker for pan‐HER inhibitors than the protein level of HER2.
Although gastric cancer is still a leading cause of cancer deaths in East Asia,39 the frequency and types of HER2 alterations in gastric cancer, especially mutations, remain unknown. In the present study, we analyzed 123 DNA samples extracted from primary gastric cancers in Japanese patients and compared the results with the TCGA and MSK‐IMPACT datasets (Table S4).40 In terms of HER2 alterations other than amplification, S310F (exon 8) is the most frequent oncogenic mutation of HER2 in gastric cancer, according to the TCGA dataset, and some mutations in the transmembrane domain or kinase domain have also been reported as oncogenic HER2 alterations.14 Thus, we undertook the direct sequencing of exons 8 and 17‐24, which contain the transmembrane domain and the kinase domain of HER2. Only information for esophagogastric cancer was obtained from the MKS‐IMPACT dataset and compared with our data, and a certain number of HER2 alterations in esophageal cancer were also included. The frequencies of HER2 amplification in the TCGA and MSK‐IMPACT databases and in our study were 14.0%, 22.6%, and 15.4%, respectively, and the frequencies of mutation were 4.8%, 3.8%, and 2.4%, respectively. Regarding HER2 mutations, we cannot conclude whether the frequency of gastric cancer in our series was higher or lower than those in the TCGA or MSK‐IMPACT datasets because we only analyzed nine of the 27 exons of HER2. Among the three mutations identified in this study, D769_splice might have a limited significance because HER2 was hardly detected using IHC, although this mutation was diagnosed as an oncogenic change by MutationTaster2.
Even though the relation between oncogenic HER2 mutations and drug sensitivity to afatinib or neratinib in gastric cancer needs further investigation, the antitumor effects of these drugs against HER2‐mutated lung cancer and breast cancer have already been reported.4, 24 The amplification and mutation of HER2 were mutually exclusive, not only in our study examining gastric cancer, but also in studies examining lung or breast cancer according to the TCGA, MSK‐IMPACT, and METABRIC databases.18 Considering the exclusiveness of HER2 alterations, patients suffering from HER2‐mutated gastric cancer might not be currently receiving anti‐HER2 drugs, and pan‐HER inhibitors could contribute to the treatment of these patients.
In conclusion, pan‐HER inhibitors exert a strong antitumor effect against HER2‐amplified gastric cancer cells and could be a promising treatment option for HER2‐positive gastric cancer patients. Our findings also suggest that IGFBP7 expression might be a useful biomarker for the selection of patients who are likely to be sensitive to pan‐HER inhibitors.
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This study was funded by Boehringer Ingelheim.
Yoshioka T, Shien K, Namba K, et al. Antitumor activity of pan‐HER inhibitors in HER2‐positive gastric cancer. Cancer Sci. 2018;109:1166–1176. https://doi.org/10.1111/cas.13546
Funding information Boehringer Ingelheim
REFERENCES
- 1. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet. 2010;376:687‐697. [DOI] [PubMed] [Google Scholar]
- 2. Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second‐line treatment of HER2‐amplified advanced gastric cancer in Asian populations: TyTAN‐a randomized, phase III study. J Clin Oncol. 2014;32:2039‐2049. [DOI] [PubMed] [Google Scholar]
- 3. Hecht JR, Bang YJ, Qin SK, et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2‐positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO‐013/LOGiC‐A randomized phase III trial. J Clin Oncol. 2016;34:443‐451. [DOI] [PubMed] [Google Scholar]
- 4. Suzawa K, Toyooka S, Sakaguchi M, et al. Antitumor effect of afatinib, as a human epidermal growth factor receptor 2‐targeted therapy, in lung cancers harboring HER2 oncogene alterations. Cancer Sci. 2016;107:45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomizawa K, Suda K, Onozato R, et al. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer. 2011;74:139‐144. [DOI] [PubMed] [Google Scholar]
- 6. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab‐based adjuvant therapy in patients with HER2‐positive breast cancer (ExteNET): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2016;17:367‐377. [DOI] [PubMed] [Google Scholar]
- 7. Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell‐like properties in cancer cells. Can Res. 2013;73:3051‐3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE. 2009;4:e7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kubo T, Yamamoto H, Lockwood WW, et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Can Res. 2008;68:6913‐6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shien K, Ueno T, Tsukuda K, et al. Knockdown of the epidermal growth factor receptor gene to investigate its therapeutic potential for the treatment of non‐small‐cell lung cancers. Clin Lung Cancer. 2012;13:488‐493. [DOI] [PubMed] [Google Scholar]
- 12. Shien K, Toyooka S, Ichimura K, et al. Prognostic impact of cancer stem cell‐related markers in non‐small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer. 2012;77:162‐167. [DOI] [PubMed] [Google Scholar]
- 13. Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Can Res. 2005;65:1642‐1646. [DOI] [PubMed] [Google Scholar]
- 14. Yamamoto H, Higasa K, Sakaguchi M, et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst. 2014;106:djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361‐362. [DOI] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI‐272, an orally active, irreversible inhibitor of the HER‐2 tyrosine kinase. Can Res. 2004;64:3958‐3965. [DOI] [PubMed] [Google Scholar]
- 22. Girnita A, Girnita L, del Prete F, Bartolazzi A, Larsson O, Axelson M. Cyclolignans as inhibitors of the insulin‐like growth factor‐1 receptor and malignant cell growth. Can Res. 2004;64:236‐242. [DOI] [PubMed] [Google Scholar]
- 23. Vasilcanu D, Girnita A, Girnita L, Vasilcanu R, Axelson M, Larsson O. The cyclolignan PPP induces activation loop‐specific inhibition of tyrosine phosphorylation of the insulin‐like growth factor‐1 receptor. Link to the phosphatidyl inositol‐3 kinase/Akt apoptotic pathway. Oncogene. 2004;23:7854‐7862. [DOI] [PubMed] [Google Scholar]
- 24. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leto SM, Sassi F, Catalano I, et al. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2‐amplified gastrointestinal carcinomas. Clin Cancer Res. 2015;21:5519‐5531. [DOI] [PubMed] [Google Scholar]
- 26. Dale OT, Aleksic T, Shah KA, et al. IGF‐1R expression is associated with HPV‐negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis. 2015;36:648‐655. [DOI] [PubMed] [Google Scholar]
- 27. Evdokimova V, Tognon CE, Benatar T, et al. IGFBP7 binds to the IGF‐1 receptor and blocks its activation by insulin‐like growth factors. Sci Signal. 2012;5:ra92. [DOI] [PubMed] [Google Scholar]
- 28. Landberg G, Ostlund H, Nielsen NH, et al. Downregulation of the potential suppressor gene IGFBP‐rP1 in human breast cancer is associated with inactivation of the retinoblastoma protein, cyclin E overexpression and increased proliferation in estrogen receptor negative tumors. Oncogene. 2001;20:3497‐3505. [DOI] [PubMed] [Google Scholar]
- 29. Ruan WJ, Lin J, Xu EP, et al. IGFBP7 plays a potential tumor suppressor role against colorectal carcinogenesis with its expression associated with DNA hypomethylation of exon 1. J Zhejiang Univ Sci B. 2006;7:929‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomimaru Y, Eguchi H, Wada H, et al. IGFBP7 downregulation is associated with tumor progression and clinical outcome in hepatocellular carcinoma. Int J Cancer. 2012;130:319‐327. [DOI] [PubMed] [Google Scholar]
- 31. Coppola D, Ferber A, Miura M, et al. A functional insulin‐like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol Cell Biol. 1994;14:4588‐4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iyer G, Price J, Bourgeois S, Armstrong E, Huang S, Harari PM. Insulin growth factor 1 like receptor (IGF‐1R). BMC Cancer. 2016;16:773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gambaro K, Quinn MC, Caceres‐Gorriti KY, et al. Low levels of IGFBP7 expression in high‐grade serous ovarian carcinoma is associated with patient outcome. BMC Cancer. 2015;15:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakraborty AK, Liang K, DiGiovanna MP. Co‐targeting insulin‐like growth factor I receptor and HER2: dramatic effects of HER2 inhibitors on nonoverexpressing breast cancer. Can Res. 2008;68:1538‐1545. [DOI] [PubMed] [Google Scholar]
- 35. Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol. 1999;26:60‐70. [PubMed] [Google Scholar]
- 36. Wong SS, Kim KM, Ting JC, et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole‐genome sequencing. Nat Commun. 2014;5:5477. [DOI] [PubMed] [Google Scholar]
- 37. Ooi WF, Xing M, Xu C, et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super‐enhancer heterogeneity. Nat Commun. 2016;7:12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao J, Wang H, Zang W, et al. Circulating tumor DNA functions as an alternative for tissue to overcome tumor heterogeneity in advanced gastric cancer. Cancer Sci. 2017;108:1881‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 40. Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering‐integrated mutation profiling of actionable cancer targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials