

Abstract
Reticulocalbin 1 (RCN1), an endoplasmic reticulum (ER)‐resident Ca2+‐binding protein, is dysregulated in cancers, but its pathophysiological roles are largely unclear. Here, we demonstrate that RCN1 is overexpressed in clinical prostate cancer (PCa) samples, associated with cyclin B, not cyclin D1 expression, compared to that of benign tissues in a Chinese Han population. Downregulation of endogenous RCN1 significantly suppresses PCa cell viability and arrests the cell cycles of DU145 and LNCaP cells at the S and G2/M phases, respectively. RCN1 depletion causes ER stress, which is evidenced by induction of GRP78, activation of PERK and phosphorylation of eIF2α in PCa cells. Remarkably, RCN1 loss triggers DU145 cell apoptosis in a caspase‐dependent manner but mainly causes necroptosis in LNCaP cells. An animal‐based analysis confirms that RCN1 depletion suppresses cell proliferation and promotes cell death. Further investigations reveal that RCN1 depletion leads to elevation of phosphatase and tensin homolog (PTEN) and inactivation of AKT in DU145 cells. Silencing of PTEN partially restores apoptotic cells upon RCN1 loss. In LNCaP cells, predominant activation of CaMKII is important for necroptosis in response to RCN1 depletion. Thus, RCN1 may promote cell survival and serve as a useful target for cancer therapy.
Keywords: apoptosis, endoplasmic reticulum, necroptosis, prostate cancer, reticulocalbin 1
1. INTRODUCTION
The endoplasmic reticulum (ER) is important for controlling the quality of a protein and maintaining calcium homeostasis. The chaperones and enzymes in the ER lumen are critical for preventing protein aggregation and ensuring normal ER function.1, 2 Disturbances in ER homeostasis result in ER stress. This leads to activation of the unfolded protein response (UPR), which exerts protective effects and promotes cell survival during the early stages of stress or induces cell death when ER function is not restored under prolonged or severe ER stress. Therefore, multiple investigations have attempted to uncover the roles and regulatory mechanisms of these ER‐resident proteins/enzymes.3, 4 For example, anterior gradient‐2 (AGR2) is an ER‐resident protein that belongs to the protein disulphide isomerase (PDI) family and has emerged as a dominant effector in the development of pathological conditions, including cancer, drug resistance and inflammatory bowel disease.3 Calumenin, a member of the CREC family that contains EF‐hand motifs that specifically coordinate the Ca2+ ion, interacts with Sec63p, which is an ER protein translocase that guides the transport of newly synthesized proteins from the cytosol to the ER lumen. In addition, extracellular calumenin binds to and stabilizes fibulin‐1, leading to inactivation of extracellular signal‐regulated kinase 1 and 2 (ERK1/2)‐mediated signaling and suppression of cell migration and tumor metastasis.5, 6 Therefore, ER‐associated proteins are not confined to the ER lumen or to the secretory pathway, and their physiological functions and roles in diseases continue to be investigated.
Reticulocalbin 1 (RCN1) is also a member of the CREC family. RCN1 localizes to the ER and contains 6 EF‐hand motifs and an ER‐retention motif, HDEL, in the carboxyl‐terminal sequence. RCN1 is a Ca2+‐binding protein that is involved in the regulation of Ca2+‐dependent activities in the ER lumen and is present throughout the whole secretory pathway of mammalian cells.4, 7 Multiple studies have shown that RCN1 is heterogeneously expressed and necessary for cell bioactivity.8 The differentially expressed RCN1 is correlated with diseases such as cancer. RCN1 overexpression has been observed in invasive breast cancer cells,9 colorectal cancer cells,10 thermoresistant gastric cancer cells,11, 12 liver cancer cells13 and prostate cancer (PCa) cells,14 suggesting a role for RCN1 in tumorigenesis and invasion.15, 16 However, the relevance of RCN1 protein levels in tumor malignancy is unclear. RCN1 is reportedly present in highly invasive breast cancer cell lines but not in poorly invasive lines.9 However, RCN1 mRNA levels are not correlated with the metastatic pattern of PCa cells.14 In addition, RCN1 is involved in regulating drug resistance, and downregulation of RCN1 is observed in cisplatin‐resistant non‐small cell lung cancer cells.17 In contrast, high RCN1 levels potently drive the formation of doxorubicin resistance.18 Thus, the definitive physiological and pathophysiological roles of RCN1 and fundamental mechanisms underlying RCN1 are poorly understood and have yet to be elucidated.
In this study, we aimed to establish the significance of RCN1 in PCa cell survival and proliferation. Our data revealed that RCN1 was overexpressed in clinical PCa tissue samples and that downregulation of RCN1 in PCa cells triggered ER stress, leading to cell apoptosis and necroptosis.
2. MATERIALS AND METHODS
2.1. Patient samples
Prostate cancer and benign prostate samples were obtained from surgical excision specimens at the Qilu Hospital of Shandong University. Utilization of the clinical samples has been approved in the Ethical Committee of School of Medicine and Qilu Hospital of Shandong University (ID No: LL‐201601032).
2.2. Cell culture
The human PCa cell lines included PC3 (China Infrastructure of Cell Line Resource), DU145 and LNCaP cells (American Type Culture Collection (ATCC), Rockville, MD, USA), which were cultured as previously described.19
2.3. Cell viability and proliferation
Cell viability was evaluated using MTT (Sigma, St Louis, MO, USA) assays. Treated cells were incubated with 10 μL of MTT (5 mg/mL) for 4 hours. The cell response was determined on a plate reader (Bio‐Rad, CA, USA). Cell proliferation was examined using the EdU (Millipore, USA) incorporation assay kit, and imaged under a fluorescence microscope (LSM‐700, Carl Zeiss, Germany).
2.4. Cell cycle and apoptosis assay
Treated cells were fixed in ice‐cold 70% EtOH overnight at 4°C. After incubation with RNase (Invitrogen, CA, USA) for 30 minutes at 37°C, treated cells were exposed to propidium iodide (PI) (Sigma, St Louis, MO, USA) for 30 minutes at 4°C in the dark. The cell cycle was examined by flow cytometry (Becton Dickinson, NJ, USA). To determine cell apoptosis, cells were incubated in an annexin V‐FITC/PI solution for flow cytometry assays. The data was analyzed using the MODFIT and CELLQUEST software (Verity Software House, Topsham, Maine, USA). To detect apoptosis in murine tumor tissues, TUNEL assays were performed using a TUNEL Apoptosis Assay Kit (Beyotime, Shanghai, China).
2.5. Transfection
Cells were transfected with the specifically targeted siRNA or a pcDNA3.1‐RCN1 plasmid using Lipofectamine 2000 Transfection Reagent. Primers included human RCN1 siRNA (RCN1HSS109143, GGAUGAGAAGCUAACUAAAGAGGAA), human AR siRNA (TRCN0000003715, CCTGCTAATCAAGTCACACAT)20 and human PTEN siRNA (PTENHSS183790, AGAUGACAAUCAUGUUGCAGCAAUU).
2.6. Measurement of mitochondrial membrane potential
The mitochondrial membrane potential (MMP, mtΔψ) was assessed using a Mitochondrial Membrane Potential Assay Kit with JC‐1 (Beyotime) according the manufacturer's instructions. The fluorescence signals of the JC‐1 aggregates (red) and monomers (green) were measured on a flow cytometer (Becton Dickinson, NJ, USA). The MMP levels were calculated as the red/green fluorescence ratio.
2.7. Caspase‐3 activity
The caspase‐3 activity in cells was evaluated using the Caspase‐3 Activity Assay Kit (Beyotime). The absorbance was measured at 405 nm, and the relative activity of caspase‐3 was determined.
2.8. Western blot
Whole cell lysates and western blotting were prepared as previously described.19 The PARP, GAPDH (SC‐47724), CaMKII, AR, PERK and p‐PERK (Thr981) antibodies were obtained from Santa Cruz Biotechnology. Anti‐p‐eIF2α (Ser51), eIF2α, AKT, p‐AKT (Thr308), phosphatase and tensin homolog (PTEN), p‐PTEN (Ser380) and p‐CaMKII (Thr286) were purchased from Cell Signaling Technology (Danvers, MA, USA). RCN1 and cyclin D1 were obtained from Abcam (UK). GRP78/BIP and cyclin B1 were purchased from Proteintech (China). Bands on the immunoblots were visualized using an enhanced chemiluminescence detection system (Millipore, DT, Germany) and exposed to X‐ray films.
2.9. In vivo tumor growth
BALB/c athymic (nu+/nu+) male mice, aged 5‐6 weeks, were purchased from the Animal Center of China Academy of Medical Sciences (Beijing, China). The research protocol complied strictly with the institutional guidelines of the Animal Care and Use Committee at Shandong University. DU145 cells (1 × 107) were injected into the left upper flank of the mouse. Three months later, tumor tissue from the mouse was inoculated into the right armpits of other mice. After the tumors grew to 5 mm in diameter, the mice were randomly assigned to different groups (n = 7) and treated with multipoint intra‐tumoral injections of siRCN1 (10 μg/30 μL/tumor) complexed with the in vivo‐jetPEI transfection reagent (Polyplus‐transfection NY, USA) every other day for 15 days. Tumor sizes were monitored daily by measuring the length (L) and width (W) using calipers, and tumor volume (V) was calculated according to the formula V = (L × W2) × 0.5.
2.10. Immunohistochemistry
Heat‐induced epitope retrieval was performed in 10‐mmol/L citric acid buffer at pH 7.2 using a microwave. The slides were incubated at 4°C overnight with the primary antibodies (RCN1, 1:1000 dilution; cyclin D1, 1:200 dilution; cyclin B1, 1:50 dilution). An HRP‐conjugated antibody and 3, 3′‐diaminobenzidine (DAB) staining (ZSGBBIO, Beijing, China) were used to visualize primary antibody binding. High resolution pictures were obtained on an Olympus digital electron microscope, and images were recorded using the OlyVIA controller software (Olympus, Tokyo, Japan). The immunohistochemistry results were assigned a mean score that considered both the intensity of the staining and the positive reactions.
2.11. Statistical analysis
The differences in protein levels among the BPH and PCa tissues were analyzed using Fisher's exact test. Kaplan‐Meier and Cox proportional hazards analyses were used for survival analysis. The data are presented as the mean ± SD of at least 3 independent experiments. The mean difference between the control and treated groups was determined using Student's t tests. Two‐way ANOVA with Bonferroni's post‐test was used to compare all groups. A value of P < .05 was considered statistically significant.
3. RESULTS
3.1. Reticulocalbin 1 is highly expressed in prostate cancer
We initiated our study by examining RCN1 expression in human tissues from the Chinese Han population. Staining of the RCN1 protein in tumor tissues was strong (7/11), and RCN1 was detectable in the BPH samples (4/15) (Figure 1A,B). To determine whether the high RCN1 level in PCa was correlated with cancer development, we examined cyclin D1 expressions in tumor and BPH samples. Cyclin D1 showed a slight increase in tumor tissues in which RCN1 was highly present (Figure 1A,C). However, RCN1 expression was clearly associated with elevated cyclin B in the PCa tissues but not in BPH (Figure 1A,D). The datasets from the Oncomine database supported the observation that RCN1 expression is enhanced in PCa (Figure 1E). However, a survival analysis using online microarray data21 revealed no significant association between the high RCN1 mRNA level and disease‐free survival of PCa patients (Figure 1F, HR = 1.13, P = .067), suggesting that RCN1 might not be associated with PCa progression or the malignant phenotype, which is consistent the observation that RCN1 levels do not correlate with the metastatic pattern of PCa.14
Figure 1.
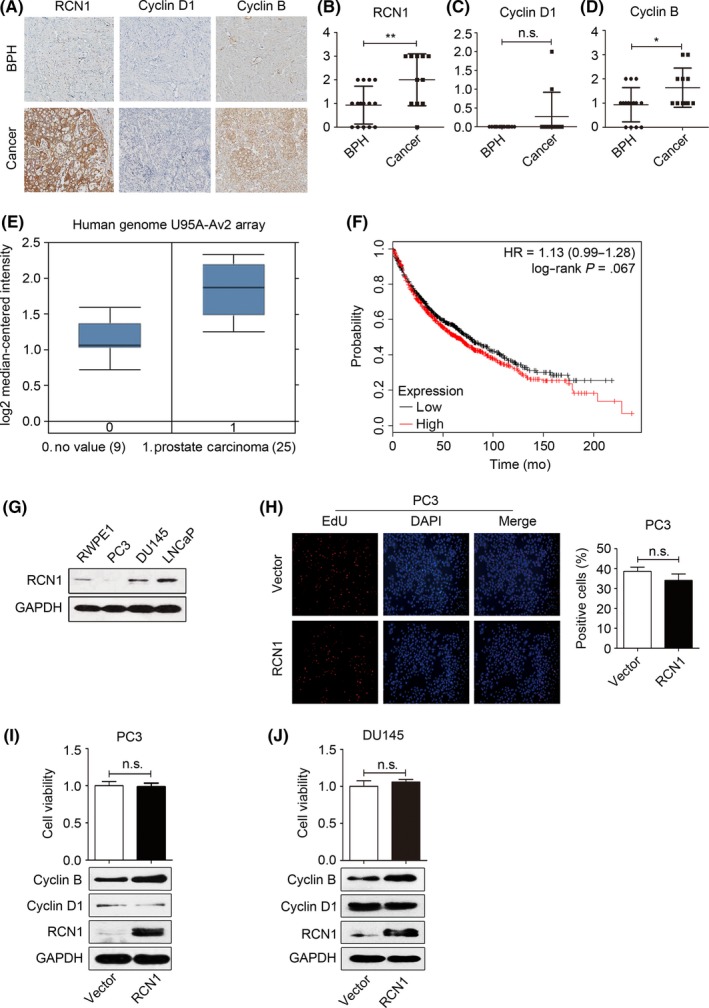
Reticulocalbin 1 (RCN1) is highly expressed in prostate tissues. A, Immunohistochemistry was used to determine RCN1, cyclin D1 and cyclin B. The statistical analysis of (B) RCN1, (C) cyclin D1 and (D) cyclin B were shown. *P < .05, **P < .01, n.s., no significance. E, Expression of RCN1 in prostate carcinoma was analyzed using the datasets from the Oncomine database. F, Survival analysis was shown using online microarray data with survival information between the high level of RCN1 mRNA with disease‐free survival in PCa patients (HR = 1.13, P = .067). G, The protein levels of RCN1 in RWPE1, PC3, DU145 and LNCaP were detected. H, Proliferation of PC3 cells transfected with pcDNA3.1‐RCN1 for 72 hours was detected using EdU. The charts indicate the number of EdU‐positive cells. I, PC3 and (J) DU145 were transfected with pcDNA3.1‐RCN1 for 72 hours. MTT assay and western blotting were performed. n.s., no significance
The basal RCN1 levels showed that RCN1 was lowest in the invasive PC‐3 cells, moderate in RWPE1 cells, but predominantly found in LNCaP and DU145 cells (Figure 1G), which is consistent with a previous report.14 We overexpressed RCN1 in PC3 cells and the results (Figure 1H) showed that ectopic expression of RCN1 had a limited effect on nuclear EdU incorporation and cell viability. In addition, RCN1 overexpression did not affect cyclin D1 and moderately increased cyclin B expression (Figure 1I). Similar results were observed in DU145 cells (Figure 1J) and bladder cancer 5637 cells after transfection of pcDNA3.1‐RCN1. The result of 5637 is not shown. Our results were consistent with Ki67 index‐based observations that RCN expression was not associated with the proliferative activities of cells that strongly expressed RCN1.8 Thus, RCN1 is broadly expressed in BPH and PCa tissues and may have a role in maintaining cell survival rather than driving cell proliferation.
3.2. Reticulocalbin 1 knockdown differentially induces apoptosis and necroptosis
To explore the role of RCN1, we performed cell cycle analysis. Depletion of endogenous RCN1 caused an accumulation of DU145 cells in the S phase (Figure 2A). However, downregulation of RCN1 in LNCaP cells arrested the cell cycle at the G2/M‐phase (Figure 2B). In addition, RCN1 knockdown significantly suppressed cell viability for both DU145 (Figure 2C) and LNCaP cells (Figure 2D), which was accompanied with induction of apoptosis (Figure 2E,F).
Figure 2.
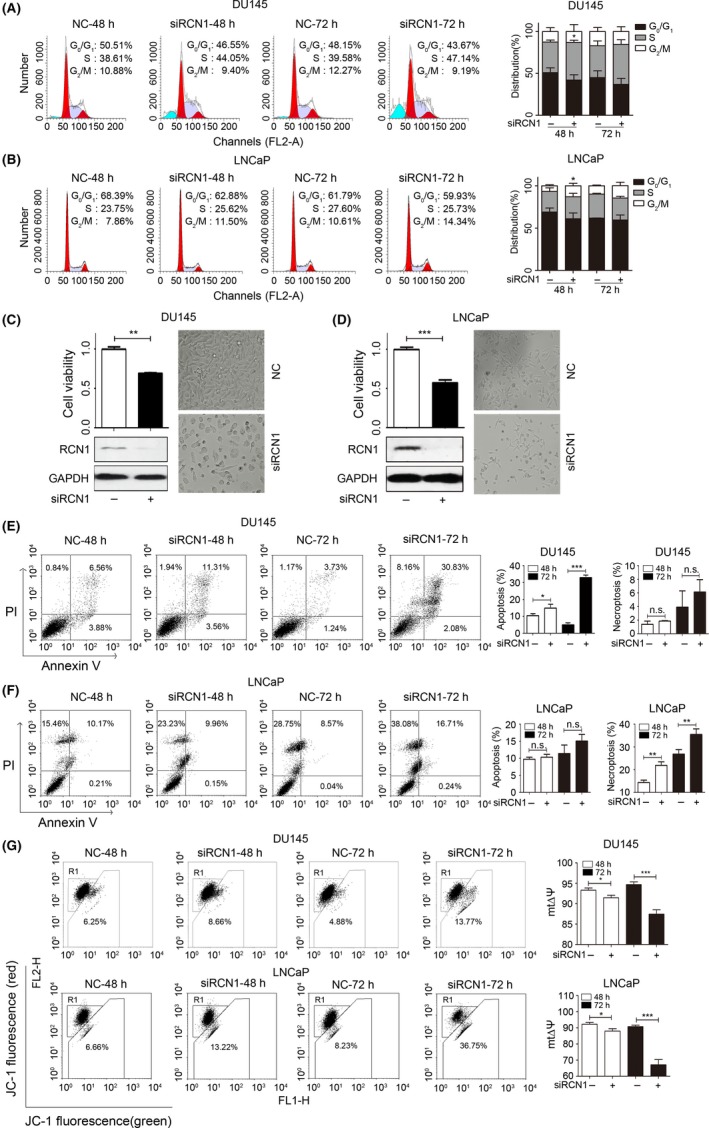
Reticulocalbin 1 (RCN1) depletion induces cell cycle arrest and cell death. A, DU145 and (B) LNCaP cells were transfected with siRCN1 for 48 or 72 hours. Cell cycle analyses were performed by flow cytometry, *P < .05. C, DU145 and (D) LNCaP cells were transfected with siRCN1 for 72 hours. Cell viability, morphological images and western blotting were obtained. **P < .01, ***P < .001. E, DU145 and (F) LNCaP cells were transfected with siRCN1 for 48 or 72 hours and stained with FITC‐conjugated annexin V and propidium iodide for flow cytometry. Statistical analysis is shown. *P < .05, **P < .01, ***P < .001, n.s., no significance. G, Changes of mitochondrial membrane potential (mtΔψ) were shown after RCN1 deletion in DU145 and LNCaP; *P < .05, ***P < .001
As we noted that sub‐G1 was shown in RCN1‐reduced DU145 cells (Figure 2A), apoptosis induced by RCN1 deficiency in time‐dependently increased (Figure 2E). Unlike the DU145 cells, necroptosis was predominant in siRCN1‐transfected LNCaP cells compared to the fraction of apoptotic cells (Figure 2F). RCN1 knockdown‐mediated cell viability inhibition and apoptosis were also observed in other cancer cells, including urinary bladder cancer and colon cancer cells (Figure S1). Changes in the mitochondrial membrane potential (mtΔψ), an indicator of the energetic state of the mitochondria and an important cell death pathway,22 revealed that the mtΔψ was time‐dependently decreased in the RCN1‐knockdown DU145 and LNCaP cells (Figure 2G). These data confirm that RCN1 knockdown leads to PCa cell death.
We subsequently investigated whether RCN1 depletion‐induced cell death was associated with caspase activation, which is a typical apoptosis pathway. RCN1 depletion in DU145 cells led to the activation of caspase‐3, whereas no significant change was observed in LNCaP cells (Figure 3A). Cleavage of the nuclear enzyme PARP, which is a hallmark of apoptosis, became noticeably higher in RCN1‐depleted DU145 cells in a time‐dependent manner. Increase in the PARP cleavage was also obvious in bladder cancer and colon cancer cells (Figure S1). However, cleaved PARP remained almost unchanged in the LNCaP cells (Figure 3B). Z‐VAD, a pan‐caspase inhibitor, partially rescued RCN1 depletion‐induced cell death in DU145 cells (Figure 3C) but did not rescue LNCaP cells (Figure 3D), supporting the observation that RCN1 knockdown mainly triggered apoptosis in DU145 cells (Figure 2). We also used necrostatin‐1, an inhibitor of necroptosis, to determine whether necroptosis was involved in RCN1 depletion‐induced cell death. Necrostatin‐1 clearly reversed the cell death in siRCN1‐treated LNCaP cells, suggesting that necroptosis, at least partially, contributed to siRCN1‐mediated LNCaP cell death (Figure 3E,F). Thus, RCN1 knockdown differentially triggered cell cycle arrest at the S phase or G2/M phase and promoted caspase‐dependent apoptosis and necroptosis in PCa cells.
Figure 3.
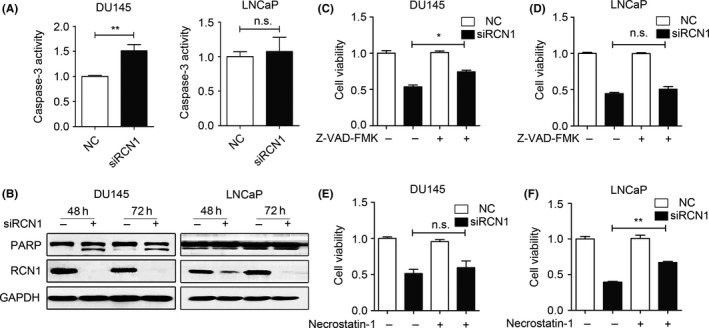
Reticulocalbin 1 (RCN1) knockdown differentially induces caspase‐dependent apoptosis and necroptosis. A, Caspase‐3 activity was evaluated after transfecting 72 hours. **P < .01, n.s, no significance. B, Protein levels of RCN1 and PARP were performed by western blotting. C, DU145 and (D) LNCaP were pre‐treated with 20 μmol/L Z‐VAD‐FMK for 5 hours and transfected with negative control (NC) or siRCN1 for 48 hours. Cell viability was monitored using MTT assays. *P < .05, n.s., no significance. E, DU145 and (F) LNCaP cells were pre‐treated with 20 μmol/L Necrostatin‐1 for 5 hours and transfected with NC or siRCN1 for 48 hours. Cell viability was monitored using MTT assays. **P < .01, n.s., no significance
3.3. Reticulocalbin 1 depletion activates endoplasmic reticulum stress and CaMKII
Because the ER‐resident RCN1 protein is a Ca2+‐binding protein, we hypothesized that RCN1 depletion triggers ER stress, which, in turn, promotes cell death. The results (Figure 4A) showed that the expression of glucose‐related protein 78 (GRP78), a sensor of the ER stress response, was elevated in RCN1‐depleted DU145 and LNCaP cells. Increases in the phosphorylation of PRKR‐like endoplasmic reticulum kinase (PERK) and its substrate, eIF2α, were evident at 24 hours, and became moderate or weak after longer siRCN1 transfection. In addition, C/EBP‐homologous protein CHOP, an indicator of ER stress‐associated apoptosis, was barely detectable in RCN1‐deficient LNCaP cells. However, induction of CHOP was initiated at 24 hours, and became stronger after prolonged depletion of RCN1 in DU145 cells (Figure 4A), reinforcing the observations that DU145 cells was prone to apoptosis when cells lose RCN1 (Figures 2 and 3). We included sodium 4‐phenylbutyrate (4‐PBA) and GSK2606414, 2 specific PERK inhibitors, which attenuate the ER stress,23 to confirm the role of ER stress in RCN1 depletion‐induced cell death. Pretreatment with either 4‐PBA or GSK2606414 alleviated the ER stress, and increased both DU145 and LNCaP cell survival when RCN1 was reduced (Figure 4B‐E).
Figure 4.
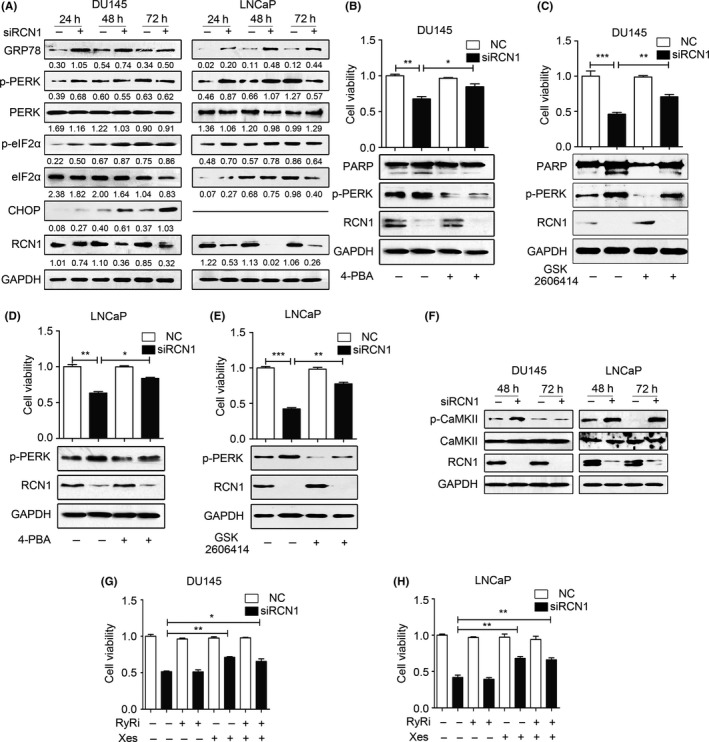
Reticulocalbin 1 (RCN1) depletion activates endoplasmic reticulum stress and CaMKII. A, Western blot analysis after DU145 and LNCaP cells transfected with siRCN1 for 24, 48 or 72 hours. B, DU145 or (D) LNCaP cells were exposed to 4‐PBA (2.5 mmol/L) for 5 hours and subsequently transfected with negative control (NC) or siRCN1 for 48 hours. Cell viability and western blotting were obtained. *P < .05. **P < .01. C, DU145 and (E) LNCaP cells were pre‐treated with GSK2606414 (10 μmol/L) for 5 hours and transfected with NC or siRCN1 for 48 hours. Cell viability and western blotting was performed. **P < .01, ***P < .001. F, DU145 and LNCaP cells were transfected with siRCN1. Then, cells were collected for western blot analysis. G, DU145 or (H) LNCaP cells were transfected with NC or siRCN1 for 24 hours and subsequently exposed to xestospongin C (5 μmol/L) or ryanodine (1 μmol/L) for 24 hours. Cell viability was monitored by using MTT assays. *P < .05, **P < .01
We examined whether loss of RCN1 rendered cells unable to survive because of Ca2+ release from the ER and a subsequent elevation in cytoplasmic Ca2+. To this end, we analyzed Ca2+‐calmodulin‐dependent protein kinase II (CaMKII) that is directly activated by increased cytoplasmic Ca2+ and calmodulin levels and acts as a causal factor in the control of cell cycle progression.24, 25 The results demonstrated that elevated p‐CaMKIIT286 were observed in DU145 cells at 48 hours; CaMKII subsequently became inactive after prolonged depletion of RCN1. Loss of RCN1 in LNCaP cells resulted in elevated p‐CaMKIIT286 in a time‐dependent manner (Figure 4F), which was consistent with a report showing that CaMKII activation triggers necroptosis.26 Because inositol 1, 4, 5‐ trisphosphate receptor (IP3R) and ryanodine receptor (RyR) control Ca2+ release from the ER to the cytoplasm in response to stimulation, we used Xestospongin C (Xes) and Ryanodine (RyRi), inhibitors of IP3R and RyR, respectively, to examine the involvement of IP3R and RyR in the RCN1‐mediated effect. Xes partially rescued RCN1 knockdown‐induced cell death. RyRi did not affect cell viability (Figure 4G), and there was no synergistic increase in the effect of Xes in RCN1‐deficient DU145 cells. Similar results were observed in RCN1‐depleted LNCaP cells (Figure 4H). Thus, RCN1 deficiency elicited the release of ER Ca2+ into the cytoplasm, leading to the activation of a Ca2+ signaling pathway involving CaMKII to trigger cell death.
3.4. Reticulocalbin 1 ablation modulates phosphatase and tensin homolog activity
We next examined the involvement of AKT pathway due to the critical role in cell survival and proliferation.27, 28 Loss of RCN1 resulted in AKT inactivation in DU145 cells. However, no significant change in p‐AKT was observed in LNCaP cells when RCN1 was reduced (Figure 5A), suggesting that AKT signaling might not be involved in RCN1 depletion‐mediated LNCaP cell death. Like LNCaP cells, RWPE1 cells, human benign prostate epithelial cells, are androgen responsive, and moderately express RCN1. We also examined changes in cell viability and AKT activation when RCN1 is deleted in RWPE1 cells. The results (Figure 5B,C) showed that depletion of RCN1 significantly suppressed cell viability, but p‐AKT had no significant change. The finding suggests that RCN1 is functional in non‐neoplastic cells and cancer cells. However, inactivation of AKT by reduced RCN1 is cell type‐dependent. Because DU145 cells are positive for wild‐type phosphatase and tensin homolog (PTEN) that antagonizes the PI3K/AKT signaling pathway,28, 29 we examined whether RCN1 affected PTEN activity. RCN1 reduction led to PTEN induction (Figure 5D), and moderately decreased p‐PTEN (Ser308, inactive form) in DU145 cells, indicating that RCN1 loss was capable of increasing PTEN activity.30, 31 Knockdown of PTEN in RCN1‐deficient DU145 cells noticeably reduced cell apoptosis, as evidenced by lower PARP cleavage, indicating the importance of PTEN/AKT signaling in RCN1 depletion‐mediated apoptosis (Figure 5E). Thus, RCN1 depletion resulted in upregulation of PTEN and inactivation of AKT signaling, which partially contributed to DU145 apoptosis.
Figure 5.
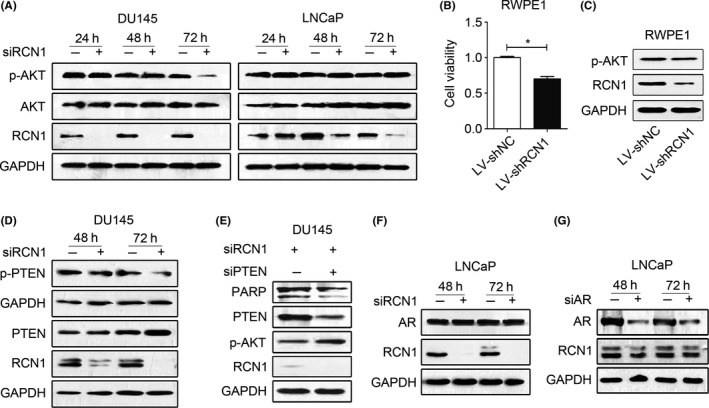
Reticulocalbin 1 (RCN1) ablation modulates PTEN activity. A, Western blotting was performed to detect the protein level after RCN1 ablation for 24, 48 and 72 hours. B, RWPE1 cells were infected with LV‐shRCN1 for 72 hours. Cell viability was monitored using MTT assay. *P < .05. C, Treated RWPE1 cells were prepared for western blot analysis. D, DU145 cells were transfected with siRCN1 for 48 and 72 hours for western blotting. E, DU145 cells were transfected with siRCN1 for 24 hours and siPTEN for another 24 hours. Western blot analysis is shown. F, LNCaP cells were transfected with siRCN1 and (G) siAR for 48 or 72 hours and western blot analysis was performed
Given the importance of androgen receptor (AR) signaling on LNCaP cell proliferation, we explored the involvement of AR in RCN1 deficiency‐mediated cell death. The results revealed that RCN1 silencing did not affect AR expression in LNCaP cells (Figure 5F). In addition, AR knockdown had a limited effect on RCN1 expression (Figure 5G), suggesting that the AR might not play a critical role in RCN1 depletion‐mediated cell death.
3.5. Reticulocalbin 1 deficiency suppresses DU145 xenograft tumor growth in mice
Driven by these observations, we validated the effect of RCN1 on DU145 xenografts in mice. After treatment with the RCN1‐targeted siRNA (Figure 6A), the body weights remained almost unchanged (Figure 6B). The tumors arising from the control mice grew to an average size of 214.8 ± 238.9 mm3 but significantly decreased to 85.3 ± 98.7 mm3 in the RCN1 knockdown group (Figure 6C,D). Similarly, the tumor weights were much higher in the control group (190.4 ± 212.8 mg) than in the RCN1‐deficient mice (76.1 ± 86.5 mg) (Figure 6E). The HE analysis clearly showed tumor‐specific tissue parameters, including loss of normal cellular morphology and enlargement and condensation of the nucleus in the control group; RCN1 knockdown was associated with smaller and more irregular nuclei, and observed cell debris in the tissues (Figure 6F). In contrast to the control group, increasing numbers of TUNEL‐positive apoptotic cells were observed in the siRCN1‐treated mice, accompanied with decreases in Ki67 positivity (Figure 6G). In addition, PARP cleavage increased in tumors with reduced RCN1. In addition, the RCN1 deficiency induced ER stress, which was evidenced by an enhancement in p‐PERK and p‐eIF2α (Figure 6H). These results demonstrated that RCN1 knockdown inhibited PCa cell proliferation and elicited apoptosis in vitro and in vivo.
Figure 6.
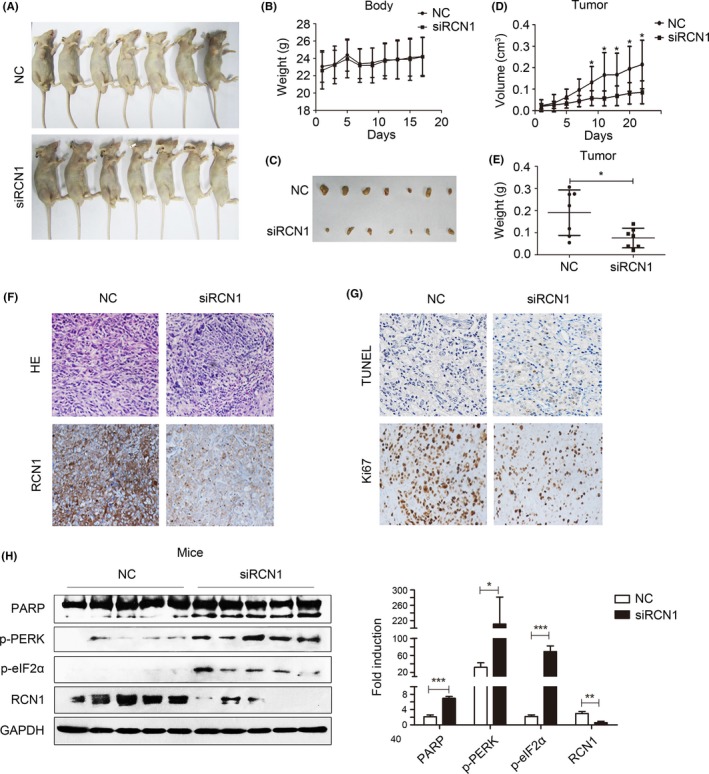
Reticulocalbin 1 (RCN1) deficiency suppresses DU145 xenograft tumor growth in mice. A, Representative photographs of the control and siRCN1 group of nude mice. B, Statistical analysis of the mice weights is shown. C, Excised tumors from euthanized nude mice from negative control and siRCN1 groups. D, Statistical analyses of the tumor volume and (E) tumor weight are shown. *P < .05. F, HE stains of the tumor tissues and immunohistochemistry of RCN1. G, Detection of apoptotic cells by TUNEL assay and Ki67 staining of tumor tissues. H, Western blotting was performed and quantified by Image J software. *P < .05, **P < .01, ***P < .001
4. DISCUSSION
This study presents evidence to support the role of RCN1 in mediating PCa cell survival. RCN1 was predominantly high in the PCa samples from the Chinese Han patients and was noticeably associated with cyclin B rather than cyclin D1. Reduced endogenous RCN1 significantly resulted in cell cycle arrest and suppressed cell viability. Furthermore, RCN1 knockdown mainly triggered DU145 cell apoptosis in a caspase‐dependent manner but caused necroptosis in LNCaP cells. Differences in the effects of RCN1 knockdown might be due to differences in cell types. Wild‐type PTEN in DU145 cells, at least in part, contributed to the induction of apoptosis because knockdown of RCN1 induced PTEN expression, leading to the inactivation of AKT and an increase in PARP cleavage. In LNCaP cells, predominant activation of CaMKII was important toward mediating necroptosis upon RCN1 depletion. In addition, RCN1 depletion initiated ER stress, which partially resulted from disruption of the ER Ca2+ channel IP3R.
Reticulocalbin 1 is reportedly overexpressed in cancer cells and correlates with malignancy in several types of cancer.9, 10, 11, 12, 13, 14, 17 We also demonstrated that RCN1 was significantly high in the Chinese Han PCa population. However, high RCN1 expression in the tumor was not correlated with the survival probability of the PCa patients who were analyzed based on the available online expression database, indicating that RCN1 expression may not be associated with PCa progression. We noted that RCN1 expression was lowest in the highly invasive PC3 cell line, which is consistent with a previous observation that RCN1 cell surface expression is more evident on LNCaP cells than in PC3 and C4‐2B4 cells.14 These observations suggest that RCN1 may not be directly correlated with the metastatic potential of PCa. High RCN1 and RCN2 expression has recently been associated with shorter recurrence‐free survival in breast cancer patients.32 Therefore, the contribution of RCN1 to carcinogenesis and cancer progression may be dependent on the cancer type.
RCN1 expression in PCa tissues was noticeably accompanied by strong staining of Cyclin B but not Cyclin D. Re‐expression of RCN1 in PC3 cells was unable to promote cell proliferation, whereas knockdown of endogenous RCN1 resulted in cell cycle arrest at the S‐phase and G2/M ‐phase in DU145 and LNCaP cells, respectively. These results indicate that RCN1 may be more important for maintaining cell survival than for initiating cell cycle progression and cell proliferation. Consistent with our results, the study by Fukuda et al8 revealed that Ki67, an indicator of proliferative cells, was low in normal human cells that strongly expressed RCN, suggesting that RCN expression is closely associated with synthesis and/or secretory functions but not with proliferative functions in cells. Furthermore, the elevated RCN1 that promotes cell survival attenuates the inhibitory effect of radiation on epithelial carcinoma cells,33 reinforcing the role of RCN1 in cell survival.
Several studies have demonstrated that downregulation of RCN1 in some cancer cells significantly suppresses cell proliferation.32, 34 Our findings reinforce these observations that RCN1 deletion significantly suppresses PCa cell viability. Knockdown of RCN1 causes PTEN activated in DU145 cells, leading to inactivation of AKT and cellular apoptosis. Thus, upregulation of PTEN contributes to the switch from cell survival to cell apoptosis upon RCN depletion. Further investigations are required to explore the regulatory mechanisms by which RCN1 negatively modulates PTEN expression and activity.
As an ER‐resident Ca2+‐binding protein, RCN1 downregulation induced ER stress, which was associated with CaMKII activation. Inhibition of IP3R, instead of RyR, partially restored RCN1 depletion‐induced cell death. Therefore, CaMKII activation might be a consequence of interrupted IP3R signaling upon RCN1 depletion. This notion is supported by a recent study showing that RCN1 specifically interacts with IP3R1 and inhibits Ca2+ release from the ER, thereby suppressing ER stress‐induced apoptosis.34 CaMKII overexpression or activation is believed to promote cancer cell proliferation and serves as a target for chemotherapy.35, 36 However, CaMKII‐dependent apoptosis has also been observed in multiple cultured cell lines.37 Recently, Nomura et al38 provided evidence that increased cytoplasmic Ca2+ triggers necroptosis by activating CaMKII/RIP1. We also found that CaMKII activation was sustained up to 72 hours in RCN1‐deficient LNCaP cells and may be responsible for a process involved in necroptosis. CaMKII‐independent mechanisms may also contribute to the induction of cell death in response to RCN1 depletion. In summary, RCN1 depletion promotes PCa cell apoptosis and necroptosis, at least in part, through CaMKII activation and AKT suppression, suggesting a role for RCN1 in maintaining cancer cell survival and its potential usefulness in cancer therapy.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
Liu X, Zhang N, Wang D, et al. Downregulation of reticulocalbin‐1 differentially facilitates apoptosis and necroptosis in human prostate cancer cells. Cancer Sci. 2018;109:1147–1157. https://doi.org/10.1111/cas.13541
Funding information
National Natural Science Foundation of China Grant Number 81473238; Natural Science Foundation of Shandong Province Grant Numbers ZR2014HM087, ZR2015HM027 and BS2015YY034
Liu and Zhang equally contributed to this study.
Contributor Information
Keli Tian, Email: tiankeli@sdu.edu.cn.
Huiqing Yuan, Email: lyuanhq@sdu.edu.cn.
REFERENCES
- 1. Kim H, Bhattacharya A, Qi L. Endoplasmic reticulum quality control in cancer: friend or foe. Semin Cancer Biol. 2015;33:25‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89‐102. [DOI] [PubMed] [Google Scholar]
- 3. Brychtova V, Mohtar A, Vojtesek B, Hupp TR. Mechanisms of anterior gradient‐2 regulation and function in cancer. Sem Cancer Biol. 2015;33:16‐24. [DOI] [PubMed] [Google Scholar]
- 4. Honore B. The rapidly expanding CREC protein family: members, localization, function, and role in disease. BioEssays. 2009;31:262‐277. [DOI] [PubMed] [Google Scholar]
- 5. Wu W, Tang X, Hu W, Lotan R, Hong WK, Mao L. Identification and validation of metastasis‐associated proteins in head and neck cancer cell lines by two‐dimensional electrophoresis and mass spectrometry. Clin Exp Metastasis. 2002;19:319‐326. [DOI] [PubMed] [Google Scholar]
- 6. Wang Q, Shen B, Chen L, et al. Extracellular calumenin suppresses ERK1/2 signaling and cell migration by protecting fibulin‐1 from MMP‐13‐mediated proteolysis. Oncogene. 2015;34:1006‐1018. [DOI] [PubMed] [Google Scholar]
- 7. Arahata K, Hayashi YK, Mizuno Y, Yoshida M, Ozawa M. Dystrophin‐associated glycoprotein and dystrophin co‐localisation at sarcolemma in Fukuyama congenital muscular dystrophy. Lancet. 1993;342:623‐624. [DOI] [PubMed] [Google Scholar]
- 8. Fukuda T, Oyamada H, Isshiki T, et al. Distribution and variable expression of secretory pathway protein reticulocalbin in normal human organs and non‐neoplastic pathological conditions. J Histochem Cytochem. 2007;55:335‐345. [DOI] [PubMed] [Google Scholar]
- 9. Liu Z, Brattain MG, Appert H. Differential display of reticulocalbin in the highly invasive cell line, MDA‐MB‐435, versus the poorly invasive cell line, MCF‐7. Biochem Biophys Res Commun. 1997;231:283‐289. [DOI] [PubMed] [Google Scholar]
- 10. Nimmrich I, Erdmann S, Melchers U, et al. Seven genes that are differentially transcribed in colorectal tumor cell lines. Cancer Lett. 2000;160:37‐43. [DOI] [PubMed] [Google Scholar]
- 11. Sinha P, Poland J, Schnolzer M, Celis JE, Lage H. Characterization of the differential protein expression associated with thermoresistance in human gastric carcinoma cell lines. Electrophoresis. 2001;22:2990‐3000. [DOI] [PubMed] [Google Scholar]
- 12. Poland J, Schadendorf D, Lage H, Schnolzer M, Celis JE, Sinha P. Study of therapy resistance in cancer cells with functional proteome analysis. Clin Chem Lab Med. 2002;40:221‐234. [DOI] [PubMed] [Google Scholar]
- 13. Yu LR, Zeng R, Shao XX, Wang N, Xu YH, Xia QC. Identification of differentially expressed proteins between human hepatoma and normal liver cell lines by two‐dimensional electrophoresis and liquid chromatography‐ion trap mass spectrometry. Electrophoresis. 2000;21:3058‐3068. [DOI] [PubMed] [Google Scholar]
- 14. Cooper CR, Graves B, Pruitt F, et al. Novel surface expression of reticulocalbin 1 on bone endothelial cells and human prostate cancer cells is regulated by TNF‐alpha. J Cell Biochem. 2008;104:2298‐2309. [DOI] [PubMed] [Google Scholar]
- 15. Giribaldi G, Barbero G, Mandili G, et al. Proteomic identification of Reticulocalbin 1 as potential tumor marker in renal cell carcinoma. J Proteomics. 2013;91:385‐392. [DOI] [PubMed] [Google Scholar]
- 16. Yoshida Y, Yamashita T, Nagano K, et al. Limited expression of reticulocalbin‐1 in lymphatic endothelial cells in lung tumor but not in normal lung. Biochem Biophys Res Commun. 2011;405:610‐614. [DOI] [PubMed] [Google Scholar]
- 17. Hirano T, Kato H, Maeda M, et al. Identification of postoperative adjuvant chemotherapy responders in non‐small cell lung cancer by novel biomarker. Int J Cancer. 2005;117:460‐468. [DOI] [PubMed] [Google Scholar]
- 18. May EW, Lin ST, Lin CC, et al. Identification of up‐ and down‐regulated proteins in doxorubicin‐resistant uterine cancer cells: reticulocalbin‐1 plays a key role in the development of doxorubicin‐associated resistance. Pharmacol Res. 2014;90:1‐17. [DOI] [PubMed] [Google Scholar]
- 19. Guo YX, Lin ZM, Wang MJ, et al. Jungermannenone A and B induce ROS‐ and cell cycle‐dependent apoptosis in prostate cancer cells in vitro. Acta Pharmacol Sin. 2016;37:814‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun NK, Huang SL, Lu HP, Chang TC, Chao CC. Integrative transcriptomics‐based identification of cryptic drivers of taxol‐resistance genes in ovarian carcinoma cells: analysis of the androgen receptor. Oncotarget. 2015;6:27065‐27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. PLoS ONE. 2013;8:e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139‐176. [DOI] [PubMed] [Google Scholar]
- 23. Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4‐phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45‐52. [DOI] [PubMed] [Google Scholar]
- 24. Fujisawa H. Regulation of the activities of multifunctional Ca2+/calmodulin‐dependent protein kinases. J Biochem. 2001;129:193‐199. [DOI] [PubMed] [Google Scholar]
- 25. de Diego I, Kuper J, Bakalova N, Kursula P, Wilmanns M. Molecular basis of the death‐associated protein kinase‐calcium/calmodulin regulator complex. Sci Signal 2010;3:ra6. [DOI] [PubMed] [Google Scholar]
- 26. Zhang T, Zhang Y, Cui M, et al. CaMKII is a RIP3 substrate mediating ischemia‐ and oxidative stress‐induced myocardial necroptosis. Nat Med. 2016;22:175‐182. [DOI] [PubMed] [Google Scholar]
- 27. Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13:234‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xie Y, Naizabekov S, Chen Z, Tokay T. Power of PTEN/AKT: molecular switch between tumor suppressors and oncogenes. Oncol Lett. 2016;12:375‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;62:6141‐6145. [PubMed] [Google Scholar]
- 30. Bolduc D, Rahdar M, Tu‐Sekine B, et al. Phosphorylation‐mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife. 2013;2:e00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627‐48630. [DOI] [PubMed] [Google Scholar]
- 32. Nakakido M, Tamura K, Chung S, et al. Phosphatidylinositol glycan anchor biosynthesis, class X containing complex promotes cancer cell proliferation through suppression of EHD2 and ZIC1, putative tumor suppressors. Int J Oncol. 2016;49:868‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren Y, Yeoh KW, Hao P, Kon OL, Sze SK. Irradiation of epithelial carcinoma cells upregulates calcium‐binding proteins that promote survival under hypoxic conditions. J Proteome Res. 2016;15:4258‐4264. [DOI] [PubMed] [Google Scholar]
- 34. Xu S, Xu Y, Chen L, et al. RCN1 suppresses ER stress‐induced apoptosis via calcium homeostasis and PERK‐CHOP signaling. Oncogenesis. 2017;6:e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rokhlin OW, Taghiyev AF, Bayer KU, et al. Calcium/calmodulin‐dependent kinase II plays an important role in prostate cancer cell survival. Cancer Biol Ther. 2007;6:732‐742. [DOI] [PubMed] [Google Scholar]
- 36. Britschgi A, Bill A, Brinkhaus H, et al. Calcium‐activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Nat Acad Sci USA. 2013;110:E1026‐E1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Timmins JM, Ozcan L, Seimon TA, et al. Calcium/calmodulin‐dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925‐2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014;74:1056‐1066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials