

Abstract
Abrogating tumor angiogenesis by inhibiting vascular endothelial growth factor receptor‐2 (VEGFR2) has been established as a therapeutic strategy for treating cancer. However, because of their low selectivity, most small molecule inhibitors of VEGFR2 tyrosine kinase show unexpected adverse effects and limited anticancer efficacy. In the present study, we detailed the pharmacological properties of anlotinib, a highly potent and selective VEGFR2 inhibitor, in preclinical models. Anlotinib occupied the ATP‐binding pocket of VEGFR2 tyrosine kinase and showed high selectivity and inhibitory potency (IC 50 <1 nmol/L) for VEGFR2 relative to other tyrosine kinases. Concordant with this activity, anlotinib inhibited VEGF‐induced signaling and cell proliferation in HUVEC with picomolar IC 50 values. However, micromolar concentrations of anlotinib were required to inhibit tumor cell proliferation directly in vitro. Anlotinib significantly inhibited HUVEC migration and tube formation; it also inhibited microvessel growth from explants of rat aorta in vitro and decreased vascular density in tumor tissue in vivo. Compared with the well‐known tyrosine kinase inhibitor sunitinib, once‐daily oral dose of anlotinib showed broader and stronger in vivo antitumor efficacy and, in some models, caused tumor regression in nude mice. Collectively, these results indicate that anlotinib is a well‐tolerated, orally active VEGFR2 inhibitor that targets angiogenesis in tumor growth, and support ongoing clinical evaluation of anlotinib for a variety of malignancies.
Keywords: angiogenesis, anlotinib, tyrosine kinase inhibitor, VEGF, VEGFR2
Abbreviations
- Akt
v‐Akt murine thymoma viral oncogene
- EGFR
epidermal growth factor receptor
- PDGF‐BB
platelet‐derived growth factor‐BB
- PDGFRβ
platelet‐derived growth factor receptor β
- RTK
receptor tyrosine kinase
- SCF‐1
stem cell factor‐1
- TKI
tyrosine kinase inhibitor
- VEGFR1
vascular endothelial growth factor receptor‐1
- VEGFR2
vascular endothelial growth factor receptor‐2
- VEGFR3
vascular endothelial growth factor receptor‐3
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Angiogenesis—the sprouting of new capillaries from pre‐existing blood vessels—is a complex process involving dissolution of the basement membrane, migration of endothelial cells, and formation of new capillary loops.1 Although vascular proliferation generally occurs only during embryonic development, angiogenesis has been shown to be a crucial step in tumor growth, invasion, and metastasis.2, 3 Persistent and unregulated angiogenesis is observed during tumor progression.4 Indeed, it has been shown that tumors cannot continue to grow without angiogenesis after they reach a size of ~1 mm3.5 Tumor cells easily acquire resistance during chemotherapy, whereas endothelial cells rarely acquire resistance because of their genetic stability.6 Thus, anti‐angiogenesis therapy targeting vascular endothelial cells in tumors is a promising therapeutic approach.7, 8 Accordingly, there has been continuing interest in studying and developing compounds that can inhibit the process of angiogenesis.9
Vascular endothelial growth factor is the best‐characterized modulator of tumor angiogenesis, metastasis, and growth among all known angiogenic factors.10, 11, 12 The production of VEGF is associated with abnormal angiogenesis in many types of cancers. Vascular endothelial growth factor is capable of regulating angiogenic processes, including endothelial cell migration and proliferation, neovascular survival, capillary tube formation and permeability, among others.13 Vascular endothelial growth factor exerts its biological effects by activating RTK, VEGFR1, VEGFR2, and VEGFR3.12 Of these receptors, VEGFR2 plays the major role in regulating angiogenesis.14 Inhibition of VEGF/VEGFR signaling has been considered a promising therapeutic approach against solid tumors.
Some monoclonal antibodies against VEGF or VEGFR, such as bevacizumab (anti‐VEGF) and ramucirumab (anti‐VEGFR2), have recently been approved for cancer therapy and shown to exhibit sustained target inhibition and good safety profiles owing to their high specificity.15, 16, 17 However, monoclonal antibodies have drawbacks that limit their application in the clinic, including the requirement for i.v. dosing, their immunogenicity and potential to induce autoimmune diseases after long‐term treatment, as well as their high cost for patients. A growing number of small‐molecule VEGFR TKI, including sunitinib, sorafenib and pazopanib, have been described in the past 10 years;18 however, these drugs have poor kinase selectivity. As a consequence, they require high daily dosing, and thus lead to unexpected adverse effects.19 Accordingly, small‐molecule inhibitors that are more potent and selective would hopefully reduce off‐target effects and overall drug exposure, and circumvent the limitations of antibodies.
Anlotinib (1‐[[[4‐(4‐fluoro‐2‐methyl‐1H‐indol‐5‐yloxy)‐6‐methoxyquinolin‐7‐Yl] oxy] methyl]cyclopropanamine dihydrochloride) (Figure 1A), characterized as a highly selective and potent VEGFR2 inhibitor, was identified through screens based on inhibition of VEGFR2 kinase. Clinical phase III trials of anlotinib have been completed in China, and clinical phase II trials against a variety of malignancies are currently underway in the USA. At a once‐daily dose of 12 mg, anlotinib showed manageable toxicity, a long circulation time, and broad‐spectrum antitumor potential in clinical trials.20
Figure 1.
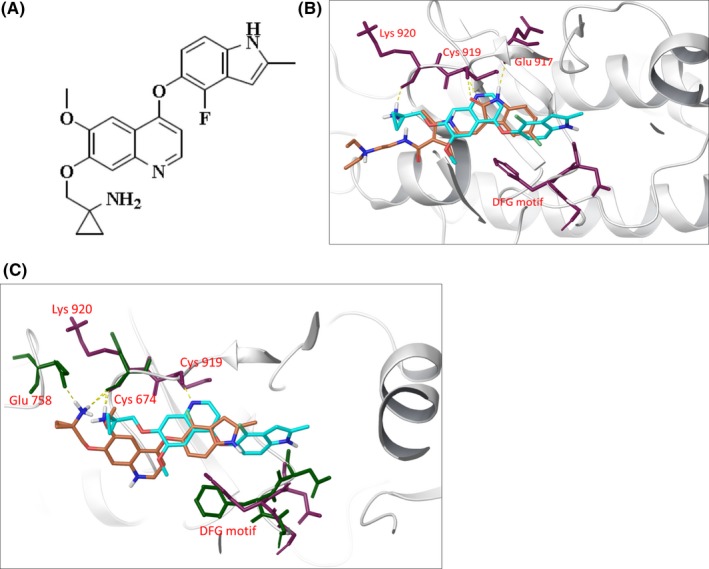
Characterization of anlotinib as a vascular endothelial growth factor receptor‐2 (VEGFR2) inhibitor. A, Chemical structure of anlotinib. B, Molecular modeling of the VEGFR2–anlotinib/sunitinib complex. Hydrogen bonds are presented as yellow dashed lines, and critical residues are presented as maroon sticks. Anlotinib (cyan, docked pose with structure PDB code 4ASD) and sunitinib (orange, from crystallographic structure PDB code 4AGD). C, Molecular modeling of the c‐Kit–anlotinib complex (orange) and VEGFR2–anlotinib complex (cyan). Hydrogen bonds are presented as yellow dashed lines, and critical residues are presented as maroon sticks (VEGFR2) and dark green sticks (c‐Kit)
In the present study, we carried out pharmacological characterization of anlotinib in preclinical models. Anlotinib inhibited tumor development mainly through highly potent and specific suppression of VEGFR2, resulting in potent anti‐angiogenesis and broad‐spectrum antitumor activity. Thus, these studies provide a rationale for the ongoing clinical evaluation of anlotinib as an anticancer agent.
2. MATERIALS AND METHODS
2.1. Materials
Anlotinib was provided by Chia Tai Tianqing Pharmaceutical Group Co., Ltd (Nanjing, China). Sunitinib and sorafenib were purchased from Selleckchem (Houston, TX, USA). Each of these drugs was prepared as a 10‐mmol/L stock solution in DMSO (in vitro studies) or normal saline (in vivo studies).
Recombinant human VEGF, SCF, and PDGF‐BB were purchased from R&D Systems (Minneapolis, MN, USA). Antibodies to c‐Kit, p‐c‐Kit, β‐tubulin, CD31, p‐ERK1/2, Akt, p‐Akt, PDGFRβ, p‐PDGFRβ, HER2, p‐HER2, VEGFR2, and p‐VEGFR2 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies to PY99 and ERK1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HRP‐conjugated secondary anti‐rabbit/mouse IgG antibodies were purchased from Calbiochem (Millipore, Bedford, MA, USA).
2.2. Animals and cell lines
All animal experiments were approved by the Institute Animal Review Boards of Shanghai Institute of Materia Medica, Chinese Academy of Sciences, with firm adherence to the ethical guidelines for the care and use of animals.
Human mast cell line‐1 (HMC‐1) was a generous gift from Dr J. H. Butterfield of the Mayo Clinic (Rochester, MN, USA). The Mo7e acute myeloid leukemia cell line was provided by the Genetics Institute (Boston, MA, USA). All other tumor cell lines were purchased from the ATCC (Manassas, VA, USA). Cells were cultured according to instructions provided by the ATCC. HUVEC were isolated from human umbilical cord veins as described previously21 and kept in medium 199 (Gibco, Waltham, MA, USA) supplemented with 20% FBS (Gibco) and endothelial cell growth supplement (Sigma, St Louis, MO, USA). All cells were incubated in a humidified atmosphere with 5% CO2 at 37°C.
2.3. Enzyme‐linked immunosorbent assay
Inhibitory activity of anlotinib against tyrosine kinases was determined using ELISA, as described previously.22 Reaction of ATP with tyrosine kinase was initiated in reaction buffer (50 mmol/L HEPES pH 7.4, 50 mmol/L MgCl2, 0.5 mmol/L MnCl2, 0.2 mmol/L Na3VO4, 1 mmol/L DTT) and incubated for 1 hour at 37°C in 96‐well plates precoated with 20 μg/mL Poly(Glu,Tyr)4:1 (Sigma). The plate was incubated with PY99 antibody and then with HRP‐conjugated anti‐mouse IgG. After reaction with o‐phenylenediamine solution and then termination with the addition of 2N H2SO4, absorbance was measured at 490 nm using a Synergy H4 Hybrid reader (Bio‐Tek Instruments, Winooski, VT, USA).
2.4. Molecular modeling
Molecular modeling calculations were based on crystallographic data for the structure 4ASD, 4AGD (for VEGFR‐2) and 3G0E (for KIT), and were carried out using Schrödinger Suite 2015‐2. Three‐dimensional structures of anlotinib and sunitinib were constructed using the Maestro module23 and prepared using the LigPrep 3.424 module in the Schrödinger suite of tools. Docking studies were carried out using Glide 6.725 in the Maestro application. Ligand docking simulations were carried out using the default standard precision scoring function and flexible docking, and adding Epik26 state penalties to docking scores in Glide. The obtained docked poses were analyzed with Maestro version 10.223 and PyMOL version 1.8.27
2.5. Western blot analysis
Cells were collected at the end of treatment and lysed in SDS sample buffer (100 mmol/L Tris‐HCl pH 6.8, 2% SDS, 20% glycerol, 1 mmol/L DTT). Cell lysates containing equal amounts of protein were separated by SDS‐PAGE and electroblotted onto PVDF membranes (Millipore). Blots were probed with specific primary antibodies and then with HRP‐conjugated secondary antibodies. Proteins were detected by immunoblotting using the western blot imaging System (Clinx Science Instruments, Shanghai, China).
2.6. Endothelial cell migration assay
HUVEC (2 × 105/mL) were suspended in M199 medium containing 1% FBS and different concentrations of drugs, and then seeded into the top Transwell chamber. M199 containing 1% FBS, with or without VEGF‐A (20 ng/mL) supplementation, was added into the lower compartment. After incubation for 8 hours at 37°C, cells that had migrated to the bottom of the membrane were fixed with 90% ethanol and stained with a 0.1% crystal violet solution. The migrated cells were then imaged using an inverted microscope (Olympus, Osaka, Japan). The dye was extracted with a 10% acetic acid solution and absorbance was measured at 590 nm.
2.7. Tube‐formation assay
An in vitro capillary tube‐formation assay was carried out as described previously.28 Briefly, Matrigel (80 μL/well) was added to a prechilled 96‐well plate and incubated at 37°C for 30 minutes. HUVEC (1 × 105/mL) were then suspended in M199 culture medium containing 20% FBS and different concentrations of drugs, and added to each well. After incubating for 6 hours, cells were imaged for capillary tube formation at high magnification (Olympus).
2.8. Rat aortic ring assay
Rat aortic ring assays were carried out as described previously.29 The thoracic aorta was dissected from male Sprague‐Dawley rats (6 weeks old) and cut into 1‐mm‐long rings. The sections were placed in a 96‐well plate embedded with Matrigel (80 μL/well) and incubated at 37°C for 1 hour. M199 medium containing VEGF‐A (50 ng/mL) and different concentrations of drugs was subsequently added to each well. After 7 days, sprouting microvessels in 5 randomly chosen fields were counted and photographed under an inverted microscope (Olympus).
2.9. Cell viability inhibition assay
Cells were seeded in 96‐well plates and treated with serial dilutions of drugs. After a 72‐hour incubation, cell proliferation was evaluated by sulforhodamine B (SRB; Sigma) assay.30 Potency of drugs in inhibiting cell proliferation was expressed as IC50 values, determined using GraphPad Prism version 5 curve‐fitting software (GraphPad Software, San Diego, CA, USA).
2.10. In vivo study
Female nude mice (Balb/cA‐nude, 5‐6 weeks old), purchased from Shanghai Laboratory Animal Center (Chinese Academy of Sciences, Shanghai, China), were housed in sterile cages under laminar airflow hoods in a specific pathogen‐free room with a 12‐hour light/12‐hour dark schedule, and fed autoclaved chow and water ad libitum. Human tumor xenografts were established by s.c. inoculating cells into the left axilla of nude mice. When tumor volumes reached 100‐200 mm3, mice were divided randomly into control and treatment groups. Control groups were given vehicle alone, and treatment groups received oral anlotinib or sunitinib daily. Tumor volume was calculated as (length × width2)/2. Tumor growth inhibition was calculated from the start of treatment by comparing changes in tumor volumes for control and treatment groups.
2.11. Immunohistochemistry
Vessel density was determined by analyzing the expression of CD31, an endothelial marker, using immunohistochemistry. Briefly, nude mice harboring SW620 tumor xenografts were treated with oral anlotinib or sunitinib daily for 14 days. Tumor sections were subsequently prepared from formalin‐fixed and paraffin‐embedded tumor tissues. Expression of CD31 in tumor tissues was assessed using a rabbit anti‐mouse antibody in conjunction with an UltraSensitive S‐P kit (Maixin‐Bio, Fuzhou, China) according to the manufacturer's instructions.
2.12. Statistical analysis
All data were expressed as means ± SD or means ± SEM. Statistical analyses were carried out using an unpaired, two‐tailed Student's t test. Differences were considered significant at P‐values <.05.
3. RESULTS
3.1. Anlotinib directly binds to VEGFR2 and strongly inhibits its activity
Inhibitory effect of anlotinib (Figure 1A) against a panel of tyrosine kinases was measured using ELISA. As shown in Table 1, anlotinib showed high selectivity for VEGF family members, especially VEGFR2 and VEGFR3, with IC50 values of 0.2 and 0.7 nmol/L, respectively. Anlotinib was 20‐fold more potent than sunitinib for inhibition of VEGF2/3, but generally exhibited inhibitory activity similar to that of sunitinib against other tyrosine kinases. The inhibitory potency of anlotinib against VEGFR1 was lower, with an IC50 value of 26.9 nmol/L. The IC50 values of anlotinib for inhibition of the PDGFR‐related kinases c‐Kit and PDGFRβ were 14.8 and 115.0 nmol/L, respectively. Anlotinib had little effect on the activity of other kinases, including c‐Met, c‐Src, EGFR and HER2, even at a concentration of 2000 nmol/L.
Table 1.
In vitro kinase inhibition profile of anlotinib
| Kinase | IC50 (nmol/L, mean ± SD) | |
|---|---|---|
| Anlotinib | Sunitinib | |
| VEGFR2 | 0.2 ± 0.1 | 4.0 ± 2.9 |
| c‐Kit | 14.8 ± 2.5 | 11.0 ± 1.5 |
| PDGFRβ | 115.0 ± 62.0 | 7.7 ± 2.2 |
| VEGFR1 | 26.9 ± 7.7 | 71.5 ± 12.8 |
| VEGFR3 | 0.7 ± 0.1 | 15.7 ± 2.1 |
| c‐Met | >2000 | >2000 |
| c‐Src | >2000 | >2000 |
| HER2 | >2000 | >2000 |
| EGFR | >2000 | >2000 |
Potency of anlotinib against recombinant tyrosine kinases in vitro, expressed as IC50. Values are presented as mean ± SD (n = 3).
EGFR, epidermal growth factor receptor; PDGFRβ, platelet‐derived growth factor receptor β; VEGFR1, vascular endothelial growth factor receptor‐1; VEGFR2, vascular endothelial growth factor receptor‐2; VEGFR3, vascular endothelial growth factor receptor‐3.
Given the high inhibitory potency of anlotinib toward VEGFR2 in enzymatic assays, we carried out a molecular docking approach to investigate the potential binding sites of anlotinib in VEGFR2 and its possible binding mode. According to previous reports, the ATP‐binding pocket of VEGFR2 is defined as including a hinge region and a hydrophobic region.31, 32, 33 As shown Figure 1B, residues of the hinge region (Cys919 and Glu917) can form hydrogen bonds with adenine mimics. The hydrophobic region lies deep in the ATP‐binding pocket, near the DFG motif (Asp1046‐Phe1047‐Gly1048). The indole group of anlotinib is located in the hydrophobic region, a region not occupied by sunitinib, indicating that anlotinib may bind deeper into the ATP‐binding pocket of VEGFR2 than sunitinib. Next, the binding modes of anlotinib in the ATP‐binding pocket of VEGFR2 were compared with that of c‐KIT. As shown in Figure 1C, the hydrophobic region of VEGFR2 is larger than that of c‐KIT; thus, binding to the indole group of anlotinib occurs deeper in this region of VEGFR2 than was the case in c‐KIT.
3.2. Anlotinib selectively inhibits VEGF‐stimulated receptor phosphorylation
Next, we further determined the effects of anlotinib on different kinds of RTK by measuring growth factor‐stimulated receptor autophosphorylation in intact cells. Ligand‐dependent kinase receptor phosphorylation was evaluated using cell lines that overexpress RTK of interest, respectively.34, 35 As shown in Figure 2A, anlotinib inhibited VEGF‐stimulated intracellular phosphorylation of VEGFR2 in a concentration‐dependent way in HUVEC with a subnanomolar IC50 value; ERK1/2, which has been reported to be downstream of VEGF,36 was phosphorylated by stimulating cells with VEGF, and was also inhibited by anlotinib. Even at a concentration of 0.1 nmol/L, anlotinib produced a clear inhibitory effect. Anlotinib inhibited SCF‐1‐stimulated phosphorylation of c‐Kit, AKT and ERK in Mo7e cells (Figure 2B). Anlotinib also inhibited PDGF‐BB‐stimulated phosphorylation of PDGFRβ, AKT and ERK in U‐87MG cells (Figure 2C). However, these inhibitory activities were lower than that of VEGFR2. Neither EGFR‐mediated signaling stimulated by EGF in A431 cells (Figure 2D) nor the constitutive HER2 signaling in BT‐474 cells was affected by anlotinib, even at a concentration of 1000 nmol/L (Figure 2E). In accordance with the kinase selectivity profile, these results indicate that anlotinib shows a high degree of selectivity for inhibition of VEGF/VEGFR2 signaling.
Figure 2.
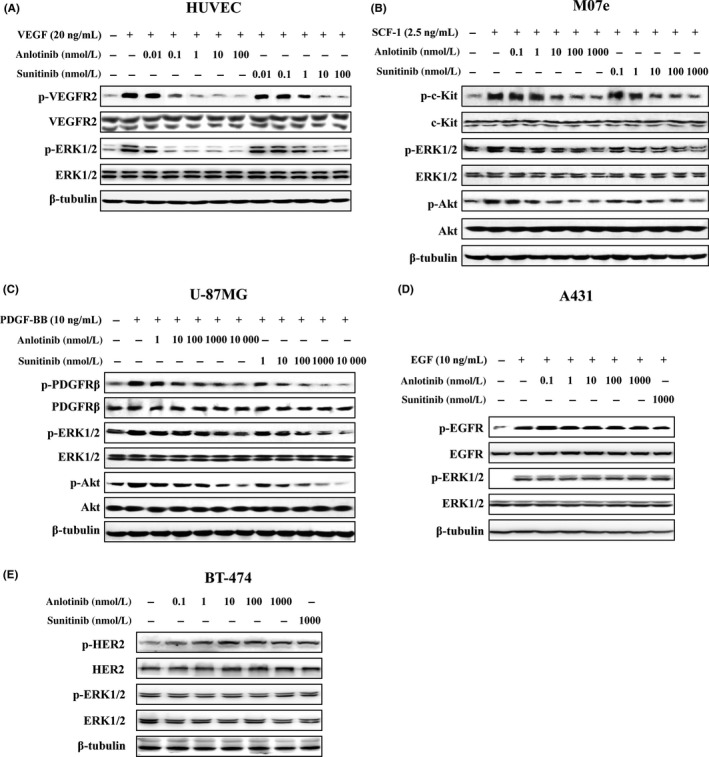
Effects of anlotinib on growth factor‐stimulated receptor phosphorylation. Serum‐starved (A) HUVEC, (B) Mo7e, (C) U‐87MG and (D) A431 cells were treated with different concentrations of test agents for 1.5 h and then stimulated with vascular endothelial growth factor (VEGF; 20 ng/mL), stem cell factor‐1 (SCF‐1; 2.5 ng/mL), platelet‐derived growth factor‐BB (PDGF‐BB; 10 ng/mL), or epidermal growth factor (EGF; 10 ng/mL) for 10 min, respectively. E, BT‐474 cells which have constitutive HER2 autophosphorylation and downstream signaling activation were treated with test agents for 1.5 h. Cell lysates were probed with the indicated antibodies
3.3. Anlotinib selectively inhibits VEGF‐stimulated proliferation of HUVEC
Given the importance of VEGF/VEGFR2 signaling in endothelial cell proliferation, we next evaluated the antiproliferative activity of anlotinib. Consistent with the aforementioned results, anlotinib potently inhibited VEGF‐stimulated HUVEC proliferation, with an IC50 value of 0.0002 μmol/L (Figure 3A). By comparison, sunitinib and sorafenib inhibited VEGF‐stimulated HUVEC proliferation with IC50 values of 0.0185 and 0.195 μmol/L, respectively. Notably, the IC50 value of anlotinib for FBS‐stimulated HUVEC proliferation was 2.0 μmol/L (Figure 3A), indicating that anlotinib selectively inhibits VEGFR‐dependent HUVEC proliferation.
Figure 3.
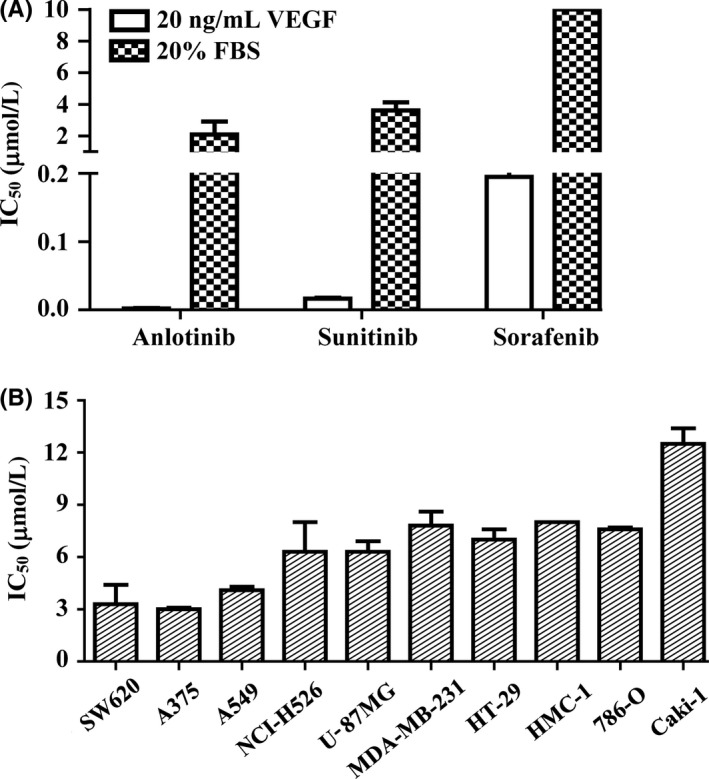
Inhibitory effects of anlotinib on cell proliferation. A, Inhibitory effect of anlotinib on vascular endothelial growth factor (VEGF)‐ or FBS‐stimulated HUVEC proliferation. HUVEC were incubated with different concentrations of drugs together with FBS (20%) or VEGF (20 ng/mL). B, Effects of anlotinib on tumor cell proliferation. Tumor cells were cultured with 10% FBS and then treated with anlotinib. Cell viability was determined by sulforhodamine B (SRB) assay. IC 50 values are presented as means ± SD of 3 independent experiments
We next examined the ability of anlotinib to inhibit tumor cell growth in vitro in a panel of tumor cells with different gene backgrounds,34, 35 focusing specifically on whether such inhibitory action was attributable to a direct antiproliferative effect. As shown in Figure 3B, anlotinib only inhibited FBS‐stimulated tumor cell proliferation at comparatively high concentrations in all these tested tumor cell lines which showed different mRNA expression levels of VEGF/VEGFR2 tested by RT‐PCR analysis (Figure S1; Data S1) or other RTK,34, 35 with IC50 values ranging from 3.0 to 12.5 μmol/L. These values are more than 10 000‐fold greater than that for VEGF‐stimulated HUVEC proliferation, suggesting that the antitumor effects of anlotinib are primarily attributable to inhibition of VEGFR2‐dependent signaling.
3.4. Anlotinib inhibits angiogenesis in in vitro bioassays
The VEGFR signaling cascade is known to play important roles in angiogenesis, a process mediated mainly by endothelial cells that results in the formation of new blood capillaries from existing vessels.37, 38 To investigate the anti‐angiogenic activity of anlotinib, we examined the effect of anlotinib on the migration of HUVEC in Transwell assays. As shown in Figure 4A, anlotinib inhibited the migration of HUVEC to the lower side of the filter in the Transwell chamber in response to VEGF stimulation. This effect was concentration dependent, with an IC50 value of 0.1 nmol/L. At a concentration of 100 nmol/L, sunitinib also significantly inhibited the migration of HUVEC.
Figure 4.
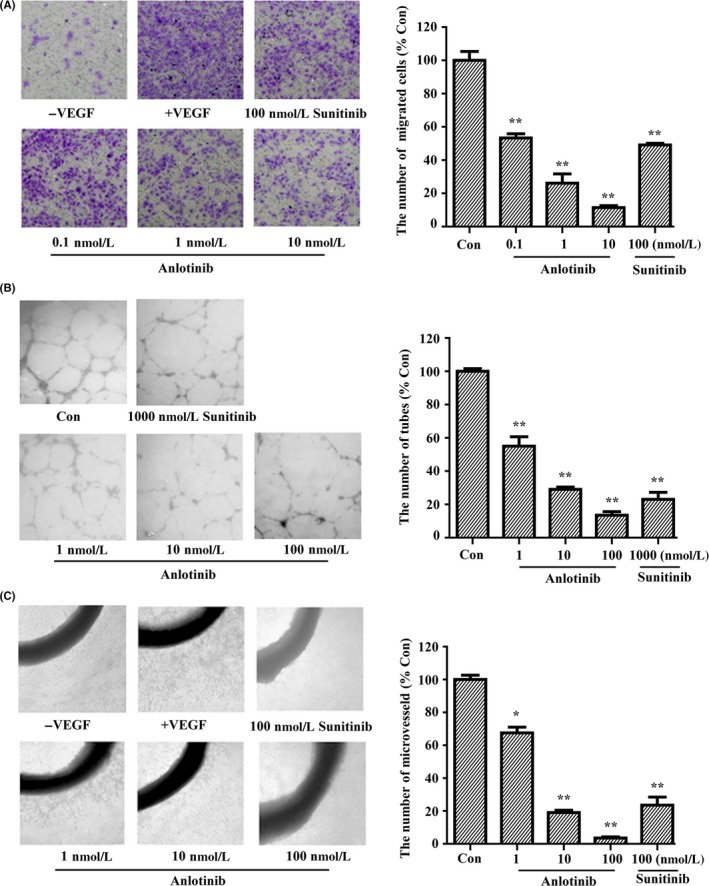
Inhibitory effects of anlotinib on angiogenesis in vitro. A, Effect of anlotinib on HUVEC migration induced by vascular endothelial growth factor (VEGF)‐A. B, Effect of anlotinib on FBS‐stimulated HUVEC tube formation. C, Effect of anlotinib on VEGF‐stimulated microvessel sprouting from rat aortic rings. Representative images are shown and data are presented as the mean ± SD of 3 independent experiments. *P < .05, **P < .01 compared with VEGF‐ or FBS‐treated groups
It has been reported that VEGF cannot induce tube formation of HUVEC when cultured on Matrigel, so 20% FBS was selected as a stimulating factor to evaluate the effect of anlotinib on the tube formation of HUVEC.34, 39 As shown in Figure 4B, anlotinib inhibited the ability of HUVEC to form tubes in a concentration‐dependent way. Following treatment with 100 nmol/L anlotinib, few enclosed tubes were detected.
Inhibition of tumor angiogenesis by anlotinib was further confirmed in the rat aortic ring culture model, which mimics several stages of angiogenesis, including endothelial cell proliferation, migration, and tube formation.28 As expected, stimulation with 50 ng/mL VEGF resulted in much greater formation of microvessels surrounding the rat aortic ring (Figure 4C). Treatment of aortic ring cultures with 1 nmol/L anlotinib inhibited microvessel formation by 38.1%; at a concentration of 100 nmol/L, anlotinib caused near complete inhibition of budding of the aortic ring.
3.5. Antitumor efficacy of anlotinib in human xenograft models
Given the encouraging anti‐angiogenic effects of anlotinib activity in vitro, we next evaluated the in vivo antitumor potential of anlotinib in the human colon cancer SW620 xenograft model. Once‐daily oral dose of anlotinib caused dose‐dependent inhibition of tumor growth (Figure 5A,C), inhibiting tumor growth by 83% compared with controls at a dose of 3 mg/kg. By comparison, the dose of sunitinib required to achieve comparable efficacy was 50 mg/kg in this model. Moreover, anlotinib had little effect on bodyweight in mice during the course of the experiment in all groups (Figure 5B). We further assessed tumor angiogenesis by measuring microvessel density in extracted tumors using an immunohistochemical analysis for CD31, an endothelial cell marker. Anlotinib induced a significant decrease in CD31‐positive microvessels, yielding inhibition rates of 48.9%, 76.3% and 91.2% at doses of 0.75, 1.5 and 3 mg/kg, respectively (Figure 5D). By comparison, sunitinib at a dose of 50 mg/kg inhibited microvessel density by 63.6%.
Figure 5.
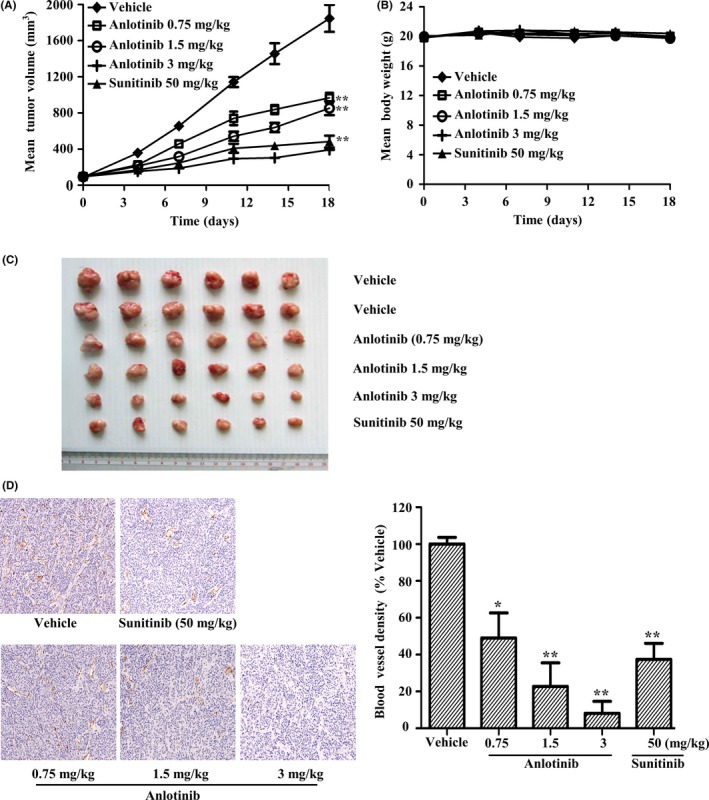
In vivo antitumor efficacy of anlotinib in SW620 tumor xenografts. (A,B) SW620 tumor‐bearing mice were orally given vehicle (n = 12) or the indicated doses of anlotinib or sunitinib (n = 6) daily for 18 d. A, Tumor volumes and (B) mouse bodyweights were determined twice weekly during the course of the experiment. C, Photographs of tumors on the final day. D, Immunohistochemical detection of the endothelial cell‐specific marker, CD31, in tumor tissue sections of SW620 xenografts. Data are presented as means ± SEM. *P < .05, **P < .01 vs vehicle
The in vivo antitumor potential of anlotinib was further investigated in multiple xenograft models, created by inoculating human cancer cell lines with the different mRNA expression levels of VEGF/VEGFR2 (Figure S1; Data S1) or other RTK.35, 40 Our study and other reports40, 41 cannot establish the relationship between VEGF/VEGFR2 expression in tumor cells and the antitumor activity of VEGFR inhibitors including anlotinib/sunitinib. Once‐daily oral dosage of anlotinib produced a dose‐dependent inhibition of tumor growth in all tumor models tested (Table 2; Figure 6). At a dose of 3 mg/kg, anlotinib inhibited tumor growth by 55%, 80%, 91% and 97% in U‐87MG, Caki‐1, Calu‐3 and SK‐OV‐3 xenografts, respectively, measured on the final treatment day. Moreover, it caused tumor regression in both Calu‐3 and SK‐OV‐3 tumor xenograft models. Treatment with a higher dose of anlotinib (6 mg/kg) inhibited tumor growth by 95% in these latter xenograft models; importantly, tumors did not rebound within 12 days after termination of anlotinib. As was the case at 3 mg/kg, anlotinib at 6 mg/kg caused tumor regression in both Calu‐3 and SK‐OV‐3 tumor xenograft models.
Table 2.
In vivo efficacy of anlotinib against a panel of human tumor xenografts
| Tumor xenograft | Tumor origin | Compound | Dose (mg/kg) | %TGI (%) | % Regression |
|---|---|---|---|---|---|
| SW620 | Colon | Anlotinib | 0.75 | 50** | — |
| Anlotinib | 1.5 | 62** | — | ||
| Anlotinib | 3 | 83** | — | ||
| Sunitinib | 50 | 78** | — | ||
| U‐87MG | Glioma | Anlotinib | 1.5 | 43** | — |
| Anlotinib | 3 | 55** | — | ||
| Anlotinib | 6 | 88** | — | ||
| Sunitinib | 50 | 44* | — | ||
| Caki‐1 | Kidney | Anlotinib | 0.75 | 48 | — |
| Anlotinib | 1.5 | 27 | — | ||
| Anlotinib | 3 | 80** | — | ||
| Sunitinib | 50 | 78** | — | ||
| SK‐OV‐3 | Ovarian | Anlotinib | 1.5 | 86** | — |
| Anlotinib | 3 | 97** | 17 | ||
| Anlotinib | 6 | 95** | 17 | ||
| Sunitinib | 50 | 88** | — | ||
| Calu‐3 | Lung | Anlotinib | 1.5 | 57** | — |
| Anlotinib | 3 | 91** | 17 | ||
| Anlotinib | 6 | 95** | 50 | ||
| Sunitinib | 50 | 92** | — |
Tumor‐bearing mice received vehicle (n = 12), anlotinib or sunitinib (n = 6) p.o. daily at the indicated doses, with the exception of anlotinib at 6 mg/kg in SK‐OV‐3 and Calu‐3 xenografts, which was orally given daily for 9 d only. Tumor growth inhibition (TGI) was calculated from the start of treatment by comparing changes in tumor volume for vehicle and treatment groups. “—” means no regression.
*P < .05, ** P < .01 compared with control groups.
Figure 6.
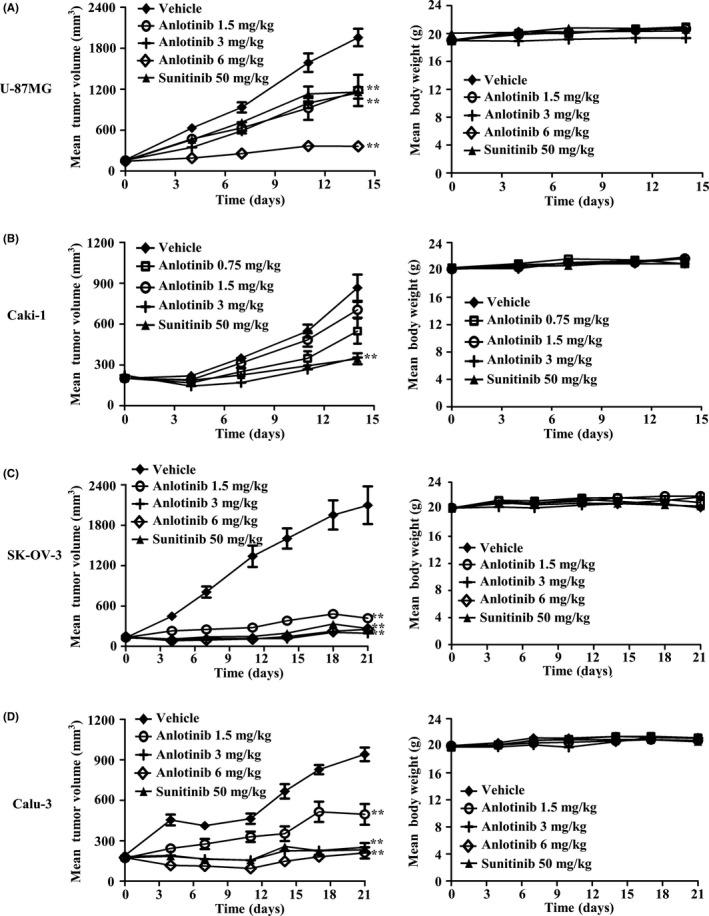
In vivo antitumor activity of anlotinib against a panel of tumor xenografts. A, U‐87MG, (B) Caki‐1, (C) SK‐OV‐3, and (D) Calu‐3 tumor‐bearing mice were orally given vehicle (n = 12) or the indicated doses of anlotinib or sunitinib (n = 6) daily. In SK‐OV‐3 and Calu‐3 xenograft models, anlotinib at 6 mg/kg was orally given daily for only 9 d. Data are presented as means ± SEM. **P < .05, **P < .01 vs vehicle
4. DISCUSSION
Angiogenesis plays a central role in tumor growth and metastasis. Among the factors involved in tumor angiogenesis, the one most closely linked to this process is VEGFR2, which drives angiogenesis through binding to its natural ligand VEGF. Neutralizing VEGF/VEGFR2 interactions with monoclonal antibodies and blocking VEGFR kinase activity with small‐molecule inhibitors are major approaches for targeting VEGF/VEGFR signaling in the treatment of solid tumors.42, 43 Several oral, non‐specific VEGFR2 TKI have been approved for cancer therapy in the past 10 years.44 Moreover, the selective anti‐VEGFR2 antibody ramucirumab was recently shown to exert beneficial effects in gastric and lung cancers.16, 17 Ultimately, it would be desirable to develop VEGFR2 inhibitors that combine the advantages of sustained target inhibition and associated long half‐life produced by antibodies with the convenient oral dosing and lower cost of small‐molecule kinase inhibitors.
In the current study, we characterized anlotinib as a highly potent and specific small‐molecule VEGFR2 inhibitor that shows significant anti‐angiogenesis and broad antitumor activity against human tumor xenografts. In assays for VEGFR2 tyrosine kinase activity, anlotinib had an IC50 value of 0.2 nmol/L, making this agent extremely potent compared with other well‐known VEGFR2 inhibitors.36 Anlotinib was also highly selective, showing a preference for inhibition of VEGFR2 compared with other tyrosine kinases (except for VEGFR3) that ranged from >74‐fold to >10 000‐fold. Most of these other clinical small‐molecule inhibitors of VEGFR2 have poor kinase selectivity. In fact, many of them inhibit more than 10 kinases with similar potency.18 Docking simulations revealed that anlotinib may form an extra interaction with VEGFR2, binding to the hydrophobic region that lies deep in the ATP‐binding pocket. As hydrophobic regions are not structurally conserved across different tyrosine kinases, the interaction with this region may account for the higher potency and selectivity of anlotinib toward VEGFR2. This improved selectivity and potency against VEGFR2 may enhance the tolerability of the drug in the clinic, and allow anlotinib to be delivered at higher doses with less toxicity. Anlotinib also showed highly potent (low nanomolar) inhibitory activity towards the VEGFR3 enzyme, which has a critical role in lymphangiogenesis.45, 46 Vascular endothelial growth factor receptor‐3 does not bind to VEGF or VEGF‐B, but binds instead to the homologous peptides, VEGF‐C and VEGF‐D. Notably, a prognostic link between expression of VEGF‐C/VEGF‐D and nodal metastasis has been identified in some tumor types.47, 48 Therefore, inhibition of VEGFR3 signaling may also have therapeutic benefit in limiting subsequent tumor dissemination.
Anlotinib inhibited VEGF‐stimulated phosphorylation of VEGFR2 and its downstream target ERK1/2 in HUVEC at low nanomolar levels, and resulted in inhibition of HUVEC proliferation in a VEGFR‐dependent way. Overall, micromolar concentrations of anlotinib were required to inhibit FBS‐stimulated proliferation of HUVEC or tumor cells in vitro, concentrations that were not attained in the in vivo setting of tumor xenograft experiments. Effective concentrations of anlotinib in plasma and tumor tissues were both at the nanomolar level—well above the concentration of anlotinib required to inhibit angiogenesis in vitro. Thus, the antiproliferative effects on vascular endothelial cells that underlie the antitumor efficacy of anlotinib likely reflect blocking VEGF/VEGFR2 signaling rather than direct cytotoxic effects on cancer cells. Accordingly, anlotinib may lead to lower toxicity compared with other well‐known receptor TKI or conventional chemotherapeutic drugs, which directly kill normal and cancerous cells by inhibiting proliferation and/or inducing apoptosis.
Endothelial cell migration and proliferation are critical events in angiogenesis.49 In the present study, anlotinib showed potent inhibition of its targets, leading to disruption of cellular processes that have been implicated in angiogenesis and tumorigenesis, including migration and tube formation. Anlotinib also inhibited microvessel outgrowth from rat aortic rings in vitro at low nanomolar levels. In an in vivo setting, this translated into profound changes in tumor physiology and disruptions in tumor vasculature. Because of their genetic instability, cancer cells easily acquire drug resistance during chemotherapy. For example, EGFR mutations in lung adenocarcinoma may cause acquired resistance to erlotinib or gefitinib,50 and rearrangements in the ALK (anaplastic lymphoma kinase) gene in advanced non‐small cell lung cancer lead to resistance to crizotinib.51 Angiogenesis is a phenomenon observed in many kinds of tumors, and endothelial cells rarely acquire resistance because of genetic stability. Thus, it is expected that anlotinib would also be effective in these multidrug‐resistant cancer patients. Notable in this context, a recent randomized, double‐blind, placebo‐controlled phase III study (NCT02388919) in patients with advanced non‐small cell lung cancer harboring EGFR or ALK mutations, and who failed previous match‐targeted therapies, showed that median overall survival following anlotinib treatment was 9.6 months compared with 6.3 months for patients in control groups.52
In the present study, we used multiple tumor xenograft models with different genetic backgrounds. Anlotinib was efficacious at doses (1.5‐6 mg/kg daily) that are significantly lower than effective doses of other TKI, which require doses of 20‐100 mg/kg to achieve significant inhibition of tumor growth in mice.53, 54 In all tumor xenograft models tested, anlotinib at a dose of 3 mg/kg showed antitumor activity comparable to that produced by the well‐known TKI sunitinib at a dose of 50 mg/kg. Importantly, tumors did not rebound after treatment with 6 mg/kg anlotinib, measured 12 days after termination of anlotinib treatment, and tumor regression was observed in some tumor xenograft models, suggesting sustained target inhibition. Our preclinical results suggest that anlotinib exerts its antitumor activity mainly by abrogating angiogenesis through specific effects on VEGFR2, and thus may lead to a wider anticancer spectrum and fewer side‐effects. Notably, it has been shown that anlotinib is clinically effective against a wide variety of tumors and shows manageable toxicity, while decreasing gastrointestinal toxicity compared with other receptor TKI.20 Hypoxia has traditionally been viewed as a consequence of malignant tumor growth, and it is now widely appreciated to play a critical role in the development and progression of tumors.41 It has been reported that long‐term treatment with VEGFR2 kinase inhibitors may cause tumor progression, partially because of hypoxia‐induced epithelial to mesenchymal transition (EMT).55, 56 However, some reports have shown that VEGFR inhibitors significantly inhibit hypoxia‐induced EMT and tumor progression.57, 58, 59 Because the preclinical study of anlotinib was conducted for a short time, the long‐term effect of anlotinib is currently unclear and further studies are needed.
In summary, our study showed that anlotinib, characterized as a novel selective VEGFR2 inhibitor, has potent anti‐angiogenic and broad‐spectrum antitumor activity in preclinical models. These favorable pharmaceutical properties support ongoing clinical evaluations of anlotinib in patients with a variety of cancers.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This research was supported by grants from the National Natural Science Foundation of China (No. 81273546) and the Shanghai Science and Technology Committee (No. 14DZ2294100).
Xie C, Wan X, Quan H, et al. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor‐2 inhibitor. Cancer Sci. 2018;109:1207–1219. https://doi.org/10.1111/cas.13536
Funding information
National Natural Science Foundation of China (Grant/Award Number: ‘81273546‘), the Shanghai Science and Technology Committee (Grant/Award Number: ‘14DZ2294100‘).
REFERENCES
- 1. Bussolino F, Mantovani A, Persico G. Molecular mechanisms of blood vessel formation. Trends Biochem Sci. 1997;22:251‐256. [DOI] [PubMed] [Google Scholar]
- 2. Harper J, Moses MA. Molecular regulation of tumor angiogenesis: mechanisms and therapeutic implications In: Bignold LP, ed. Cancer: Cell Structures, Carcinogens and Genomic Instability. Experientia Supplementum, vol. 96, Basel, Switzerland: Birkhäuser; 2006:223‐268. [DOI] [PubMed] [Google Scholar]
- 3. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653‐660. [DOI] [PubMed] [Google Scholar]
- 4. Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757‐1763. [DOI] [PubMed] [Google Scholar]
- 5. Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1‐18. [DOI] [PubMed] [Google Scholar]
- 6. Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404‐407. [DOI] [PubMed] [Google Scholar]
- 7. Al‐Husein B, Abdalla M, Trepte M, Deremer DL, Somanath PR. Antiangiogenic therapy for cancer: an update. Pharmacotherapy. 2012;32:1095‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kerbel RS. A decade of experience in developing preclinical models of advanced‐ or early‐stage spontaneous metastasis to study antiangiogenic drugs, metronomic chemotherapy, and the tumor microenvironment. Cancer J. 2015;21:274‐283. [DOI] [PubMed] [Google Scholar]
- 9. Wang Z, Dabrosin C, Yin X, et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35(suppl):S224‐S243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia‐initiated angiogenesis. Nature. 1992;359:843‐845. [DOI] [PubMed] [Google Scholar]
- 11. Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor‐induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841‐844. [DOI] [PubMed] [Google Scholar]
- 12. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669‐676. [DOI] [PubMed] [Google Scholar]
- 13. Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull. 2011;34:1785‐1788. [DOI] [PubMed] [Google Scholar]
- 15. Keating GM. Bevacizumab: a review of its use in advanced cancer. Drugs. 2014;74:1891‐1925. [DOI] [PubMed] [Google Scholar]
- 16. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro‐oesophageal junction adenocarcinoma (RAINBOW): a double‐blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224‐1235. [DOI] [PubMed] [Google Scholar]
- 17. Cooper MR, Binkowski C, Hartung J, Towle J. Profile of ramucirumab in the treatment of metastatic non‐small‐cell lung cancer. Onco Targets Ther. 2016;9:1953‐1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fontanella C, Ongaro E, Bolzonello S, Guardascione M, Fasola G, Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Transl Med. 2014;2:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elice F, Rodeghiero F. Bleeding complications of antiangiogenic therapy: pathogenetic mechanisms and clinical impact. Thromb Res. 2010;125(suppl 2):S55‐S57. [DOI] [PubMed] [Google Scholar]
- 20. Sun Y, Niu W, Du F, et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi‐target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J Hematol Oncol. 2016;9:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745‐2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lazaro I, Gonzalez M, Roy G, Villar LM, Gonzalez‐Porque P. Description of an enzyme‐linked immunosorbent assay for the detection of protein tyrosine kinase. Anal Biochem. 1991;192:257‐261. [DOI] [PubMed] [Google Scholar]
- 23. Maestro Schrödinger. New York, NY: LLC; 2015. [Google Scholar]
- 24. LigPrep Schrödinger. New York, NY: LLC; 2015. [Google Scholar]
- 25. Glide Schrödinger. New York, NY: LLC; 2015. [Google Scholar]
- 26. Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK a prediction and protonation state generation for drug‐like molecules. J Comput Aided Mol Des. 2007;21:681‐691. [DOI] [PubMed] [Google Scholar]
- 27. Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015.
- 28. Min JK, Han KY, Kim EC, et al. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004;64:644‐651. [DOI] [PubMed] [Google Scholar]
- 29. Lee CC, Liu KJ, Wu YC, Lin SJ, Chang CC, Huang TS. Sesamin inhibits macrophage‐induced vascular endothelial growth factor and matrix metalloproteinase‐9 expression and proangiogenic activity in breast cancer cells. Inflammation. 2011;34:209‐221. [DOI] [PubMed] [Google Scholar]
- 30. Xie CY, Xu YP, Jin W, Lou LG. Antitumor activity of lobaplatin alone or in combination with antitubulin agents in non‐small‐cell lung cancer. Anticancer Drugs. 2012;23:698‐705. [DOI] [PubMed] [Google Scholar]
- 31. Li J, Zhou N, Luo K, et al. In silico discovery of potential VEGFR‐2 inhibitors from natural derivatives for anti‐angiogenesis therapy. Int J Mol Sci. 2014;15:15994‐16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanphanya K, Wattanapitayakul SK, Phowichit S, Fokin VV, Vajragupta O. Novel VEGFR‐2 kinase inhibitors identified by the back‐to‐front approach. Bioorg Med Chem Lett. 2013;23:2962‐2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang TH, Lee CI, Huang WH, Lee AR. Synthesis and evaluation of Novel 2‐pyrrolidone‐fused (2‐oxoindolin‐3‐ylidene)methylpyrrole derivatives as potential multi‐target tyrosine kinase receptor inhibitors. Molecules. 2017;22:913‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian S, Quan H, Xie C, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor‐2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102:1374‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor‐2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389‐4400. [DOI] [PubMed] [Google Scholar]
- 36. Amino N, Ideyama Y, Yamano M, et al. YM‐359445, an orally bioavailable vascular endothelial growth factor receptor‐2 tyrosine kinase inhibitor, has highly potent antitumor activity against established tumors. Clin Cancer Res. 2006;12:1630‐1638. [DOI] [PubMed] [Google Scholar]
- 37. Shahneh FZ, Baradaran B, Zamani F, Aghebati‐Maleki L. Tumor angiogenesis and anti‐angiogenic therapies. Hum Antibodies. 2013;22:15‐19. [DOI] [PubMed] [Google Scholar]
- 38. Petrova TV, Makinen T, Alitalo K. Signaling via vascular endothelial growth factor receptors. Exp Cell Res. 1999;253:117‐130. [DOI] [PubMed] [Google Scholar]
- 39. Ilan N, Mahooti S, Madri JA. Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J Cell Sci. 1998;111:3621‐3631. [DOI] [PubMed] [Google Scholar]
- 40. Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327‐337. [PubMed] [Google Scholar]
- 41. Heinrich EL, Walser TC, Krysan K, et al. The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron. 2012;5:5‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312:1171‐1175. [DOI] [PubMed] [Google Scholar]
- 43. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58‐62. [DOI] [PubMed] [Google Scholar]
- 44. De Falco S. Antiangiogenesis therapy: an update after the first decade. Korean J Intern Med. 2014;29:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karkkainen MJ, Haiko P, Sainio K, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74‐80. [DOI] [PubMed] [Google Scholar]
- 46. Arinaga M, Noguchi T, Takeno S, Chujo M, Miura T, Uchida Y. Clinical significance of vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 in patients with nonsmall cell lung carcinoma. Cancer. 2003;97:457‐464. [DOI] [PubMed] [Google Scholar]
- 47. Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kuroda T, Chayama K. Regulation of vascular endothelial growth factor (VEGF)‐C and VEGF‐D expression by the organ microenvironment in human colon carcinoma. Eur J Cancer. 2004;40:1604‐1609. [DOI] [PubMed] [Google Scholar]
- 48. Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K. Expression of VEGF‐C and VEGF‐D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci. 2004;95:32‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Y, Feng L, Wang S, et al. Calycosin protects HUVECs from advanced glycation end products‐induced macrophage infiltration. J Ethnopharmacol. 2011;137:359‐370. [DOI] [PubMed] [Google Scholar]
- 50. Han JY, Lee KH, Kim SW, et al. A phase II study of poziotinib in patients with epidermal growth factor receptor (EGFR)‐mutant lung adenocarcinoma who have acquired resistance to EGFR‐tyrosine kinase inhibitors. Cancer Res Treat. 2017;49:10‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Casaluce F, Sgambato A, Sacco PC, et al. Resistance to crizotinib in advanced non‐small cell lung cancer (NSCLC) with ALK rearrangement: mechanisms, treatment strategies and new targeted therapies. Curr Clin Pharmacol. 2016;11:77‐87. [DOI] [PubMed] [Google Scholar]
- 52. Han B, Li K, Wang Q, et al. Efficacy and safety of third‐line treatment with anlotinib in patients with refractory advanced non‐small‐cell lung cancer (ALTER‐0303): a randomised, double‐blind, placebo‐controlled phase 3 study. Lancet Oncol. 2017;18:S3. [Google Scholar]
- 53. Wood JM, Bold G, Buchdunger E, et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor‐induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178‐2189. [PubMed] [Google Scholar]
- 54. Beebe JS, Jani JP, Knauth E, et al. Pharmacological characterization of CP‐547,632, a novel vascular endothelial growth factor receptor‐2 tyrosine kinase inhibitor for cancer therapy. Cancer Res. 2003;63:7301‐7309. [PubMed] [Google Scholar]
- 55. Mizumoto A, Yamamoto K, Nakayama Y, et al. Induction of epithelial‐mesenchymal transition via activation of epidermal growth factor receptor contributes to sunitinib resistance in human renal cell carcinoma cell lines. J Pharmacol Exp Ther. 2015;355:152‐158. [DOI] [PubMed] [Google Scholar]
- 56. Marijon H, Dokmak S, Paradis V, et al. Epithelial‐to‐mesenchymal transition and acquired resistance to sunitinib in a patient with hepatocellular carcinoma. J Hepatol. 2011;54:1073‐1078. [DOI] [PubMed] [Google Scholar]
- 57. Martinelli E, Troiani T, Morgillo F, et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin Cancer Res. 2010;16:4990‐5001. [DOI] [PubMed] [Google Scholar]
- 58. Li Y, Yang X, Su LJ, Flaig TW. VEGFR and EGFR inhibition increases epithelial cellular characteristics and chemotherapy sensitivity in mesenchymal bladder cancer cells. Oncol Rep. 2010;24:1019‐1028. [DOI] [PubMed] [Google Scholar]
- 59. Sosman JA, Puzanov I, Atkins MB. Opportunities and obstacles to combination targeted therapy in renal cell cancer. Clin Cancer Res. 2007;13:764s‐769s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials