

Abstract
Tunicamycin (TM) is an N‐linked glycosylation (NLG) inhibitor with strong antitumor activity, the exact underlying molecular mechanism of which remains to be elucidated. In our previous studies, we found that TM reversed drug resistance and improved the efficacy of combination treatments for hepatocellular carcinomas (HCC). Here, we investigated the effects of TM on HCC cell proliferation and migration as well as the mechanism of those effects. Our results showed that TM inhibited cell proliferation and migration as well as induced apoptosis of hepatocellular carcinoma cells. TM inhibited proliferation of HCC cells by inducing cell apoptosis and cell cycle arrest at the G2/M phase. Meanwhile, TM inhibited migration of HCC cells by suppressing CD44s‐mediated epithelial‐mesenchymal transition (EMT). TM inhibited migration and invasion of HCC cells by decreasing CD44 expression and altering its glycosylation. In addition, CD44s is involved in promoting EMT and is associated with a poor prognosis in HCC patients. Overexpression of CD44s promoted tumor migration and activated phosphorylation of ERK1/2 in HCC cells, whereas TM inhibited CD44s overexpression‐associated cell migration. The ability of TM to inhibit cell migration and invasion was enhanced or reversed in CD44s knockdown cells and cells overexpressing CD44s, respectively. The MEK/ERK inhibitor U0126 and TM inhibited hyaluronic acid‐induced cell migration in HCC cells. Furthermore, TM inhibited exogenous transforming growth factor beta (TGF‐β)‐mediated EMT by an ERK1/2‐dependent mechanism and restored the TGF‐β‐mediated loss of E‐cadherin. In summary, our study provides evidence that TM inhibits proliferation and migration of HCC cells through inhibition of CD44s and the ERK1/2 signaling pathway.
Keywords: CD44s, EMT, ERK1/2, hepatocellular carcinoma, tunicamycin
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most prevalent and the third most deadly type of malignant tumor worldwide. Surgical resection and liver transplantation are available options for the treatment of early‐stage HCC.1 However, the prognosis of HCC remains poor because of a high level of frequent intrahepatic spread and extrahepatic metastasis.2 Cancer cell migration is one of the crucial events in HCC metastasis.3 Therefore, identification of factors that affect migration and elucidation of the underlying molecular mechanism involved in the progression of metastasis have become critical concerns.
Acquisition of migratory and invasive capability is the initial step in metastasis. Epithelial‐mesenchymal transition (EMT) is proposed to be a crucial mechanism that regulates the initial steps of metastatic progression.4 CD44, a major adhesion molecule of the extracellular matrix, has been implicated in a wide variety of physiological processes, including leukocyte homing and activation, wound healing, and cell migration.5 A previous study showed that CD44 alternative splicing is differentially regulated during EMT, resulting in a switch in expression from the variable exon‐containing CD44v isoform to the standard isoform, CD44s, which is devoid of all CD44 variable exons.6 Furthermore, a previous study showed that up‐regulation of CD44 isoforms is associated with poorly differentiated HCC and shortened survival.7, 8
N‐Linked glycosylation (NLG) is believed to play a central role in sustaining protein stability, trafficking, and function. Tunicamycin (TM) potently inhibits NLG by competitively inhibiting DPAGT1 activity and can target several types of tumors by reducing angiogenesis, inhibiting colony formation, and enhancing TRAIL‐induced apoptosis.9 Recently, we found that TM can reverse drug resistance and improve the efficacy of combination treatments for HCC by targeting the DPAGT1/Akt/ABCG2 pathway.10 However, the effects of TM on the proliferation and migration of HCC cells are unknown.
In the present study, we investigated the effect of TM on the proliferation and migration of HCC cells as well as the underlying molecular mechanisms of these processes. We found that TM inhibits the proliferation and migration of HCC cells through CD44s and the ERK1/2 pathway.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
MHCC‐97L and MHCC‐97H were kindly provided by the Liver Cancer Institute of Zhongshan Hospital, Fudan University (Shanghai, China). SMMC‐7721 and L02 were obtained from the Cell Bank of the Institute of Biochemistry and Cell Biology, China Academy of Sciences (Shanghai, China). HEK‐293T was purchased from the ATCC (Manassas, VA, USA). Human primary HCC‐LY5 cell lines established in our laboratory were cultured in Williams’ Medium E supplemented with 10% FBS. The other HCC cell lines used in this study were cultured in DMEM (Sigma‐Aldrich, St Louis, MO, USA) containing 10% heat‐inactivated FBS (HyClone) and incubated at 37°C in a humidified atmosphere with 5% CO2. Chemicals and other reagents were purchased from Sigma‐Aldrich unless otherwise specified.
2.2. Cell cycle analysis
For cell cycle analysis, cells were plated in a 6‐well culture plate and grown for 24 hours. Cells were then incubated with 1 mM thymidine (Sigma‐Aldrich) or 100 ng/mL nocodazole for 24 hours to synchronize cells at the G1/S or G2/M boundary. Cells were then treated with fresh media containing 2.5 μg/mL TM for different lengths of time. Next, cells were trypsinized, washed twice with cold PBS and fixed with cold 70% ethanol at −20°C overnight. Cells were then washed twice with PBS and incubated with 10 mg/mL RNase A, 400 mg/mL propidium iodide and 0.1% Triton X in PBS at room temperature (RT) for 30 minutes. Cells were subsequently analyzed by flow cytometry.
2.3. Apoptotic assay
Annexin V and 7‐AAD staining was used to visualize apoptotic cells according to the manufacturer's instructions. Briefly, cells were seeded in 6‐well plates and treated with TM at 2.5 μg/mL for 24 hours. Cells were then collected, washed twice with PBS and resuspended in 400 mL of 1 × binding buffer. Next, 5 mL Annexin V‐PE and 7‐AAD solution was added, and the samples were incubated for 15 minutes at RT and analyzed by flow cytometry.
2.4. MTT assay
Cells (5000/well) were seeded on 96‐well plates for 24 hours and then exposed to different concentrations of TM. After incubation for 48 hours, 20 mL of 5 mg/mL MTT was added to the medium, and cells were incubated at 37°C for another 4 hours. Then the culture medium was discarded and 100 mL DMSO was added to each well to dissolve the precipitate. Absorbance (A) was measured at 570 nm using an ELISA plate reader, with background subtraction measurements done at 630 nm. The inhibition rate was calculated as follows:
2.5. Western blotting
Cell lysates were separated by SDS‐PAGE and transferred onto nitrocellulose membranes. The membranes were incubated with a primary antibody overnight at 4°C and subsequently probed with HRP‐conjugated secondary antibodies. Immunoreactive blots were visualized using an ECL reagent (Pierce, Rockford, IL, USA). Antibody information is listed in Table S1.
2.6. Quantitative real‐time RT‐PCR
Total RNA extraction, reverse transcription, and quantitative real‐time RT‐PCR (qRT‐PCR) analyses were carried out as previously described using an ABI Prism 7500 System (Applied Biosystems, Carlsbad, CA, USA) with SYBR® Premix Ex Taq (Takara, Dalian, China).11 Primer information is provided in Table S2.
2.7. Vector constructs, lentivirus production, and cell transduction
The CD44s ORF sequence was PCR amplified using specific primers (forward: 5′‐ ATGGACAAGTTTTGGTGGCAC‐3′; reverse: 5′‐TTACACCCCAATCTTCATGTCC‐3′) and cloned into the lentiviral expression vector pWPXL (Addgene, Cambridge, MA, USA) by replacing the GFP fragment. Viral packaging was carried out in HEK‐293T cells after cotransfection of the pWPXL‐CD44s vector with the packaging plasmid psPAX2 and envelope plasmid pMD2.G (Addgene) using Lipofectamine 2000 (Invitrogen). The viruses were harvested 72 hours after transfection, and the viral titers were determined. MHCC‐97L cells were infected with 1 × 106 recombinant lentivirus‐transducing units in the presence of 6 μg/mL polybrene (Sigma).
2.8. RNA interference
Small‐interfering RNA (siRNA) oligos targeting CD44s and a negative control were synthesized and annealed by GenePharma (Shanghai, China). Two target sequences of CD44s siRNA, 518 and 688, were selected. These sequences were: 5′‐CCGCTTTGCAGGTGTATTC‐3′; and 5′‐AAATGGTCGCTACAGCATC‐3′, respectively. An siRNA with a non‐targeting sequence (scrambled sequence) was used as a negative control (shNC) in our experiment. RNA interference was carried out as described previously.11
2.9. In vitro migration and invasion assays
For in vitro migration and invasion assays, cells were seeded onto the upper chamber of a transwell or on a Matrigel‐coated transwell (BD Biosciences, Franklin Lakes, NJ, USA) in serum‐free media. The lower chamber contained DMEM with 10% FBS as a chemoattractant. After 12 or 48 hours of incubation, non‐migrated cells were gently removed from the upper chamber with a cotton swab. Cells were fixed and stained using Giemsa solution and counted in 5 randomly chosen visual fields.
2.10. Statistical analysis
Statistical analyses were carried out using SPSS 16.0 software. All data are presented as the mean ± SD. Two‐group comparisons were analyzed using the two‐tailed Student's t test. Three or more group comparisons were analyzed using one‐way ANOVA. P < .05 was considered statistically significant.
3. RESULTS
3.1. TM suppresses the proliferation of HCC cells by inducing G2/M arrest
To determine the effects of TM on the proliferation of cells, HCC cells were treated with different concentrations of TM for 48 hours. Cell proliferation was determined by the MTT assay. As shown in Figure 1A, TM inhibited cell proliferation of HCC cell in a dose‐dependent way. To exclude the possibility that the effect of TM on cell proliferation was occurring as a result of drug toxicity, the effect of TM on the immortalized human liver cell line L02 was also investigated. Results showed that L02 cells had a lower sensitivity compared to HCC cells treated with TM (Figure 1A).
Figure 1.
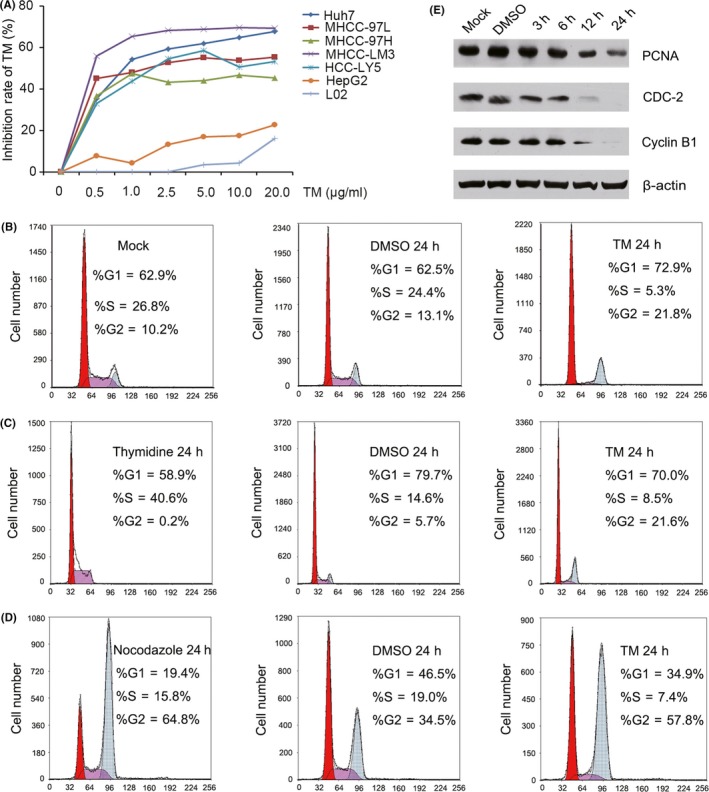
Effect of tunicamycin (TM) on hepatocellular carcinoma (HCC) cell growth and cell cycle‐related protein expression. A, Growth inhibition rates of Huh7, MHCC‐97L, MHCC‐97H, MHCC‐LM3, HCC‐LY5, HepG2 and L02 cells resulting from treatment with TM for 48 h. B, Cell cycle distribution of MHCC‐97L cells that were treated with 2.5 μg/mL TM for 48 h. C, After the cells were synchronized with thymidine, cell cycle distribution of MHCC‐97L cells was determined by flow cytometry. D, After the cells were synchronized with nocadazole, cell cycle distribution of MHCC‐97L cells was determined by flow cytometry. E, Expression of proliferating cell nuclear antigen (PCNA), CDC‐2 and cyclin B1 was detected by western blotting in MHCC‐97L cells treated with 2.5 μg/mL TM for the indicated length of time
To further investigate the mechanism by which TM affects HCC proliferation, cell cycle distributions of MHCC‐97L cells were determined by flow cytometry. Our results showed that TM induced G2/M arrest in MHCC‐97L cells (Figure 1B). To further confirm the effects of TM on the cell cycle, MHCC‐97L cells were synchronized with thymidine or nocadazole. Results showed that TM induced G2/M arrest in MHCC‐97L cells (Figure 1C,D). In addition, we also determined the expression of proteins related to G2/M cell cycle arrest. Our results showed that TM inhibited CDC‐2, cyclin B1 and proliferating cell nuclear antigen (PCNA) expression in a dose‐dependent way (Figure 1E). Therefore, these results showed that TM suppresses the proliferation of HCC cell by inducing G2/M arrest.
3.2. Tunicamycin induces HCC cell apoptosis by Bcl‐2 family proteins
Apoptosis is also widely believed to be the major antiproliferative mechanism of anticancer drugs in many tumor cell types. Therefore, we also investigated the effect of TM on HCC cell apoptosis. Increased apoptosis was observed in MHCC‐97L cells treated with TM, implying that an increased rate of apoptosis could be one of the mechanisms of TM inhibition of cell proliferation (Figure 2A). To understand the mechanism by which TM induces cell apoptosis, we assessed the expression of Bcl‐2 family proteins using western blotting. Results showed that the proapoptotic Bcl‐2 family proteins Bim and Bid were up‐regulated, and that the concomitant anti‐apoptosis proteins Bcl‐xL and Mcl‐1 were down‐regulated in TM‐treated MHCC‐97L cells (Figure 2B). However, the expression of Bcl‐2 and PDCD4 did not change after TM treatment.
Figure 2.
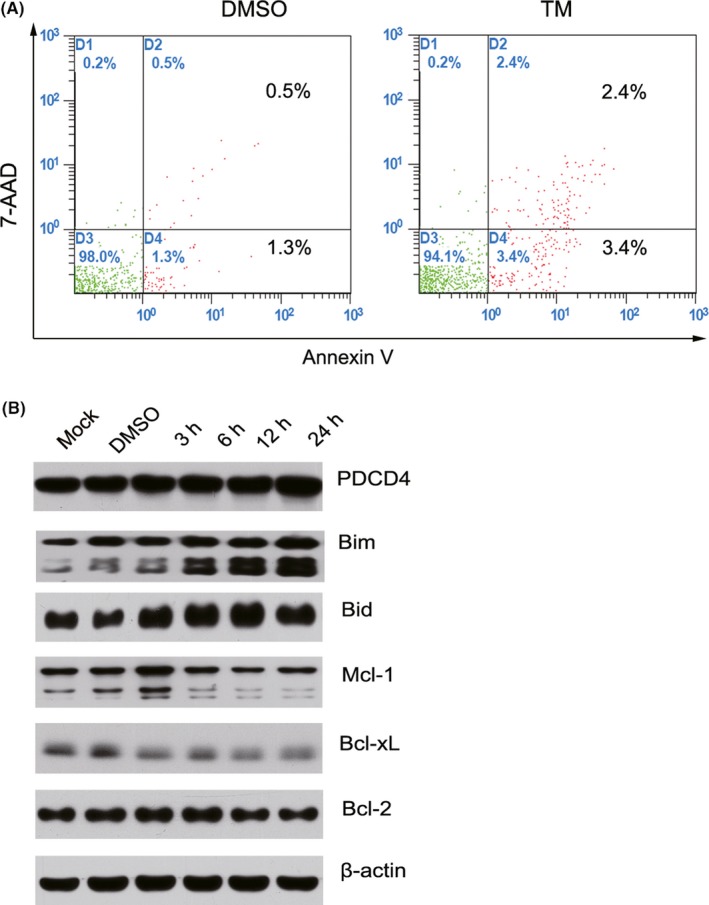
Tunicamycin (TM) induces cell apoptosis and regulates the expression of Bcl‐2 family proteins in hepatocellular carcinoma (HCC) cells. A, Following treatment of MHCC‐97L cells with 2.5 μg/mL TM for 48 h, apoptotic cells were detected by Annexin V and 7‐AAD double staining. B, Expression of PDCD4, Bim, Bid, Mcl‐1, Bcl‐xL and Bcl‐2 was detected by western blotting in MHCC‐97L cells treated with 2.5 μg/mL TM for the indicated length of time
3.3. Tunicamycin inhibits HCC cell migration through CD44s and the ERK1/2 pathway
Cancer cell migration is one of the crucial events in HCC metastasis. Therefore, we detected the effect of TM on cell migration. Our results showed that TM inhibited HCC cell migration (Figure 3A).
Figure 3.
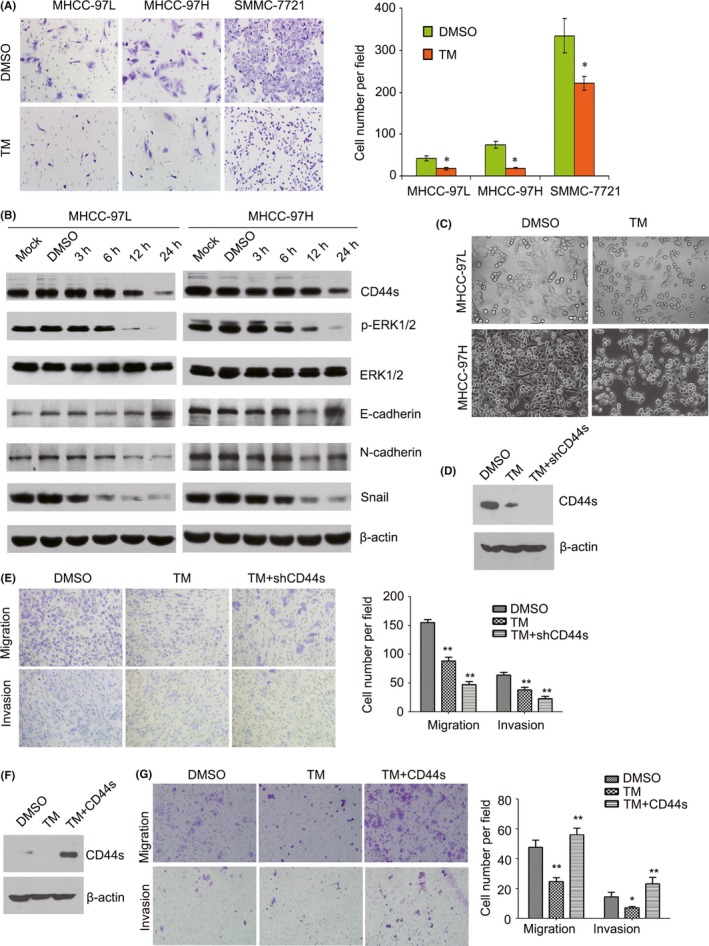
Tunicamycin (TM) inhibits hepatocellular carcinoma (HCC) cell migration through the CD44s and ERK1/2 pathway. A, Following treatment of MHCC‐97L, MHCC‐97H and SMMC‐7721 cells with 2.5 μg/mL TM for 48 h, cell migration was detected by transwell assay. *P < .05. B, Expression of CD44s, p‐ERK1/2, ERK1/2, E‐cadherin, N‐cadherin and Snail was detected by western blotting in MHCC‐97L and MHCC‐97H cells treated with 2.5 μg/mL TM for the indicated length of time. C, Phase‐contrast images (200×) show morphological changes in TM‐treated MHCC‐97L and MHCC‐97H cells and control cells. D, Expression of CD44s was detected by western blotting in CD44s knockdown MHCC‐97L cells or control cells treated with 2.5 μg/mL TM for 12 h. E, Migration and invasion were measured using transwell assays with CD44s knockdown MHCC‐97L cells or control cells treated with 2.5 μg/mL TM. **P < .01. F, Expression of CD44s was detected by western blotting in CD44s overexpressing PLC/PRF/5 cells or control cells treated with 2.5 μg/mL TM for 12 h. G, Migration and invasion were measured using transwell assays with CD44s overexpressing PLC/PRF/5 cells or control cells treated with 2.5 μg/mL TM. *P < .05; **P < .01
The adhesion molecule CD44 plays a role in cell migration, metastasis and EMT.12 Therefore, we detected the effect of TM on CD44s expression in HCC cells. The results showed that TM inhibited expression of CD44s in a time‐dependent way in MHCC‐97L and MHCC‐97H cells (Figure 3B). In addition, we found that TM treatment of cells partially resulted in morphological changes from scattered growth structures to tightly packed colonies, suggesting the inhibition of EMT (Figure 3C). To explore the role of CD44s in HCC migration, we first detected the migratory potential of several HCC cell lines using a BD transwell chamber. The results showed that the rates of cell migration were much higher in Huh7, MHCC‐97L, MHCC‐97H and SMMC‐7721 cells than in PLC/PRF/5 and Hep3B cells (Figure 4A). Notably, most of the highly metastatic cell lines (Huh7, MHCC‐97L, MHCC‐97H and SMMC‐7721) expressed higher levels of CD44s as well as the EMT‐related markers Snail and N‐cadherin, whereas the low CD44s‐expressing cell lines PLC/PRF/5 and Hep3B showed high expression levels of E‐cadherin, as determined by western blotting and real‐time PCR (Figure 4B; Figure S1). Furthermore, we analyzed the expression levels of CD44 (total CD44) and Snail (a major transcription factor governing EMT) using data sets from TCGA (The Cancer Genome Atlas) and GEO (Gene Expression Omnibus). Our results showed that patients in the high CD44 expression group had shorter overall survival time than patients in the low expression group (log‐rank, P = .017, Figure 5A) in publicly available gene expression array datasets (TCGA) using prognostic database PROGgene V2.13 In addition, we found that there was a significant positive correlation between the expression of CD44 and Snail in HCC tissue using data sets from TCGA and GEO (Figure 5B). Furthermore, we found that there was a significant negative correlation between the expression of CD44 and E‐cadherin in HCC tissue using data sets from TCGA and GEO (Figure 5C). Therefore, all of these results suggested that high levels of CD44s expression are related to a high metastatic potential and EMT.
Figure 4.
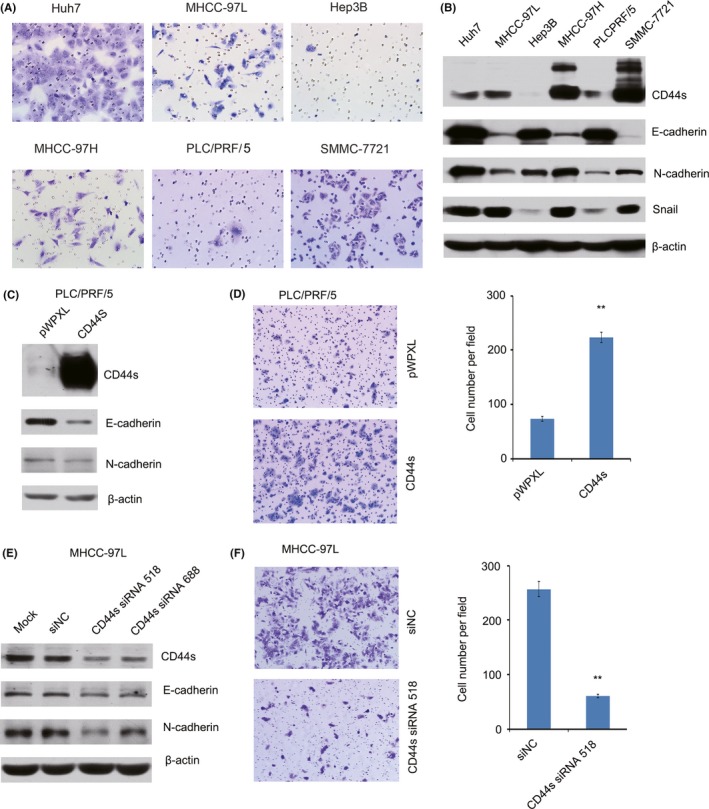
CD44s expression is associated with a mesenchymal phenotype and migratory ability in hepatocellular carcinoma (HCC) cells. A, Highly metastatic cell lines have higher migration potential than less metastatic cell lines. B, Expression of CD44s, E‐cadherin, N‐cadherin and Snail was detected by western blotting in HCC cell lines. C, Expression levels of CD44s, E‐cadherin and N‐cadherin were detected by western blotting in CD44s‐overexpressing PLC/PRF/5 cells. D, Cell migration was measured using transwell assays in CD44s‐overexpressing PLC/PRF/5 cells. E, Expression levels of CD44s, E‐cadherin and N‐cadherin were detected by western blotting in CD44s‐knockdown MHCC‐97L cells. F, Cell migration was measured using transwell assays in CD44s‐knockdown MHCC‐97L cells. **P < .01
Figure 5.
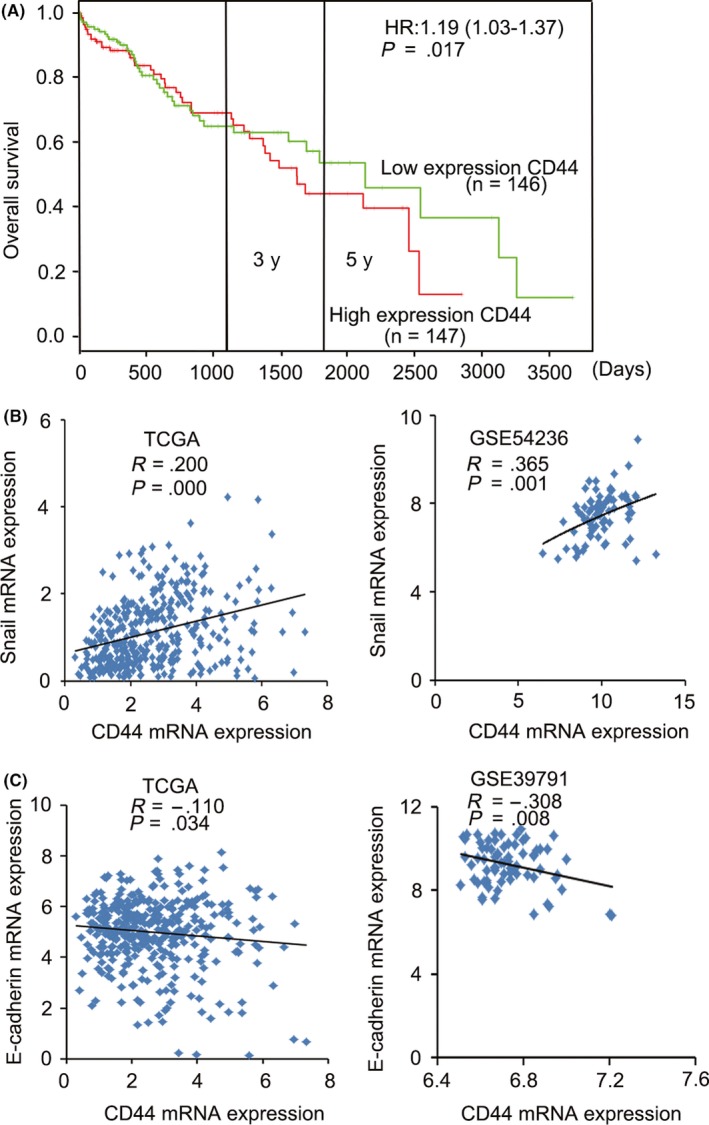
CD44 is associated with poor prognosis in hepatocellular carcinoma (HCC) patients. A, Patients with high expression levels of CD44 had shorter overall survival than patients with low expression levels. B, Correlation between CD44 expression and level of Snail in HCC tissues was analyzed using data sets from TCGA (The Cancer Genome Atlas) and GEO (Gene Expression Omnibus). C, Correlation between CD44 and E‐cadherin expression in HCC tissues was analyzed using data sets from TCGA and GEO
To investigate whether the migratory potential of HCC cells was determined by CD44s, a CD44s expression vector was stably transfected into PLC/PRF/5 cells. Overexpression of CD44s increased the in vitro migration of the transfected cells (Figure 4C,D). Conversely, CD44s knockdown inhibited cell migration in HCC cells (Figure 4E,F). To confirm the role of CD44s in TM‐induced inhibition of the migration of HCC cells, a CD44s siRNA was transfected into MHCC‐97L cells. Our results showed that the ability of TM to inhibit cell migration and invasion was enhanced in CD44s knockdown cells (Figure 3D,E). To exclude the role of endogenous CD44s, a CD44s expression vector was stably transfected into PLC/PRF/5 cells (low expression of CD44s). Conversely, the ability of TM to inhibit cell migration and invasion was reversed in cells overexpressing CD44s (Figure 3F,G). Furthermore, results showed that up‐regulation of E‐cadherin and concomitant down‐regulation expression of N‐cadherin as well as the EMT‐related transcription factor Snail was observed in TM treated cells (Figure 3B). Therefore, these results indicated that TM inhibited cell migration by regulating CD44s‐mediated EMT in HCC cells.
The ERK pathway has been implicated in malignant transformation and in the regulation of cellular proliferation and metastasis of several tumor types.14, 15 Furthermore, we found that overexpression of CD44s increased ERK1/2 activation in HCC cells (Figure S2). Therefore, we detected the expression of ERK1/2 in TM‐treated HCC cells. Results showed that TM inhibited ERK1/2 activation in a time‐dependent way in MHCC‐97L and MHCC‐97H cells (Figure 3B). Hyaluronic acid (HA) is a major component of the extracellular matrix and can bind to its specific cell surface receptor, CD44.16 Our results showed that HA increased ERK1/2 activation in a dose‐dependent way in MHCC‐97L cells (Figure 6A). The MEK/ERK inhibitor U0126 inhibited HA‐induced cell migration in MHCC‐97L cells. Furthermore, TM also suppressed HA‐induced cell migration in MHCC‐97L cells (Figure 6B,C). Therefore, these results suggested that TM inhibited migration of cells through the CD44s‐ERK1/2 pathway and inhibition of EMT.
Figure 6.
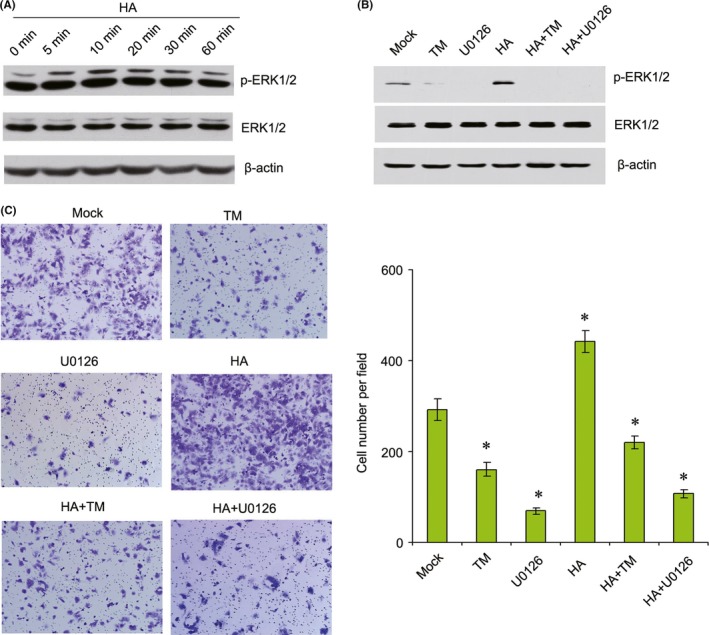
Tunicamycin (TM) inhibits hyaluronic acid (HA)‐induced cell migration by suppressing ERK1/2 activity in MHCC‐97L cells. A, Expression levels of p‐ERK1/2 and ERK1/2 were detected by western blotting in MHCC‐97L cells treated with 50 μg/mL HA for the indicated length of time. B, Expression levels of p‐ERK1/2 and ERK1/2 were detected by western blotting in MHCC‐97L cells treated with 2.5 μg/mL TM, 50 μg/mL HA or 10 μmol/L U0126 alone or in combination for 12 h. C, MHCC‐97L cells were treated with 2.5 μg/mL TM, 50 μg/mL HA or 10 μmol/L U0126 alone or in combination for 12 h, and migration was measured using transwell assays. *P < .05
3.4. Tunicamycin inhibits CD44s glycosylation and reduced its expression
CD44 is a glycosylated protein localized to the apical domain of plasma membranes.17 Following digestion with the glycosidase PNGase F, western blotting using an anti‐CD44 antibody showed the appearance of a new band of approximately 70 kDa, showing that CD44 is glycosylated in HCC cells (Figure 7A,B). Then, we detected the expression of CD44s in TM‐treated cells. A band of about 70 kDa was also observed in TM‐treated cells (Figure 7A,B). These results showed that the inhibitory effect of TM on the proliferation and migration of HCC cells was partly a result of inhibition of CD44s glycosylation. In addition, we also detected the expression of CD44s mRNA in TM‐treated HCC cells. Our results showed that TM suppressed the expression of CD44s mRNA in HCC cells (Figure 7C). Therefore, these results suggest that TM inhibits the proliferation and migration of HCC cells by decreasing CD44s expression and altering its glycosylation.
Figure 7.
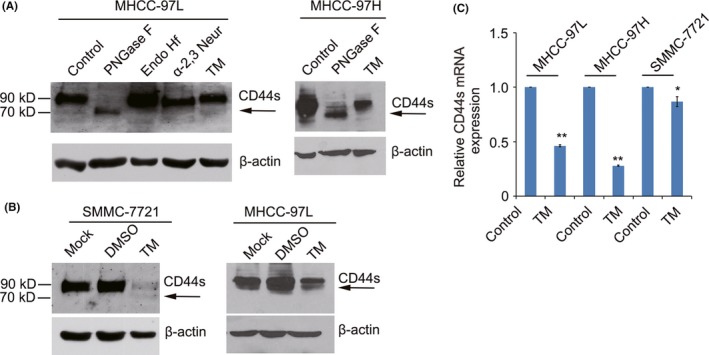
Tunicamycin (TM) inhibits CD44 expression and alters its glycosylation. A, MHCC‐97L and MHCC‐97H cell lysates were treated with PNGase F, Endo Hf, or α‐2,3‐neuraminidase at 37°C according to the datasheets. Shift in CD44 molecular weight was detected by western blotting. B, Expression level of CD44s was detected by western blotting in SMMC‐7721 and MHCC‐97L cells treated with 2.5 μg/mL TM for 24 h. C, Expression of CD44s was detected by real‐time PCR in MHCC‐97L, MHCC‐97H and SMMC‐7721 cells treated with 2.5 μg/mL TM for 24 h. *P < .05; **P < .01
3.5. Tunicamycin inhibits transforming growth factor beta‐mediated cell migration
Because transforming growth factor beta (TGF‐β) signaling is one of the major driving factors of EMT in HCC,18 we speculated that inhibition of TM during EMT might be attributed to a blockade of endogenous TGF‐β signaling. Our results showed that TGF‐β induced EMT of HCC cells by increasing expression of N‐cadherin and Snail, decreasing E‐cadherin expression. At the same time, expression of CD44s and p‐ERK1/2 also was up‐regulated in TGF‐β‐treated cells (Figure 8A). Transwell assays also showed that TGF‐β promoted cell migration in MHCC‐97L cells (Figure 8B). However, TGF‐β‐promoted migration was reversed following TM treatment (Figure 8B). Furthermore, TM also inhibited the expression of CD44s, p‐ERK1/2, N‐cadherin and Snail in TGF‐β‐treated MHCC‐97L cells (Figure 8A). Therefore, these results showed that TM inhibited TGF‐β‐mediated cell migration in HCC cells.
Figure 8.
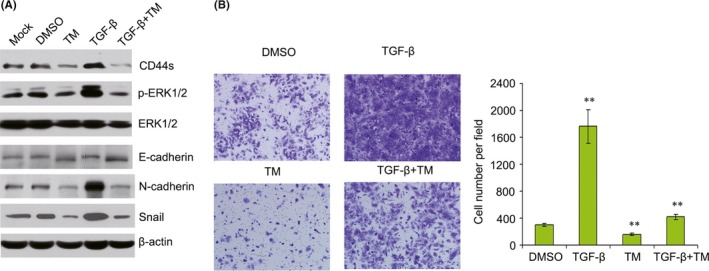
Tunicamycin (TM) inhibits transforming growth factor (TGF)‐β‐induced cell migration by suppressing ERK1/2 activity and CD44 expression in MHCC‐97L cells. A, Expression levels of CD44s, p‐ERK1/2, ERK1/2, E‐cadherin, N‐cadherin and Snail were detected by western blotting in MHCC‐97L cells treated with 2.5 μg/mL TM or 5 μg/L TGF‐β for 12 h. B, Following the treatment of MHCC‐97L cells with 2.5 μg/mL TM or 5 μg/L TGF‐β for 12 h, cell migration was detected by transwell assay. ** P < .01
4. DISCUSSION
Hepatocellular carcinoma is the most common primary malignancy of the liver in adults and the third most common cause of cancer death worldwide. In most cases, death results from metastasis at secondary sites. Tumor metastasis is a complex and multistage process, and tumor cells are required to express a variety of properties including altered adhesiveness, increased motility and invasive capacity to complete the metastatic process.19 EMT is proposed to be a crucial mechanism that regulates the initial steps of metastatic progression.20 Therefore, interruption of this step may be a strategy for the prevention and treatment of HCC metastasis.4 In the present study, we showed that TM inhibited HCC cell proliferation by inducing G2/M cell cycle arrest and cell apoptosis. TM inhibited the migration of HCC cells by decreasing activation of the CD44s‐ERK1/2 signaling pathway.
Tunicamycin exerts a direct effect on HCC cells partially through inhibition of CD44. CD44 is a transmembrane glycoprotein that includes an extracellular region that interacts with growth factors and HA as well as a cytoplasmic moiety that is capable of interacting with cytoskeletal components.21, 22 In this study, we found that TGF‐β‐promoted migration was reversed following TM treatment. Previous studies have also shown that inhibition of CD44 by an anti‐CD44 mAb blocks tumor growth, metastasis, and invasion.23 Furthermore, our previous results also showed that CD44s expression was upregulated in HCC tissues.24 In addition, a previous study has indicated a shift in CD44 expression from variant isoforms (CD44v) to the standard isoform (CD44s) during EMT.6 Our results showed that CD44v expression was decreased in TM‐treated cells. However, CD44s expression was also decreased in TM‐treated cells (Figure S3). Therefore, we did not observe the shift in CD44 expression from CD44v to CD44s in TM‐treated cells. Considering the accumulating body of evidence that highlights the important role of CD44s signaling in tumor progression, disrupting CD44s‐mediated signaling pathways may lead to improved treatment of HCC.
Glycosylation of the CD44 molecule causes conformational changes that may affect its ability to bind to HA. Katoh et al. found that treatment of ovarian cells with a deglycosylating enzyme decreased the binding of CD44 to HA.25 Bartolazzi et al. reported that in melanoma cells, mutation of the CD44 N‐linked glycosylation sites inhibited CD44 binding to HA.26 Interactions between CD44, HA, and epidermal growth factor receptor (EGFR) promote the progression of various tumors by cross‐activation of several signaling pathways. In head and neck squamous cell carcinoma, HA mediates the formation of a complex including CD44 and EGFR.27 After the HA/CD44 interaction that recruits and forms a CD44‐EGFR complex, multiple downstream signaling pathways are activated, further promoting diverse tumor progression behaviors. We also found that HA could activate phosphorylation of EGFR. In addition, CD44 and EGFR were colocalized in MHCC‐97L cells examined by confocal microscopy (Figure S4). EGFR is a glycosylated protein localized to the apical domain of plasma membranes.28 Our results showed that EGFR is glycosylated in hepatocellular carcinoma cells (Figure S4). In addition, we found that TM inhibited EGFR expression by regulating its glycosylation. Furthermore, a recent study also showed that tunicamycin effectively enhanced TRAIL‐induced apoptosis possibly through the inhibition of the EGFR pathway in human colon cancer cells.29 Bourguignon et al. previously reported the notion that CD44 can participate in the activation of EGFR in response to HA in squamous cell carcinoma cells.30 Therefore, we speculated that TM inhibited HCC cell proliferation and migration possibly through regulation of the CD44s‐mediated EGFR signaling pathways. However, these results need to be confirmed in the future.
Here we determined that TM inhibited the proliferation and migration of HCC cells. Moreover, we found that TM inhibited the proliferation and migration of HCC cells through attenuated activation of ERK1/2. These results will provide supportive evidence to uncover the antitumor mechanisms of TM. The ERK1/2 signaling pathway is believed to play an important role in cancer proliferation, apoptosis and migration.31 The activation of ERK1/2 pathways in the progression of EMT in several cell lines has been widely established.32, 33 Levels of phosphorylated forms of ERK1/2 are significantly higher in HCC models and human HCC tissue specimens than in healthy liver samples.34, 35 Moreover, MAPK/ERK activity has been shown to be positively correlated with tumor size and aggressive tumor activity,36, 37 suggesting that ERK1/2 activation reflects aggressive tumor activity in clinical conditions. Furthermore, we found that CD44s can activate the phosphorylation of ERK1/2 in HCC cells. Therefore, we showed that TM inhibited the CD44‐ERK1/2 signaling pathway to regulate the proliferation and migration of HCC cells. Therefore, TM could be used as a potential agent against HCC metastasis.
In conclusion, our study suggests that the glycosylation inhibitor TM attenuates the proliferative and migratory abilities of HCC through the CD44s and ERK1/2 dependent pathway. CD44s or its downstream effector ERK1/2 might be able to be used as novel therapeutic targets for HCC therapy.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Key Program for Basic Research of China (973) (2015CB553905), National Natural Science Foundation of China (81602113, 81472570, 81272438, 81472726), the SKLORG Research foundation (91‐14‐09, 91‐15‐03), Research Project of Shanghai Municipal Health and Family Planning Commission (201540107), and the Foundation of Renji Hospital, School of Medicine, Shanghai Jiao Tong University (RJZZ16‐014).
Hou H, Ge C, Sun H, Li H, Li J, Tian H. Tunicamycin inhibits cell proliferation and migration in hepatocellular carcinoma through suppression of CD44s and the ERK1/2 pathway. Cancer Sci. 2018;109:1088–1100. https://doi.org/10.1111/cas.13518
Funding informationNational Key Program for Basic Research of China (973), (Grant/Award Number: ‘2015CB553905’). National Natural Science Foundation of China (Grant/Award Numbers: ‘81602113, 81472570, 81472726, 81272438’), the SKLORG Research Foundation (Grant/Award Number: ‘91‐14‐09, 91‐15‐03’), Research Project of Shanghai Municipal Health and Family Planning Commission (Grant/Award Number: ‘201540107’), the Foundation of Renji Hospital, School of Medicine, Shanghai Jiao Tong University (Grant/Award Number: ‘RJZZ16‐014’).
REFERENCES
- 1. Wang X, Zhang A, Sun H. Power of metabolomics in diagnosis and biomarker discovery of hepatocellular carcinoma. Hepatology. 2013;57:2072‐2077. [DOI] [PubMed] [Google Scholar]
- 2. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559‐1564. [DOI] [PubMed] [Google Scholar]
- 3. Kawada K, Hasegawa S, Murakami T, et al. Molecular mechanisms of liver metastasis. Int J Clin Oncol. 2011;16:464‐472. [DOI] [PubMed] [Google Scholar]
- 4. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97‐110. [DOI] [PubMed] [Google Scholar]
- 5. Marhaba R, Zoller M. CD44 in cancer progression: adhesion, migration and growth regulation. J Mol Histol. 2004;35:211‐231. [DOI] [PubMed] [Google Scholar]
- 6. Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial‐mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Endo K, Terada T. Protein expression of CD44 (standard and variant isoforms) in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, p53 expression, and patient survival. J Hepatol. 2000;32:78‐84. [DOI] [PubMed] [Google Scholar]
- 8. Zhao Q, Zhou H, Liu Q, et al. Prognostic value of the expression of cancer stem cell‐related markers CD133 and CD44 in hepatocellular carcinoma: from patients to patient‐derived tumor xenograft models. Oncotarget. 2016;7:47431‐47443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parodi AJ. Role of N‐oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J. 2000;348:1‐13. [PMC free article] [PubMed] [Google Scholar]
- 10. Hou H, Sun H, Lu P, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther. 2013;12:2874‐2884. [DOI] [PubMed] [Google Scholar]
- 11. Tian H, Ge C, Li H, et al. Ribonucleotide reductase M2B inhibits cell migration and spreading by early growth response protein 1‐mediated phosphatase and tensin homolog/Akt1 pathway in hepatocellular carcinoma. Hepatology. 2014;59:1459‐1470. [DOI] [PubMed] [Google Scholar]
- 12. Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20:561‐573. [DOI] [PubMed] [Google Scholar]
- 13. Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014;14:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang L, Yan Q, Fang S, et al. Calcium binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating ERK signaling pathway. Hepatology. 2017;66:1529‐1545. [DOI] [PubMed] [Google Scholar]
- 15. Sankpal NV, Fleming TP, Sharma PK, Wiedner HJ, Gillanders WE. A double‐negative feedback loop between EpCAM and ERK contributes to the regulation of epithelial‐mesenchymal transition in cancer. Oncogene. 2017;36:3706‐3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ahrens T, Assmann V, Fieber C, et al. CD44 is the principal mediator of hyaluronic‐acid‐induced melanoma cell proliferation. J Invest Dermatol. 2001;116:93‐101. [DOI] [PubMed] [Google Scholar]
- 17. Whelan SA, Lu M, He J, et al. Mass spectrometry (LC‐MS/MS) site‐mapping of N‐glycosylated membrane proteins for breast cancer biomarkers. J Proteome Res. 2009;8:4151‐4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mima K, Okabe H, Ishimoto T, et al. CD44s regulates the TGF‐beta‐mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012;72:3414‐3423. [DOI] [PubMed] [Google Scholar]
- 19. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reichl P, Haider C, Grubinger M, Mikulits W. TGF‐beta in epithelial to mesenchymal transition and metastasis of liver carcinoma. Curr Pharm Des. 2012;18:4135‐4147. [DOI] [PubMed] [Google Scholar]
- 21. Chen L, Bourguignon LY. Hyaluronan‐CD44 interaction promotes c‐Jun signaling and miRNA21 expression leading to Bcl‐2 expression and chemoresistance in breast cancer cells. Mol Cancer. 2014;13:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourguignon LY. Hyaluronan‐CD44 interaction promotes microRNA signaling and RhoGTPase activation leading to tumor progression. Small GTPases. 2012;3:53‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weidle UH, Maisel D, Klostermann S, Weiss EH, Schmitt M. Differential splicing generates new transmembrane receptor and extracellular matrix‐related targets for antibody‐based therapy of cancer. Cancer Genomics Proteomics. 2011;8:211‐226. [PubMed] [Google Scholar]
- 24. Tian H, Ge C, Zhao F, et al. Downregulation of AZGP1 by Ikaros and histone deacetylase promotes tumor progression through the PTEN/Akt and CD44s pathways in hepatocellular carcinoma. Carcinogenesis. 2017;38:207‐217. [DOI] [PubMed] [Google Scholar]
- 25. Katoh S, Zheng Z, Oritani K, Shimozato T, Kincade PW. Glycosylation of CD44 negatively regulates its recognition of hyaluronan. J Exp Med. 1995;182:419‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartolazzi A, Nocks A, Aruffo A, Spring F, Stamenkovic I. Glycosylation of CD44 is implicated in CD44‐mediated cell adhesion to hyaluronan. J Cell Biol. 1996;132:1199‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang SJ, Bourguignon LY. Role of hyaluronan‐mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am J Pathol. 2011;178:956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Azimzadeh Irani M, Kannan S, Verma C. Role of N‐glycosylation in EGFR ectodomain ligand binding. Proteins. 2017;85:1529‐1549. [DOI] [PubMed] [Google Scholar]
- 29. Guo X, Meng Y, Sheng X, et al. Tunicamycin enhances human colon cancer cells to TRAIL‐induced apoptosis by JNK‐CHOP‐mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs. 2017;28:66‐74. [DOI] [PubMed] [Google Scholar]
- 30. Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan‐CD44 interaction with leukemia‐associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co‐activation, phospholipase C epsilon‐Ca2 + signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J Biol Chem. 2006;281:14026‐14040. [DOI] [PubMed] [Google Scholar]
- 31. Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105‐143. [DOI] [PubMed] [Google Scholar]
- 32. Chen X, Ye S, Xiao W, Wang W, Luo L, Liu Y. ERK1/2 pathway mediates epithelial‐mesenchymal transition by cross‐interacting with TGFbeta/Smad and Jagged/Notch signaling pathways in lens epithelial cells. Int J Mol Med. 2014;33:1664‐1670. [DOI] [PubMed] [Google Scholar]
- 33. Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial‐mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McKillop IH, Schmidt CM, Cahill PA, Sitzmann JV. Altered expression of mitogen‐activated protein kinases in a rat model of experimental hepatocellular carcinoma. Hepatology. 1997;26:1484‐1491. [DOI] [PubMed] [Google Scholar]
- 35. Ito Y, Sasaki Y, Horimoto M, et al. Activation of mitogen‐activated protein kinases/extracellular signal‐regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951‐958. [DOI] [PubMed] [Google Scholar]
- 36. Schmitz KJ, Wohlschlaeger J, Lang H, et al. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J Hepatol. 2008;48:83‐90. [DOI] [PubMed] [Google Scholar]
- 37. Dominguez D, Montserrat‐Sentis B, Virgos‐Soler A, et al. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol Cell Biol. 2003;23:5078‐5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials