

Abstract
Radiotherapy (RT) can be used as preoperative treatment to downstage initially unresectable locally rectal carcinoma, but radioresistance and recurrence remain significant problems. Retinoblastoma binding protein 6 (RBBP6) has been implicated in the regulation of cell cycle, apoptosis and chemoresistance both in vitro and in vivo. The present study investigated whether the inhibition of RBBP6 expression would improve radiosensitivity in human colorectal cancer cells. After SW620 and HT29 cells were exposed to radiation, the levels of RBBP6 mRNA and protein increased over time in both cells. Moreover, a significant reduction in clonogenic survival and a decrease in cell viability in parallel with an obvious increase in cell apoptosis were demonstrated in irradiated RBBP6‐knockdown cells. Transfection with RBBP6 shRNA improved the levels of G2‐M phase arrest, which blocked the cells in a more radiosensitive period of the cell cycle. These observations indicated that cell cycle and apoptosis mechanisms may be connected with tumor cell survival following radiotherapy. In vivo, the tumor growth rate of nude mice in the RBBP6‐knockdown group was significantly slower than that in other groups. These results indicated that RBBP6 overexpression could resist colorectal cancer cells against radiation by regulating cell cycle and apoptosis pathways, and inhibition of RBBP6 could enhance radiosensitivity of human colorectal cancer.
Keywords: apoptosis, cell cycle, preoperative radiotherapy, RBBP6, rectal cancer
1. INTRODUCTION
Colorectal cancer (CRC) is still the third leading cause of cancer death among young adults, after breast and lung cancer, despite tremendous advances in cancer screening and treatment in the USA.1 Surgical resection is thought to be the predominant therapy for the early stage CRC and total mesorectal excision (TME) is the standard surgery for rectal cancer. However, radical surgery is difficult to perform, especially for initially unresectable advanced colon cancer and mid‐low rectal cancer.2 Preoperative radiotherapy is widely used to downstage tumors to lower treatment failure, but the local recurrence and outcome remain variable, which indicates significant differences in tumor response to radiation.3, 4, 5, 6 Therefore, it is essential to explore the molecular mechanisms of CRC cell survival following irradiation treatment, and, furthermore, there is an urgent need to identify useful molecular targets to improve the response of CRC to local irradiation.
p53 is thought to be mutated in approximately 50% of human malignancies. p53 mutations are found in 5% of adenomas, 50% of malignant polyps and 75% of invasive CRC, with an increasing frequency positively correlating with the extent of malignancy.7 In addition to DNA damage, p53 is activated by many factors, including chemicals, viruses, oxidation, osmotic pressure and ultraviolet radiation.8 Acting as a key tumor suppressor, the p53 protein plays an important role in DNA transcription as well as in suppressing the genesis and development of tumors. When DNA damage occurs, p53 causes cell cycle arrest, so that the cells have enough time to repair DNA damage. If the cells fail to repair DNA damage, p53 will activate the apoptotic pathway to maintain the integrity of chromosomal DNA.9 Cyclin‐dependent kinases (CDK), formed at particular stages of the cell cycle progression, are responsible for regulating critical checkpoints involved in the G2/M and G1/S phase transitions.10 Activation of p53 drives the expression of many Cyclin Dependent Kinase inhibitors (CDKi), especially for p21. p21, as a crucial inhibitory factor for CDK, can block the cell cycle in the G2 phase by inhibiting the M phase checkpoint CyclinB1/CDK1 complex.11
Retinoblastoma binding protein 6 (RBBP6) gene, located on chromosome 16p22.2, is also referred to as PACT, P2P‐R or RBQ‐1. Three major transcripts (transcript 6.1, 6.0 and 1.1 kb), encoding proteins of 1792, 1758 and 118 amino acids, respectively, are expressed by the RBBP6 gene as a consequence of alternative splicing.12 RBBP6 has been found to be highly expressed in esophagus cancer, lung cancer, breast cancer and cervical cancer,13, 14, 15 suggesting that RBBP6 may play an important role in the progression of human malignant tumors. RBBP6 is a multi‐domain protein that interacts with p53 and Rb using its ring finger domain. It promotes the ubiquitination and degradation of p53 protein through its E3 ubiquitin ligase activity. Due to its ability to bind the tumor suppressors p53 and Rb, it has been proposed that RBBP6 plays a key role in cell cycle, cell proliferation, apoptosis and chemoresistance.16, 17, 18 Knockdown of RBBP6 expression enhanced the chemosensitivity of lung and breast cancer and resulted in the inhibition of tumor growth. However, in colorectal cancer patients who receive adjuvant radiotherapy before tumor resection, the role of RBBP6 remains unclear.
RBBP6 has an important function in the development and progression of cancer. The aim of the present study was to evaluate whether RBBP6 can influence the sensitivity of colorectal cancer cells to radiotherapy, and to determine whether the RBBP6 gene is a useful biomarker for guiding irradiation treatment before curative resection of colorectal cancer. In a previous study,19 we found that RBBP6 was markedly increased in colon cancer tissues relative to normal colon epithelium and high RBBP6 expression was associated with a poor clinical outcome in human colorectal cancer. Here, we sought to assess the effects of RBBP6 expression on radiotherapy in colorectal cells by downregulating RBBP6 mRNA and protein, and attempted to elucidate the underlying mechanisms by which RBBP6 mediates radiotherapy resistance.
2. MATERIALS AND METHODS
2.1. Cell culture and plasmids
Human cell lines SW480, HCT8, SW620 and HT29 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were maintained in DMEM with 10% FBS (Gibco, USA) at 37°C, 95% humidity and 5% CO2. The control‐shRNA plasmid and the short hairpin RNA (shRNA) plasmid for silencing RBBP6 were purchased from Genechem (Shanghai, China). SW620 and HT29 cells were transduced with 5 × 105 TU/mL of lentivirus particles. Then, stable clones of both cells expressing RBBP6 shRNA or control‐shRNA were obtained by puromycin selection and were determined by western blot and real‐time RT‐PCR. Cells transduced with shNT (SW620 shNT and HT29 shNT) were served as controls. The effective target sequences of RBBP6 shRNA were as follows:
sh‐RBBP6‐1: 5′‐ CCTTTGATGGGCTCCACAT‐3′;
sh‐RBBP6‐2: 5′‐GATGACTCTTCCGCGTCTA‐3′.
2.2. Real‐time PCR
Total RNA was isolated from SW620 and HT29 cells using an RNA extraction kit (Qiagen, Germany). After RNA was isolated, a cDNA Synthesis Kit (Promega, USA) was used for synthesizing cDNA according to protocols of the manufacturer. Quantitative PCR reaction was conducted using a ViiA 7 Real‐Time PCR System (Life Technology, USA) with SYBR Premix DimerEraser (Perfect Real Time) (Takara, Japan) in accordance with the manufacturer's protocol. Primer sequences were as follows:
RBBP6 forward 5′‐CTCCCCATACACTTCCTCTCC‐3′;
RBBP6 reverse 5′‐TTCTTTTAGTCGTCGCTGCTC‐3′;
GAPDH forward 5′‐GGCCAAGGTCATCCATGACAA‐3′;
GAPDH reverse 5′‐TCTTCTGACACCTACCGGGGA‐3′.
Cycling conditions were as follows: initial denaturation (1 minutes at 95°C) followed by 25 cycles of denaturation for 1 minutes at 94°C, annealing for 1 minutes at 94°C, and elongation for 45 seconds at 72°C, with a final extension for 5 minutes at 72°C. Each experiment was performed in triplicate.
2.3. Western blot analysis
Total protein was extracted using RIPA solution (Beyotime Biotechnology, Jiangsu, China) and a BCA Protein Assay Kit (Beyotime Biotechnology, Jiangsu, China) was used to measure the concentration of the protein. Equal amounts of the protein (40 μg) were spotted into 8% SDS‐polyacrylamide gel for electrophoresis and then transferred onto PVDF membranes. The membranes were soaked for an hour in 5% milk in TBST at room temperature. Subsequently, membranes were incubated with primary antibodies against RBBP6, cleaved Caspase‐3, cleaved PARP, Cyclin B1, CDK1, p21, p53, MDM2, Bax, Bcl‐2, Bcl‐XL, β‐actin or GAPDH overnight (1:1000, Cell Signaling Technology, USA). Then, the membranes were incubated with HRP‐tagged secondary antibody for 1 hour. The targeted proteins were detected by ECL reagent (Pierce Biotechnology, USA). GAPDH levels served as loading controls.
2.4. Clonogenic survival assay
SW620 and HT29 cells were plated into culture dishes with the appropriate number of cells and irradiated with single doses of 0, 2, 4, 8 or 10 Gy of gamma radiation. After approximately 14 days, colonies were stained with crystal violet and counted. Colonies containing fewer than 50 cells were not scored. Plating efficiency (PE) was measured for each cell line. Survival fractions (SF) was calculated using the equation SF = colonies counted/(cells seeded × PE) × 100%. All assays were independently performed in triplicate.
2.5. Cell viability assay
Cell viability was analyzed using the Cell Counting Kit‐8 (Dojino, Japan) according to the manufacturer's protocol. SW620 and HT29 cell lines were diluted, and then 1 × 103 cells were seeded in 96‐well plates per well containing 0.1 mL of medium. Following 24 hours incubation period, cells were irradiated with 0‐10 Gy radiation in triplicate. At the appropriate time (0, 24, 48, 72 hours), each well was incubated with 10 μL CCK8 solution for 2 hours at 37°C. Then, Gen5 microplate reader (BioTek, USA) was used to detect the absorbance at 450 nm. Cell viability was measured according to the formula: cell viability (%) = A450 (sample)/A450 (control) × 100.
2.6. Cell cycle analysis
Cell cycle was analyzed using the Cell Cycle Kit (BD, CA, USA) in accordance with the manufacturer's protocol. Both attached and detached SW620 and HT29 cells were collected by trypsinization and washed in PBS for 5 minutes by centrifugation at 1500 rpm. Then, the cells were fixed in 70% ice‐cold ethanol for 30 minutes at 4°C. Following gentle centrifugation, the cells were resuspended in RNase A/PI staining solution and incubated in the dark for 30 minutes. Then the cells were measured by flow cytometry (BD Accuri C6, USA).
2.7. Apoptosis assay
Floating and adherent cells were collected and washed with PBS. Cells were then centrifuged for 2 minutes at 1500 rpm and resuspended in 1× Annexin‐V binding buffer at a concentration of 1 × 105 cells/mL. The cells were stained using the Annexin V‐APC/PI Apoptosis Kit (eBioscience, CA, USA) for 15 minutes at room temperature in the dark. Cells were analyzed by flow cytometry (BD Accuri C6, USA).
2.8. Co‐immunoprecipitation
Cells were lysed with modified RIPA buffer (10 mmol/L Tris–HCl, pH 7.5, 5 mmol/L EDTA, 150 mmol/L NaCl, 1% sodium deoxycholate, 1% Triton X‐100, 0.1% SDS) supplemented with 1% protease inhibitors for subsequent co‐IP. Endogenous protein RBBP6, MDM2 or P53 were immunoprecipitated from cell lysates using 5 μg of anti‐RBBP6, anti‐MDM2 or anti‐P53 antibody mixed with 10 μL of washed protein G (Santa Cruz, CA, USA), respectively. Then, the immunoprecipitates were washed 5 times in cold PBS and subjected to western blotting.
2.9. Mice xenograft models
HT29 cells (2 × 106) were suspended in 150 μL of PBS and injected subcutaneously into the hind legs of the 4‐week‐old male nude mice. Ten days later, healthy nude mice bearing a diameter of 8 mm HT29 tumor xenografts in the hind legs were selected and divided into 4 groups. The chosen mice were treated with lentiviral vectors (control‐shRNA), lentiviral vectors (RBBP6‐shRNA), lentiviral vectors (control‐shRNA) plus local tumor irradiation or lentiviral vectors (RBBP6‐shRNA) plus local tumor irradiation, respectively. The procedures are as follows: On day 1, the mice were administered a single intratumoral injection of lentiviral vectors (control‐shRNA or RBBP6‐shRNA) in the Laboratory Animal's SPF Housing Facility. On day 7, the gene interference effect of lentiviral vectors was significant. Meanwhile, we irradiated the animals’ tumors on day 1, 7, 14 and 21 (2 Gy/f, DT = 8 Gy). Tumor weight and volume were measured every 7 days and mice were killed on the 28th day. All procedures involving animals were in accordance with the Shanghai Jiaotong University Affiliated Shanghai General Hospital Animal Care guidelines. Efforts were made to ensure the animals suffered minimally.
2.10. Immunohistochemical analysis for RBBP6
HT29 tumors were harvested and fixed in formalin. Immunohistochemistry analysis was performed following the standard protocol as previously described.19
2.11. Statistical analysis
All data are presented as mean ± SD. The significance of the experimental values was determined using Student t test (2‐tailed). Differences with a P < .05 were considered statistically significant. All the statistical analyses were done using SPSS 19.0 statistical software (SPSS, Chicago, IL, USA).
3. RESULTS
3.1. Irradiation induced upregulation of RBBP6 expression
To investigate the role of RBBP6 in mediating sensitivity to colorectal cancer radioresistance, 4 human colorectal cancer cell lines were used to determine the radiotherapy effect. As the CCK‐8 assays showed, all cells’ viability was reduced by radiation treatment in a dose‐dependent manner; SW480 and HCT8 cells seem to be more sensitive to radiation than SW620 and HT29 cells (Figure 1A). Western blot showed that the expression of RBBP6 in SW620 and HT29 cells was higher than that in SW480 and HCT8 cells (Figure 1B). Radiation treatment was performed in SW620 and HT29 cell lines. Following 10 Gy of irradiation, the mRNA levels of RBBP6 increased over time in both SW620 and HT29 cells (Figure 1C). RBBP6 protein levels were also altered after radiation treatment, which was similar to our qPCR data (Figure 1D).
Figure 1.
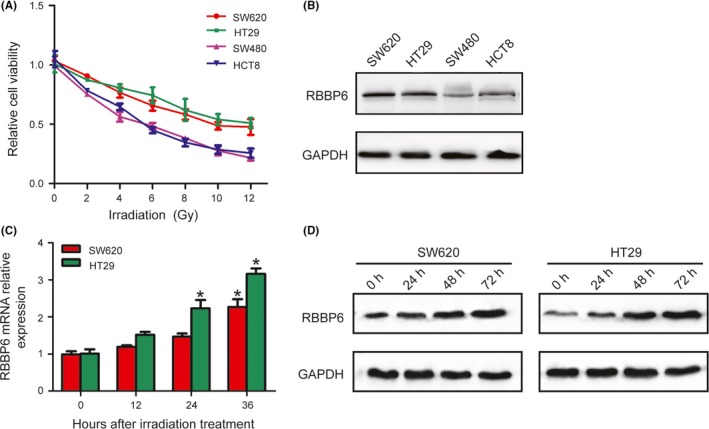
Irradiation induced upregulation of RBBP6 expression. A, CCK8 assay showed that SW480 and HCT8 cells seem to be more sensitive to radiation than SW620 and HT29 cells. B, Western blotting confirmed the expression of RBBP6 is higher in SW620 and HT29 cells than that in SW480 and HCT8 cells. C,D, Levels of RBBP6 mRNA and protein were increased in SW620 and HT29 cells over time following 10 Gy of irradiation treatment
3.2. Effect of shRNA on expression of RBBP6
To determine whether RBBP6 can increase radioresistance in colorectal cancer cells, we silenced RBBP6 by shRNA. Both SW620 RBBP6‐KD and HT29 RBBP6‐KD stable cell lines were generated (Figure 2).
Figure 2.
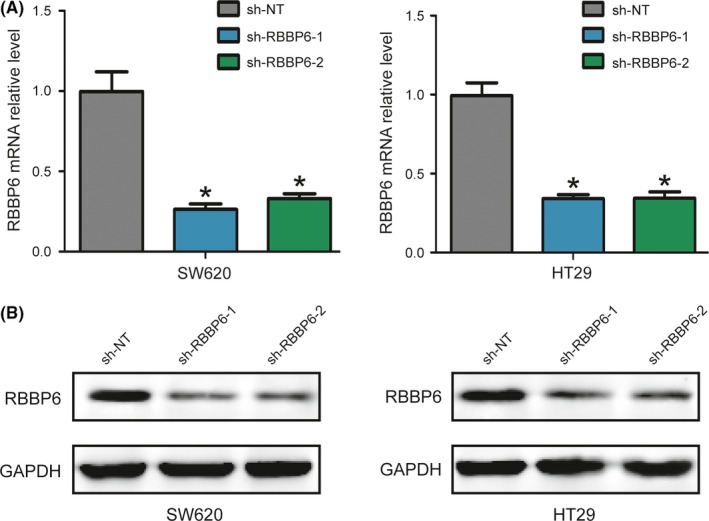
The effect of shRNA on expression of RBBP6. A,B, RBBP6 was silenced in both SW620 and HT29 cell lines
3.3. Inhibition of RBBP6 decreased cell viability
To determine how RBBP6 inhibition affects cell viability and proliferation, CCK‐8 assay was performed following 10 Gy of irradiation for 0, 24, 48 or 72 hours or 5 doses of irradiation (0, 2, 4, 6 and 10 Gy) for 48 hours. As a result, RBBP6‐KD cell lines showed a significant reduction in cell viability for all 3 incubation times (24, 48 and 72 hours) compared to control cells (Figure 3A). Moreover, cell viability was reduced by radiation treatment in a dose‐dependent manner (Figure 3B). These data indicated that RBBP6 contributed to radioresistance in SW620 and HT29 cells and targeted inhibition of RBBP6 sensitized these cells to radiation treament.
Figure 3.
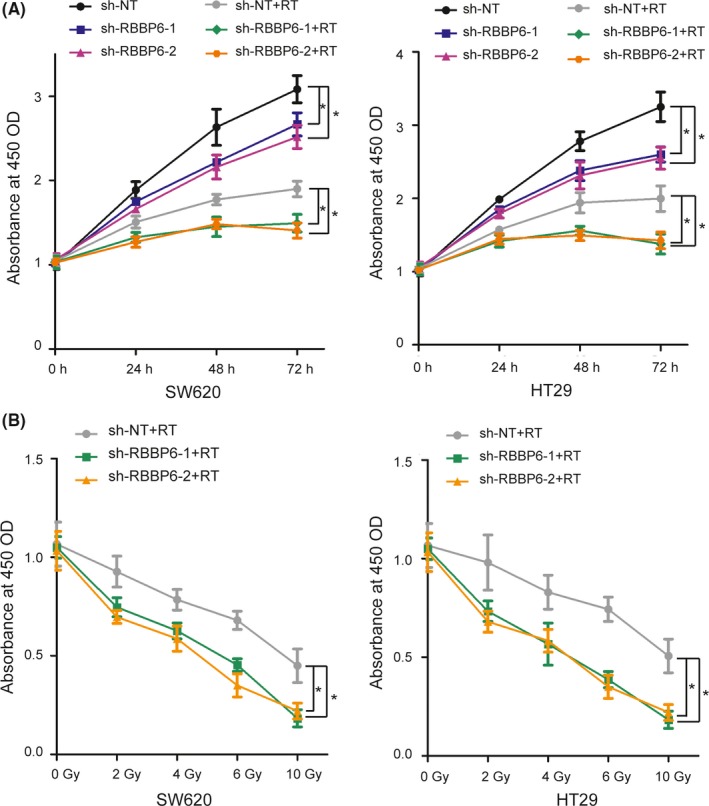
Inhibition of RBBP6 decreased cell viability. A, CCK8 assay illustrated the difference in cell growth between control and RBBP6‐KD cell lines following 10 Gy irradiation; B, CCK8 assay illustrated the difference in cell growth between control and RBBP6‐KD cell lines at 48 h following different doses of irradiation
3.4. Inhibition of RBBP6 decreased clonogenic survival
To further analyze the radiosensitizing ability of RBBP6 inhibition, the clonogenic survival assay was performed after treatment with radiation in SW620 and HT29 cell lines. Lower survival curves were demonstrated in RBBP6‐KD cells compared to the control cells (Figure 4). After silencing RBBP6, the survival rate was significantly decreased in SW620 and HT29 cell lines.
Figure 4.
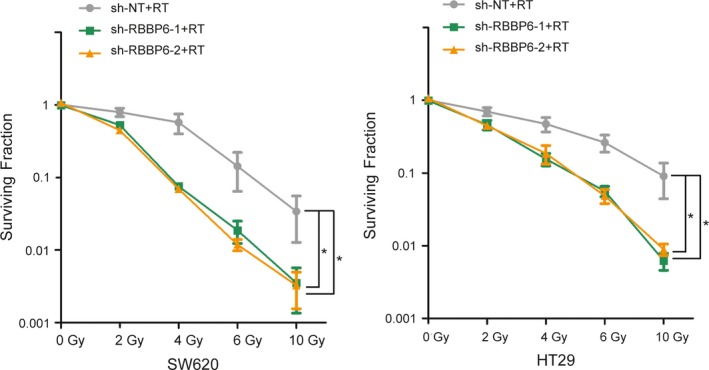
Inhibition of RBBP6 decreased clonogenic survival. Clonogenic survival assay demonstrated inhibition of RBBP6 increased the radiosensitivity of SW620 and HT29 cells
3.5. Inhibition of RBBP6 sensitized cells to radiation‐induced apoptosis and induced arrest of cells in G2‐M phase
We compared the extent of radiation‐induced apoptosis in colorectal cancer cells transfected with either RBBP6 shRNA or control‐shRNA. Annexin V analysis showed that the radiation‐induced apoptotic cells were significantly higher in RBBP6‐KD cells compared with the control cells. Following 10 Gy irradiation, the proportion of cell apoptosis increased with the prolongation of culture time. However, there was little change in the number of apoptotic cells that were not irradiated. (Figure 5A,B). Furthermore, cell cycle analysis showed a significant increase in the G2‐M fraction in RBBP6‐KD cells after irradiation compared to cells that had been transduced with a non‐targeting shRNA (Figure 5C).
Figure 5.
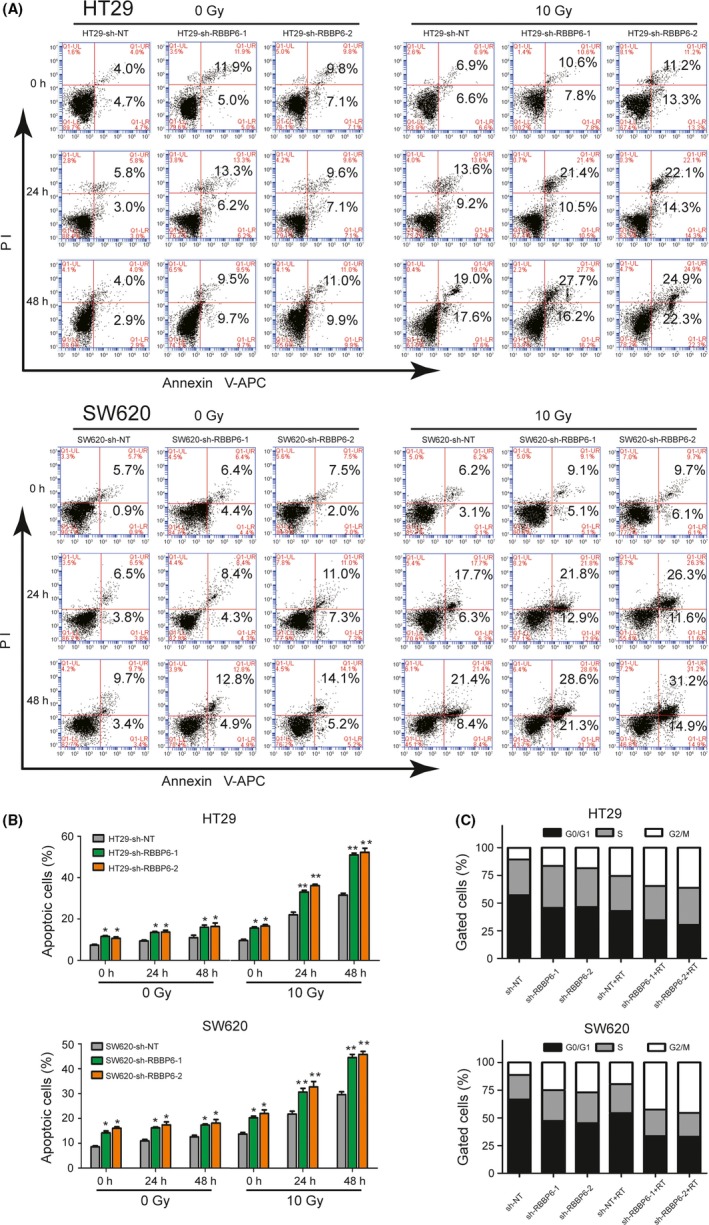
Inhibition of RBBP6 sensitized cells to radiation‐induced apoptosis and induced G2‐M arrest. A, Annexin V assay showed that the percentage of apoptotic cells was higher in RBBP6‐KD cells than that in control cells following 10 Gy irradiation. B, Apoptosis analysis showed that inhibition of RBBP6 improved the radiation sensitivity of colorectal cancer (CRC); cell cycle analysis identified G2‐M arrest in RBBP6‐KD cell lines compared to control cells at 24 h following 10 Gy irradiation
RBBP6 inhibition resulted in an increase of cleaved PARP, a well‐established apoptotic marker, in HT29 and SW620 cells. Conversely, in the control cells, which are resistant to apoptosis, the majority of the PARP protein remained full‐length and uncleaved (Figure 6A). This suggested that RBBP6‐KD may confer sensitivity to radiation‐induced apoptosis. Furthermore, RBBP6 inhibition resulted in a more dramatic decrease of Cyclin B1 in HT29 and SW620 cells than the control cells (Figure 6B); Cyclin B1 is thought to be the key protein in the control of the cell cycle at the G2/M transition. These data indicated that both RBBP6‐KD cell lines were blocked in a more radiosensitive stage of the cell cycle following 10 Gy irradiation for 48 hours.
Figure 6.
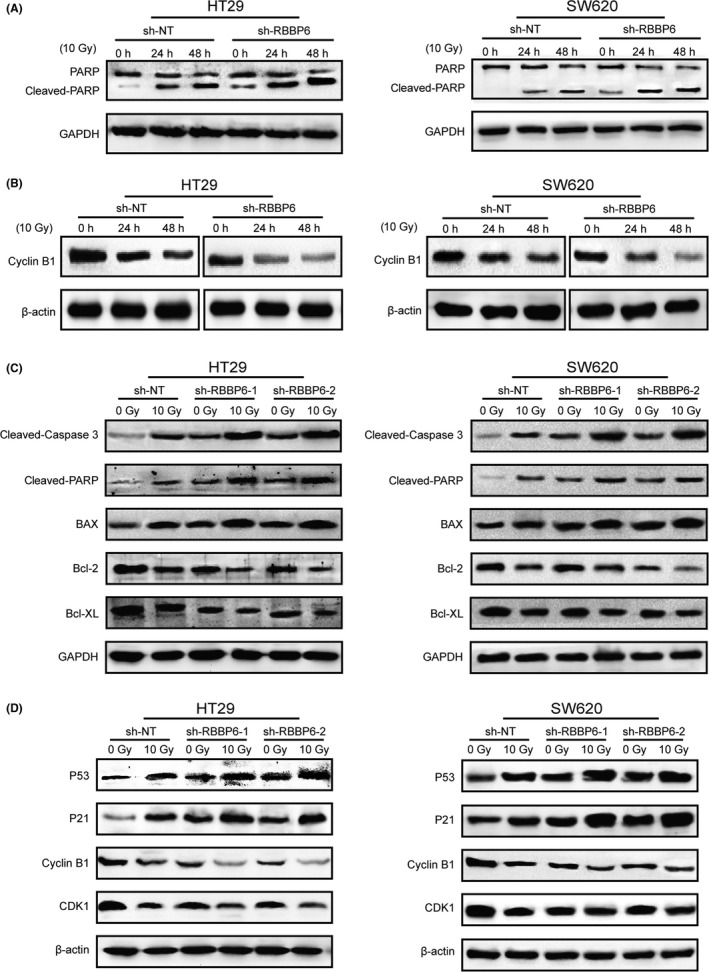
Inhibition of RBBP6 induced the expression of apoptosis and cell cycle markers changed. A, Cleaved PARP level in control and RBBP6‐KD HT29/SW620 cells at 0, 24 and 48 h following 10 Gy irradiation. B, Cyclin B1 level in control and RBBP6‐KD HT29/SW620 cells at 0, 24 and 48 h following 10 Gy irradiation. C, Expression of pro‐apoptotic proteins cleaved‐Caspase 3, cleaved‐PARP, BAX and anti‐apoptotic proteins Bcl‐2, Bcl‐XL. D, Expression of p53, p21 and cycle checkpoint proteins Cyclin B1 and CDK1
In agreement with the above results, we observed after RT an increased activation of Caspase 3, which is known to cleave PARP in RBBP6‐KD cell lines compared to the control cells (Figure 6C). We also determined the expression of anti‐apoptotic mitochondrial proteins Bcl‐2, Bcl‐XL and pro‐apoptotic mitochondrial protein BAX. The expression of BAX was significantly increased and Bcl‐2 and Bcl‐XL were decreased after RT in HT29‐KD cells and SW620‐KD cells compared to the control cells (Figure 6C). Furthermore, the expression of cell cycle checkpoint protein Cyclin B1/Cdk1 was decreased after RT in RBBP6‐KD cell lines compared to the control cells (Figure 6D).
3.6. Inhibition of RBBP6 led to both cell apoptosis and cycle arrest through attenuating the p53/MDM2 interaction
We further investigated the mechanisms of increased cell apoptosis and cycle arrest following RBBP6 silencing. By western blotting, we observed a significant increase in the level of p53 protein and p21 protein in both cells silenced of RBBP6 relative to control cells. In contrast, the level of Rb protein was not affected (Figure 7A). Acting as a tumor suppressor, p53 plays an important role in regulating cell apoptosis and cycle progression. p21 is a negative regulator of cell cycle, which affects cycle arrest at G1 and G2 through multiple mechanisms. Therefore, we conclude that p53 protein levels in RBBP6‐KD cells are higher than those in the control cells. Probably, this is the major reason for the difference in cell apoptosis and cell cycle progression after silencing RBBP6.
Figure 7.
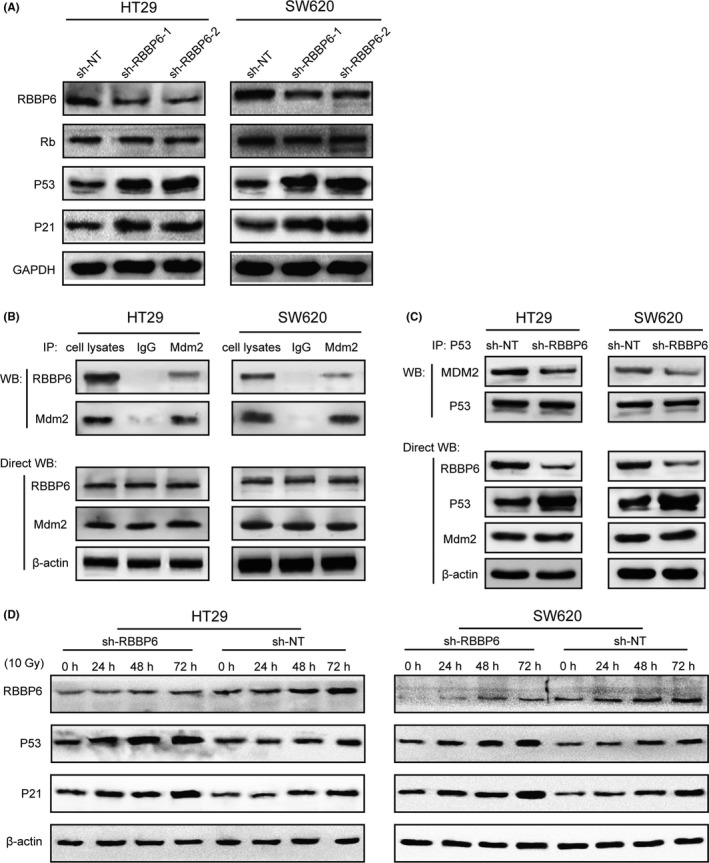
Inhibition of RBBP6 led to both cell apoptosis and cycle arrest through attenuating the p53/MDM2 interaction. A, RBBP6 inhibition does not affect Rb abundance in in SW620 and HT29 cells but increases the p53 and p21 protein level. B, Endogenous RBBP6 and MDM2 have physical interaction in SW620 and HT29 cells. C, Inhibition of RBBP6 decreases the interaction between p53 and MDM2 but increase the level of p53. D, The levels of protein RBBP6, p53 and p21 increased following 10 Gy radiation
We next explored the mechanism of elevated expression of p53 protein in the absence of RBBP6. p53 is antagonized primarily by murine double minute 2 (MDM2), which can promote the ubiquitination and degradation of p53 protein. Thus, we investigated whether RBBP6 interacted with MDM2 in colorectal cancer cells. Co‐immunoprecipitation assays suggested that endogenous RBBP6 and MDM2 have physical interaction in SW620 and HT29 cell lysates (Figure 7B). In addition, inhibition of RBBP6 decreases the interaction between p53 and MDM2 but increases the level of p53 (Figure 7C). These results demonstrated that RBBP6 can bind MDM2 and promote the assembly of p53/MDM2 complex.
We treated both the RBBP6‐KD and control cells with 10 Gy of irradiation and cultured for different periods of time (24, 48 and 72 hours) to observe the changes in p53‐related proteins. By western blotting, we found that the expression of p53 protein was gradually increased with the prolongation of culture time in both RBBP6‐KD and control cells. However, the p53 protein levels in the RBBP6‐KD cells increased more significantly (Figure 7D), which indicated that radiation treatment can increase the level of p53, while inhibition of RBBP6 can amplify this effect. Interestingly, the level of p21 protein also increased with the increase of p53 protein. As a negative regulator of cell cycle, the upregulation of p53‐inducible p21 results in downregulation of Cyclin B1/Cdk1, which eventually leads to G2/M cycle arrest.
3.7. Inhibition of RBBP6 enhanced radiosensitivity of colorectal cancer in vivo
Based on the above results in vitro, we hypothesized that RBBP6 may be a key target for radiation therapy in colorectal cancer. Then we proceeded to study whether treatment with lentiviral vectors (RBBP6‐shRNA) would enhance tumor response to radiotherapy. We established HT29 tumor xenografts into the hind legs of nude mice. When the tumors grew up to approximately 8 mm in diameter (200 mm3), the nude mice were divided into 4 groups and each group had 5 nude mice. The 4 groups of tumors were treated with lentiviral vectors (scrambled shRNA), lentiviral vectors (RBBP6‐shRNA), lentiviral vectors (scrambled shRNA) plus local tumor irradiation, and lentiviral vectors (RBBP6‐shRNA) plus local tumor irradiation, respectively. The tumor volume curves were drawn before the mice were killed on the 28th day and the harvested tumor weights were measured (Figure 8A,B). As the data revealed, RBBP6‐KD‐treated mice showed a significant suppression of tumor progression compared to the control group (Figure 8C,D).
Figure 8.
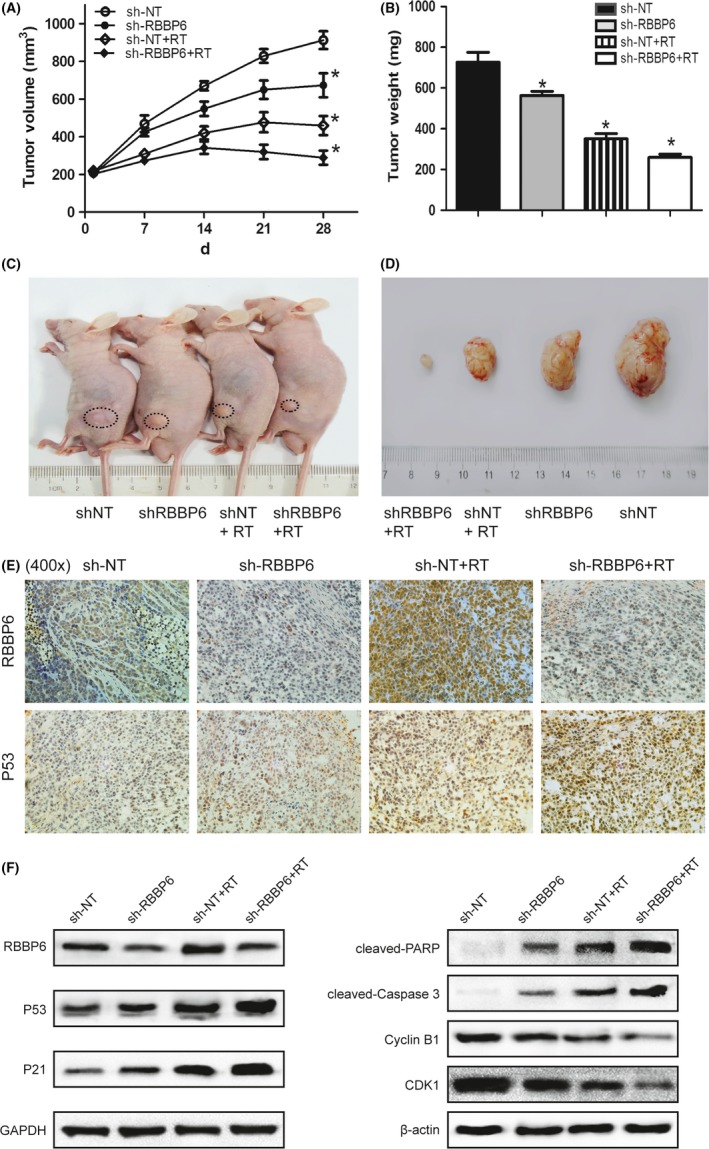
Effect of RBBP6‐KD and radiation on tumor xenograft growth. A, Tumor volume curves of nude mice in different groups. B, Average weight of tumors harvested in the 4th week. C,D, RBBP6‐KD significantly inhibited growth of tumor xenograft in nude mice. E, Expression of RBBP6 protein and p53 protein in HT29 tumor xenografts. F, Expression of p53, p21, cleaved‐PARP, cleaved‐Caspase 3 and Cyclin B1/CDK1 proteins in HT29 tumor xenografts
As the immunohistochemical analysis showed (Figure 8E), lentiviral vector transfer resulted in a decrease of RBBP6 protein in HT29 tumor xenografts. Obviously, the levels of RBBP6 protein expression in the sh‐RBBP6 group were lower than that in the sh‐NT group. Furthermore, RT could significantly increase the expression of protein p53, while silencing of RBBP6 can amplify this effect. By western blotting, we again demonstrated the upregulation of p53, p21, cleaved‐PARP and cleaved‐Caspase 3 proteins and the downregulation of Cyclin B1/CDK1 in the RBBP6‐KD group (Figure 8F). The mechanism by which RBBP6 degrades protein p53 and increases the radioresistance of CRC is shown in Figure 9.
Figure 9.
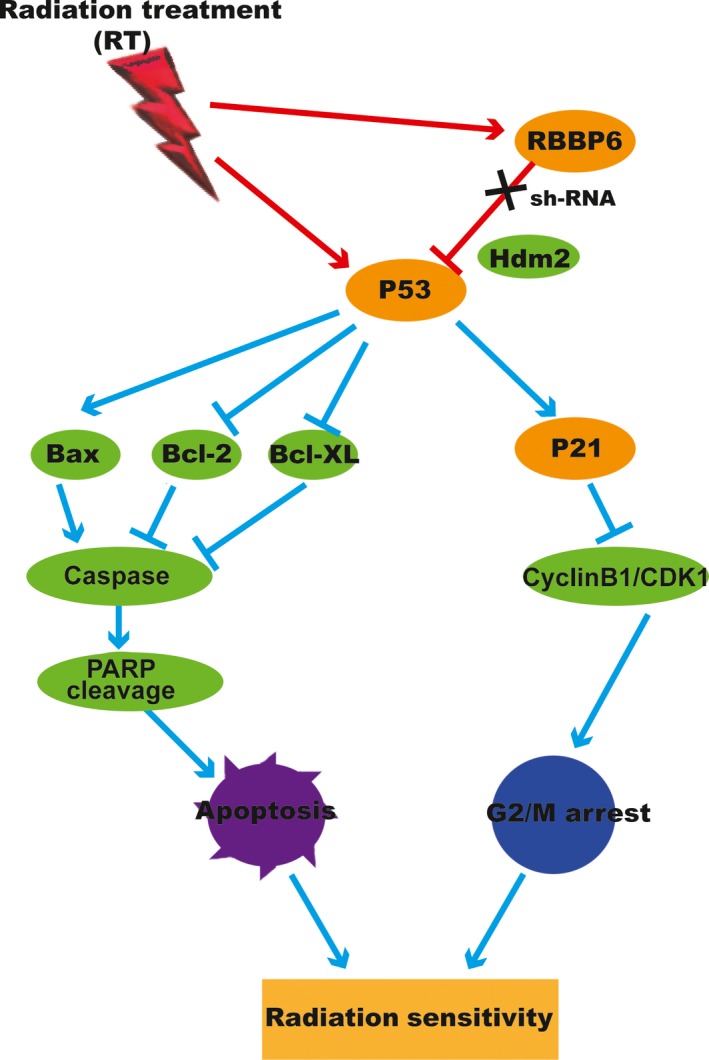
Radioresistance is associated with G2/M arrest and repression of apoptosis pathway by upregulation of Bcl‐XL, Bcl‐2 and CyclinB1/CDK1 and downregulation of p53, p21, Bax, Caspase‐3 and cleaved PARP in colorectal cancer (CRC)
4. DISCUSSION
To the best of our knowledge, this is the first report confirming that overexpression of RBBP6 endows colorectal cancer cells with resistance to apoptosis, which may compromise the therapeutic efficacy of preoperative radiotherapy. RBBP6 expression seemed to be a predictive molecular marker for radiation response and inhibition of RBBP6 could enhance the radiosensitivity of human colorectal cancer.
High expression of RBBP6 protein has been found to be related to cell cycle arrest, decreased apoptosis, and increased chemoresistance in breast cancer, lung cancer and cervical cancer.13, 14, 15 Our previous study showed that RBBP6 expression was higher in colorectal cancer samples than that of the adjacent noncancerous tissue and RBBP6 overexpression was correlated with recurrence and a poor survival rate.19 We provided clinical evidence of high expression of RBBP6 in promoting the progression of colon cancer. Based on the above evidence, in the present study, we investigated whether RBBP6 plays a key role in regulating radiation resistance. As our present results showed, RBBP6 was significantly upregulated after irradiation in both SW620 and HT29 cells. Therefore, we hypothesized that RBBP6 may be related to the radiotherapy sensitivity of colorectal cancer.
To further clarify the role of RBBP6 in radiotherapy, a shRNA strategy was applied to silence RBBP6 in radioresistant SW620 and HT29 cell lines. The data showed that the clonogenic survival rate and cell viability were significantly decreased in RBBP6‐shRNA‐treated cells compared to the control cells. Attenuation of RBBP6 protein expression by shRNA treatment resulted in an increased apoptosis rate induced by radiation. In addition, transfection with RBBP6 shRNA induced a G2‐M arrest. Thus, at the time of irradiation, the RBBP6‐knockdown cells were blocked in a more radiosensitive phase of the cell cycle.20 We further investigated the mechanism of RBBP6 inhibition to promote apoptosis and cycle arrest. We found that RBBP6 knockdown results in elevated levels of p53 and p21 protein, but there was no obvious change in Rb protein. Acting as a tumor suppressor, p53 plays an important role in regulating cell apoptosis and cycle progression. p53 is antagonized primarily by murine double minute 2 (MDM2), which can promote the ubiquitination and degradation of p53 protein.9 Activation of p53 contributes to the expression of p21, which can block the cell cycle in the G2 phase by inhibiting the M phase checkpoint CyclinB1/CDK1 complex.11 Co‐immunoprecipitation experiments confirmed the interaction between endogenous RBBP6 and MDM2 in cells and inhibition of RBBP6 decreases the interaction between p53 and MDM2. Western blotting showed that radiation treatment could increase the level of p53 protein, while RBBP6 knockdown can enhance this effect. These results demonstrated that RBBP6 can bind MDM2 and promote the assembly of p53/MDM2 complex. As the expression of p53 increased, other protein levels also changed. The proapoptotic proteins Bax and cleaved Caspase‐3 increased while antiapoptotic protein Bcl‐2 decreased, which could lead to cell apoptosis. Upregulation of p21 protein inhibits the expression of CyclinB1/CDK1 and eventually leads to G2‐M arrest. In vivo, the tumor xenograft growth of mice treated with radiation plus lentiviral vectors (RBBP6‐shRNA) was significantly inhibited compared with the single‐treatment groups. These results proved for the first time that RBBP6 plays a critical role in regulating radioresistance and downregulation of RBBP6 can increase the radiotherapy sensitivity of colorectal cancer cells.
Radiotherapy has been established as an effective local treatment for carcinomas.21, 22, 23, 24, 25 Preoperative radiotherapy is a promising treatment option to downstage initially unresectable tumors, increase the resection rate and improve the overall survival of patients, especially for patients with low rectal cancer.26 However, the rates of local and systemic recurrence vary significantly after radiotherapy, which illustrates that progression and resistance remain common events.27, 28, 29 Preoperative chemoradiotherapy results in fewer than 50% of rectal cancer patients achieving complete pathological response.30 Response to radiation is considered to be a key factor in the success of radiotherapy, but the response to radiation is determined by multifactorial etiologies (tumor pathologic type, tumor volume, tumor hypoxia, clinical stage, radiation dose and radiation modes).31, 32 Until now, no valid radiosensitizing target has been unambiguously identified to have prognostic or predictive value for colorectal cancer radiotherapy, although a few potential molecular markers, including EGFR, KRAS/NRAS, BRAF, PTEN and PIK3CA, have been investigated;33, 34 the role they played in radiotherapy remains to be elucidated. In the present study, we observed that RBBP6 overexpression could enhance cell survival following radiation exposure. The RBBP6 shRNA‐induced arrest of colorectal cancer cells in the more radiosensitive G2‐M stage may well lead to cell apoptosis. Conversely, high expression of RBBP6 in colorectal cancer cells may contribute to radioresistance.
Apoptosis is the primary mechanism for the death of colorectal cancer cells induced by radiotherapy.35, 36 In the course of radiotherapy, several apoptotic signaling pathways could be involved and the resistance to radiation may result from disruption of the pathways.37, 38 In this study, RBBP6 inhibition obviously increased the extent of cell apoptosis after irradiation treatment, supporting an anti‐apoptotic function of RBBP6. To further explore the possible mechanism by which RBBP6 promotes apoptosis evasion and radioresistance, we used western blot analysis to examine the expression of PARP and Caspase‐3. As the results showed, in RBBP6‐KD cells exposed to radiation, compared with control cells, significantly more cleaved Caspase‐3 and cleaved PARP were demonstrated. This indicates that inhibition of RBBP6 expression may contribute to the induction of apoptosis through caspase‐dependent pathways.
In summary, our results proposed that RBBP6, as demonstrated in pretreatment biopsies, could serve as a molecular biomarker for identifying patients likely to respond to preoperative radiotherapy. Inhibiting the overexpression of RBBP6 in tumors can improve the therapeutic effect of radiotherapy for colorectal cancer. The underlying mechanisms of anti‐RBBP6 strategies for improving the radiation sensitivity of colorectal cancer appear to be multifaceted, and involve alterations in cell cycle distribution as well as regulation of caspase‐dependent apoptosis. Based on the data, we hypothesize that RBBP6 may be involved in radiosensitivity of colorectal cancer, at least in part, by regulating the p53/MADM2 pathway.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Xiao C, Wang Y, Zheng M, et al. RBBP6 increases radioresistance and serves as a therapeutic target for preoperative radiotherapy in colorectal cancer. Cancer Sci. 2018;109:1075–1087. https://doi.org/10.1111/cas.13516
Funding information
National Natural Science Foundation of China (81472241 and 81270557); Special Research Fund for the Doctoral Program of Higher Education of China (20130073110018).
Chao Xiao, Yupeng Wang and Miao Zheng contributed equally to this work.
Contributor Information
Hong Zhang, Email: hong.zhang@oru.se.
Xiaoliang Wang, Email: xiaoliangwang1975@hotmail.com.
REFERENCES
- 1. Bhandari A, Woodhouse M, Gupta S. Colorectal cancer is a leading cause of cancer incidence and mortality among adults younger than 50 years in the USA: a SEER‐based analysis with comparison to other young‐onset cancers. J Investig Med. 2017;65:311‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deijen CL, Tsai A, Koedam TW, et al. Clinical outcomes and case volume effect of transanal total mesorectal excision for rectal cancer: a systematic review. Tech Coloproctol. 2016;20:811‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghiselli R, Ortenzi M, Cardinali L, Skrami E, Gesuita R, Guerrieri M. Functional outcomes after TEM in patients with complete clinical response after neoadjuvant chemoradiotherapy. Surg Endosc. 2016;31:2997‐3003. [DOI] [PubMed] [Google Scholar]
- 4. Rodel C, Hofheinz R, Fokas E. Rectal cancer: neoadjuvant chemoradiotherapy. Best Pract Res Clin Gastroenterol. 2016;30:629‐639. [DOI] [PubMed] [Google Scholar]
- 5. Hafner MF, Debus J. Radiotherapy for colorectal cancer: current standards and future perspectives. Visc Med. 2016;32:172‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shin JS, Tut TG, Yang T, Lee CS. Radiotherapy response in microsatellite instability related rectal cancer. Korean J Pathol. 2013;47:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bahnassy AA, Zekri AR, Salem SE, et al. Differential expression of p53 family proteins in colorectal adenomas and carcinomas: prognostic and predictive values. Histol Histopathol. 2014;29:207‐216. [DOI] [PubMed] [Google Scholar]
- 8. Chen TH, Huang CC, Yeh KT, et al. Human papilloma virus 16 E6 oncoprotein associated with p53 inactivation in colorectal cancer. World J Gastroenterol. 2012;18:4051‐4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amaral JD, Xavier JM, Steer CJ. The role of p53 in apoptosis. Disc Med. 2010;9:145‐152. [PubMed] [Google Scholar]
- 10. Tutone M, Almerico AM. Recent advances on CDK inhibitors: an insight by means of in silico methods. Eur J Med Chem. 2017;142:300‐315. [DOI] [PubMed] [Google Scholar]
- 11. Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579‐590. [DOI] [PubMed] [Google Scholar]
- 12. Pugh DJ, Ab E, Faro A, Lutya PT, Hoffmann E, Rees DJ. DWNN, a novel ubiquitin‐like domain, implicates RBBP6 in mRNA processing and ubiquitin‐like pathways. BMC Struct Biol. 2006;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Motadi LR, Bhoola KD, Dlamini Z. Expression and function of retinoblastoma binding protein 6 (RBBP6) in human lung cancer. Immunobiology. 2011;216:1065‐1073. [DOI] [PubMed] [Google Scholar]
- 14. Moela P, Choene MM, Motadi LR. Silencing RBBP6 (Retinoblastoma Binding Protein 6) sensitises breast cancer cells MCF7 to staurosporine and camptothecin‐induced cell death. Immunobiology. 2014;219:593‐601. [DOI] [PubMed] [Google Scholar]
- 15. Moela P, Motadi LR. RBBP6: a potential biomarker of apoptosis induction in human cervical cancer cell lines. Onco Targets Ther. 2016;9:4721‐4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miotto B, Chibi M, Xie P, et al. The RBBP6/ZBTB38/MCM10 axis regulates DNA replication and common fragile site stability. Cell Rep. 2014;7:575‐587. [DOI] [PubMed] [Google Scholar]
- 17. Huang P, Ma X, Zhao YM, Miao L. The C. elegans Homolog of RBBP6 (RBPL‐1) regulates fertility through controlling cell proliferation in the germline and nutrient synthesis in the intestine. PLoS One. 2013;8:e58736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dlamini Z, Rupnarain C, Naicker S, Hull R, Mbita Z. Expression analysis and association of RBBP6 with apoptosis in colon cancers. J Mol Histol. 2016;47:169‐182. [DOI] [PubMed] [Google Scholar]
- 19. Chen J, Tang H, Wu Z, et al. Overexpression of RBBP6, alone or combined with mutant TP53, is predictive of poor prognosis in colon cancer. PLoS One. 2013;8:e66524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herst PM, Broadley KW, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med. 2012;52:1486‐1493. [DOI] [PubMed] [Google Scholar]
- 21. Peponi E, Skloupiotis V, Tsironis D, et al. Preoperative chemoradiation in locally advanced rectal cancer: efficacy and safety. Gastroenterology Res. 2015;8:303‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu KT, Wan JF, Zhu J, et al. Role of pelvic radiotherapy for locally advanced rectal cancer and synchronous unresectable distant metastases. Cancer Radiother. 2016;20:805‐810. [DOI] [PubMed] [Google Scholar]
- 23. Garg PK, Sharma J, Jakhetiya A, Goel A, Gaur MK. Preoperative therapy in locally advanced esophageal cancer. World J Gastroenterol. 2016;22:8750‐8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jegadeesh N, Liu Y, Zhang C, et al. The role of adjuvant radiotherapy in pathologically lymph node positive prostate cancer. Cancer. 2016;123:512‐520. [DOI] [PubMed] [Google Scholar]
- 25. Rivera R, Banks A, Casillas‐Lopez A, et al. Targeted intraoperative radiotherapy for the management of ductal carcinoma in situ of the breast. Breast J. 2016;22:63‐74. [DOI] [PubMed] [Google Scholar]
- 26. Li JL, Ji JF, Cai Y, et al. Preoperative concomitant boost intensity‐modulated radiotherapy with oral capecitabine in locally advanced mid‐low rectal cancer: a phase II trial. Radiother Oncol. 2012;102:4‐9. [DOI] [PubMed] [Google Scholar]
- 27. Glimelius B. Neo‐adjuvant radiotherapy in rectal cancer. World J Gastroenterol. 2013;19:8489‐8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nahas SC, Nahas CSR, Marques CFS, et al. Pathologic complete response in rectal cancer: can we detect it? lessons learned from a proposed randomized trial of watch‐and‐wait treatment of rectal cancer. Dis Colon Rectum. 2016;59:255‐263. [DOI] [PubMed] [Google Scholar]
- 29. Li Y, Wang J, Ma XW, et al. A review of neoadjuvant chemoradiotherapy for locally advanced rectal cancer. Int J Biol Sci. 2016;12:1022‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia‐Albeniz X, Gallego R, Hofheinz RD, et al. Adjuvant therapy sparing in rectal cancer achieving complete response after chemoradiation. World J Gastroenterol. 2014;20:15820‐15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hulshof MCCM, van Laarhoven HWM. Chemoradiotherapy in tumours of the oesophagus and gastro‐oesophageal junction. Best Pract Res Cl Ga. 2016;30:551‐563. [DOI] [PubMed] [Google Scholar]
- 32. Stoker SD, Fles R, Herdini C, et al. The impact of the overall radiotherapy time on clinical outcome of patients with nasopharyngeal carcinoma. A retrospective study. PLoS One. 2016;11:e0151899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kidess‐Sigal E, Liu HE, Triboulet MM, et al. Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label‐free platform: comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget. 2016;7:85349‐85364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sievers CK, Kratz JD, Zurbriggen LD, et al. The multidisciplinary management of colorectal cancer: present and future paradigms. Clin Colon Rectal Surg. 2016;29:232‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arora H, Qureshi R, Rizvi MA, Shrivastava S, Parihar MS. Study of apoptosis‐related interactions in colorectal cancer. Tumour Biol. 2016;37:14415‐14425. [DOI] [PubMed] [Google Scholar]
- 36. Deng Q, Huang CM, Chen N, et al. Chemotherapy and radiotherapy downregulate the activity and expression of DNA methyltransferase and enhance Bcl‐2/E1B‐19‐kDa interacting protein‐3‐induced apoptosis in human colorectal cancer cells. Chemotherapy. 2012;58:445‐453. [DOI] [PubMed] [Google Scholar]
- 37. Abraha AM, Ketema EB. Apoptotic pathways as a therapeutic target for colorectal cancer treatment. World J Gastroenterol. 2016;8:583‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]