

Abstract
Radiotherapy induces anti‐tumor immunity by induction of tumor antigens and damage‐associated molecular patterns (DAMP). DNA, a representative DAMP in radiotherapy, activates the stimulator of interferon genes (STING) pathway which enhances the immune response. However, the immune response does not always parallel the inflammation associated with radiotherapy. This lack of correspondence may, in part, explain the radiation‐resistance of tumors. Additive immunotherapy is expected to revive tumor‐specific CTL facilitating radiation‐resistant tumor shrinkage. Herein pre‐administration of the double‐stranded RNA, polyinosinic‐polycytidylic acid (polyI:C), in conjunction with radiotherapy, was shown to foster tumor suppression in mice bearing radioresistant, ovalbumin‐expressing Lewis lung carcinoma (LLC). Extrinsic injection of tumor antigen was not required for tumor suppression. No STING‐ and CTL‐response was induced by radiation in the implant tumor. PolyI:C was more effective for induction of tumor growth retardation at 1 day before radiation than at post‐treatment. PolyI:C targeted Toll‐like receptor 3 with minimal effect on the mitochondrial antiviral‐signaling protein pathway. Likewise, the STING pathway barely contributed to LLC tumor suppression. PolyI:C primed antigen‐presenting dendritic cells in draining lymph nodes to induce proliferation of antigen‐specific CTL. By combination therapy, CTL efficiently infiltrated into tumors with upregulation of relevant chemokine transcripts. Batf3‐positive DC and CD8+ T cells were essential for therapeutic efficacy. Furthermore, polyI:C was shown to stimulate tumor‐associated macrophages and release tumor necrosis factor alpha, which acted on tumor cells and increased sensitivity to radiation. Hence, polyI:C treatment prior to radiotherapy potentially induces tumor suppression by boosting CTL‐dependent and macrophage‐mediated anti‐tumor responses. Eventually, polyI:C and radiotherapy in combination would be a promising therapeutic strategy for radiation‐resistant tumors.
Keywords: cytotoxic T lymphocyte, dendritic cell, radiation, Toll‐like receptor 3, tumor necrosis factor‐α
Abbreviations
- Batf3
basic leucine‐zipper ATF‐like transcription factor 3
- cGAMP
cyclic GMP‐AMP
- cGAS
cyclic GMP‐AMP synthase
- DAMP
damage‐associated molecular patterns
- DC
dendritic cells
- DLN
draining lymph node
- dsRNA
double‐stranded RNA
- IFN
interferon
- IR
ionizing radiation
- LLC
Lewis lung carcinoma
- LLC‐OVA
ovalbumin‐expressing Lewis lung carcinoma
- MAVS
mitochondrial antiviral‐signaling protein
- MDSC
myeloid derived suppressor cells
- MyD88
myeloid differentiation primary response 88
- pDC
plasmacytoid dendritic cells
- polyI:C
polyinosinic‐polycytidylic acid
- STING
stimulator of interferon genes
- TAA
tumor‐associated antigen
- TAM
tumor‐associated macrophages
- TBK
TANK‐binding kinase 1
- TICAM‐1
toll‐interleukin 1 receptor domain (TIR)‐containing adaptor molecule‐1
- TIR
toll‐interleukin 1 receptor domain
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor alpha
1. INTRODUCTION
Tumor‐directed irradiation induces biological response in both the tumor and tumor‐infiltrating immune cells. IR provokes DNA damage in the tumor and induces inflammation in the tumor microenvironment.1, 2 TAA are released from damaged cells that facilitate cross‐presentation by DC, which reside in DLN and cross‐prime antigen‐specific T cells. Simultaneously, DNA liberated from tumor cells by radiation serves as a DAMP that modulates DC maturation.3 Both DAMP and TAA liberated from irradiated tumors act on immune cells within tumors and DLN.
Endosomal detection of DNA activates the TLR 9‐MyD88 signaling pathway in pDC. Cytosolic detection of DNA activates the cGAS‐STING pathway in the cytoplasm of various cells.4 The 2 pathways are linked to activation of TBK1 and IκB kinase, leading to induction of type I IFN.5 Type I IFN signaling by STING but not by MyD88 plays an important role in DC maturation and CTL‐dependent tumor growth retardation during radiotherapy of the MC38 tumor.6 By contrast, endosomal detection of structured RNA leads to activation of the TLR3‐TICAM‐1 pathway.7 This pathway specifically resides in myeloid cells including DC and macrophages.8 CTL are effectively induced by exogenously added TAA and polyI:C, a kind of synthetic dsRNA mimic.9 Although structured RNA might be a DAMP from host cells,10 less knowledge on RNA DAMP has been reported than DNA DAMP, thereby the relationship between RNA and radiation therapy has been poorly discussed.
Nevertheless, maturation of DC by innate sensors is an essential prerequisite for induction and proliferation of tumor‐specific CTL.11 Once CTL are induced to proliferate in response to TAA, tumor regression occurs in primary as well as in metastatic tumors.12 CTL infiltration into the tumor is essential for tumor growth retardation at distant sites. With radiation therapy, an abscopal effect has been reported in rare clinical cases, which implies that CTL induction by radiotherapy is effective not only locally but also at distant tumor sites.13, 14
In experimental LLC‐OVA implanted mice, CTL are barely induced by radiation alone and host cell STING is not involved in therapeutic efficacy. Hence, radiation monotherapy is not sufficient to activate an immune response. However, when radiation was combined with a dsRNA, polyI:C, radiation‐induced anti‐tumor immunity emerged without giving TAA (OVA antigen in the case of LLC‐OVA). The TLR3 pathway was essential for the anti‐tumor effect of polyI:C.9 In the tumor microenvironment, TLR3 is expressed in Batf3‐positive DC (CD141+ DC in human, CD8α+ and CD103+ DC in mouse) and in TAM, but not in pDC.15, 16, 17 TAM are known to release TNF‐α in response to polyI:C to induce tumor cell death.16 Herein, the therapeutic effect of polyI:C in combination with radiotherapy is shown to be a result of priming of CTL by TLR3‐positive DC and enhanced radiation‐sensitivity through TNF‐α produced by intra‐tumor macrophages.
2. MATERIALS AND METHODS
2.1. Mice
Inbred wild‐type C57BL/6 (WT B6) and Batf3 −/− mice were purchased from Clea Japan and Jackson Laboratory, respectively. Tnf‐α−/− and Tlr3 −/− mice were kindly provided by Y. Iwakura (Tokyo University of Science) and S. Akira (Osaka University) respectively. Ticam‐1 −/− and Mavs −/− mice were generated in our laboratory.18, 19 STING‐deficient (Tmem173 −/−) mice were developed in our laboratory by CRISPR/Cas9 method as previously reported.20 Mice 6‐14 weeks old were used and maintained under specific pathogen‐free conditions. All animal experiments were approved by the Institutional Animal Care and Use Committee of Hokkaido University and carried out in compliance with their guidelines.
2.2. Cell culture
LLC‐OVA cells,21 kindly provided by Dr T. Nishimura and Dr H. Kitamura (Hokkaido University), were cultured in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FBS, 2 mmol/L l‐glutamine, 25 mmol/L HEPES buffer, 55 μmol/L 2‐mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin (Life Technologies), and 100 μg/mL G418 (Roche).
2.3. Tumor challenge
Mice were shaved on the back and LLC‐OVA cells (2 × 106) suspended in 200 μL PBS were s.c. injected. Tumor size was measured by calipers and volume was calculated using the following formula: tumor volume (cm3) = (long diameter) × (short diameter)2 × 0.4. When the tumor volume reached approximately 0.4 cm3, mice were i.p. injected with polyI:C (GE Healthcare) with no detectable LPS. After 24 hours, X‐irradiation was carried out using MBR‐1520R‐4 (Hitachi, Tokyo, Japan) under the condition of 150 kV, 20 mA and 1.5 Gy/min. Low‐energy radiation was filtered with a 2‐mm‐thick aluminium filter. During the irradiation, mice were anesthetized and shielded with 3‐mm‐thick lead excluding the tumor area. In vivo depletion of CD8+ T cells was achieved by i.p. injection of ascites containing mAb against CD8β.
2.4. Flow cytometry
Single‐cell suspensions isolated from tumors, spleens, or lymph nodes were stained with fluorescence‐labeled Abs after blockade with an anti‐CD16/32 Ab. For intracellular TNF‐α staining, we isolated tumors from mice 1 hour after injection of PBS or polyI:C and incubated the cells in the presence of 10 μg/mL Brefeldin A for 5 hours. Following fixation, permeabilizing and staining with anti‐TNF Ab were carried out using BD Cytofix/Cytoperm Kit (BD Biosciences). Cells were analyzed on FACS Calibur or FACS Aria II (BD Biosciences). Data analysis was done with FlowJo software (Tree Star). Abs used for the flow cytometric analysis are listed in Table S1.
2.5. Determination of TNF‐α levels in tumor
Small pieces of tumor samples were homogenized with CelLytic MT Mammalian Tissue Lysis/Extraction Reagent (Sigma, St Louis, MO, USA) supplemented with Complete Protease Inhibitor Mixture (Roche). TNF‐α levels in lysate were determined using a cytometric beads assay (BD Biosciences).22
2.6. WST‐1 assay
5 × 103 cells were cultured in 96‐well plates and treated with TNF‐α (R&D Systems). X‐irradiation was carried out using CellRad (Faxitron Bioptics) under the condition of 130 kV, 5 mA, 1.5 Gy/min. Cells were cultured for 48 hours after radiation. Then, cell viability was determined using WST‐1 assay reagent (Dojindo Laboratories, Kumamoto, Japan).22
2.7. Quantitative reverse transcription PCR (RT‐qPCR)
Total RNA was isolated from tumor samples using TRIzol Reagent (Invitrogen) after cutting samples into small pieces. High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used for reverse transcription. Quantitative PCR was carried out with Power SYBR Green PCR Master Mix (Applied Biosystems) with a StepOne Real‐Time PCR System (Applied Biosystems). Gene expression levels of targets were normalized to GAPDH levels. Data were analyzed by the ΔΔC T method. Primer pairs are listed in Table S2.
2.8. Statistical analysis
P‐values were calculated using the following statistical analysis. Mann‐Whitney U‐test and one‐way ANOVA with Bonferroni's test were carried out in the case of 2 groups‐ and multiple‐comparison, respectively.
3. RESULTS
3.1. PolyI:C enhances radio‐induced tumor shrinkage through the TLR3‐TICAM‐1 pathway
LLC‐OVA cells were implanted into mice and when tumors grew to ~0.4 cm3, tumor‐directed X‐irradiation was carried out as previously reported.23, 24 One day before the irradiation, mice were i.p. injected with polyI:C or left untreated. The LLC‐OVA tumor is known to be relatively resistant to radiotherapy and to immunotherapy.23 PolyI:C or radiation alone induced only modest tumor growth retardation (Figure 1A). However, combination of the two was more effective and induced tumor shrinkage (Figure 1A). Pretreatment with polyI:C was better than post‐treatment judged by the degree of tumor growth retardation (Figure S1). These results suggest that pretreatment with polyI:C additively enhances radiation‐induced tumor growth inhibition. Because polyI:C stimulates both the endosomal TLR3‐TICAM‐1 and the cytosolic melanoma differentiation‐associated gene 5 (MDA5) MAVS pathways,25 contribution of these 2 pathways to retardation of tumor growth was assessed in KO mice (Figure 1B). Tumor growth suppression was largely abrogated in TLR3‐ or TICAM‐1‐deficient mice with minimal effect in MAVS‐deficient animals. Hence, inhibition of tumor growth was mainly through the TLR3‐TICAM‐1 pathway in response to polyI:C‐radiation therapy.
Figure 1.
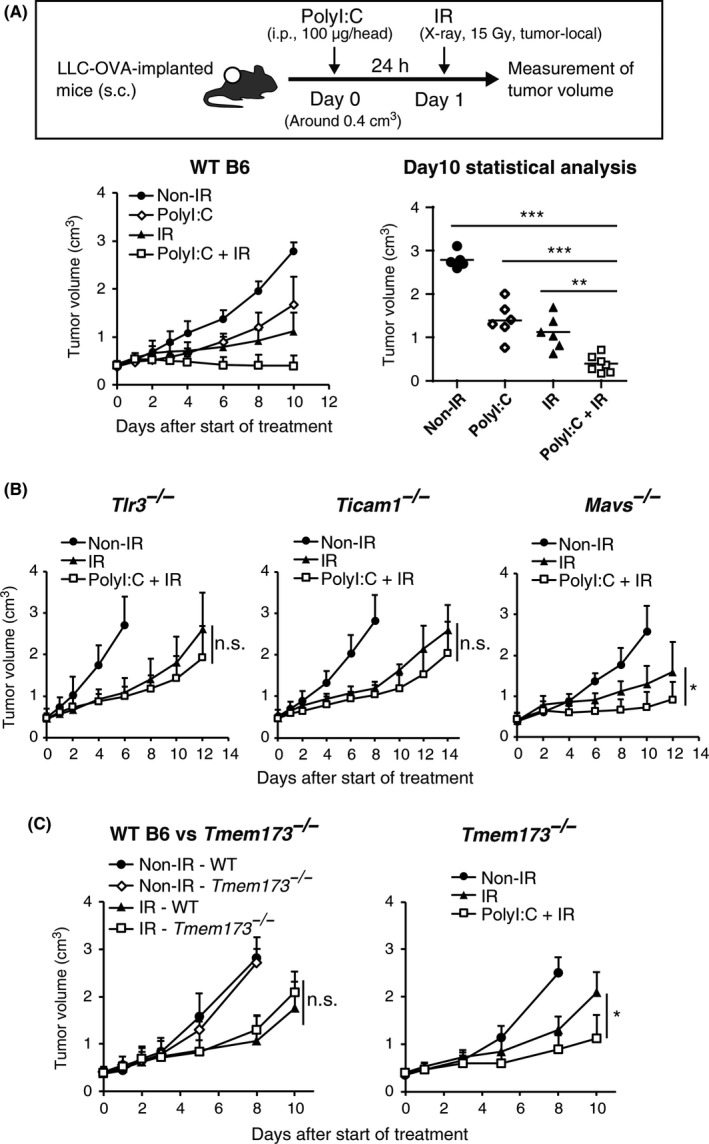
Augmentation of radiation‐induced tumor growth retardation by polyinosinic‐polycytidylic acid (polyI:C): identification of the key signaling pathway. A, Ovalbumin‐expressing Lewis lung carcinoma (LLC‐OVA)‐implanted WT B6 mice were i.p. injected with polyI:C (100 μg/head) when tumor volume reached approximately 0.4 cm3. After 24 hours, 15 Gy ionizing radiation (IR) was carried out. Data were pooled from two independent experiments with similar results. n = 5‐7 mice per group. B, Growth of LLC‐OVA tumors on WT B6, Tlr3 −/−, Ticam −/−, or Mavs −/− mice in the same treatment as in Figure 1A; n = 3‐5 mice per group (C) Growth of LLC‐OVA tumors on WT B6 or Tmem173 −/− mice in the same treatment as in (A); n = 5 mice per group. Data are shown as average (SD) tumor size. *P < .05, **P < .01, ***P < .001. n.s., not significant
3.2. STING barely associates with radio‐induced LLC‐OVA growth suppression in a mouse model
Because STING (encoded by the Tmem173 gene) has been reported to be a central mediator of radiation‐induced anti‐tumor responses,6 X‐irradiation was carried out on LLC‐OVA tumor‐bearing Tmem173 −/− mice. Radiation was as effective in Tmem173 −/− recipients as it was in WT B6 mice (Figure 1C), indicating that STING in host immune cells provides little or no therapeutic benefit. Furthermore, STING is likely not involved in DC maturation and subsequent CTL activation during radiotherapy. TLR3 stimulation of DC prior to radiation therapy significantly reduced tumor size in Tmem173 −/− mice (Figure 1C). Thus, at least in the LLC‐OVA model, polyI:C increases the therapeutic effect of radiation in a STING‐independent way. As polyI:C works as a DC‐priming adjuvant,15 the role of DC/CTL in the anti‐tumor immune response was evaluated.
3.3. DC and CD8+ T cells participate in polyI:C‐radiation‐mediated tumor shrinkage
Role of polyI:C‐induced DC priming in tumor growth retardation was evaluated in the LLC‐OVA model. Batf3 −/− mice are deficient in TLR3‐positive DC15, 26 and tumor growth retardation by combination therapy was abrogated in Batf3 −/− mice (Figure 2A). Without polyI:C, tumor growth suppression by radiation‐monotherapy was similar in WT and Batf3 −/− mice (Figure 2A). Thus, unlike radiation monotherapy, combination therapy induced immunogenic tumor growth retardation governed by Batf3‐positive DC.
Figure 2.
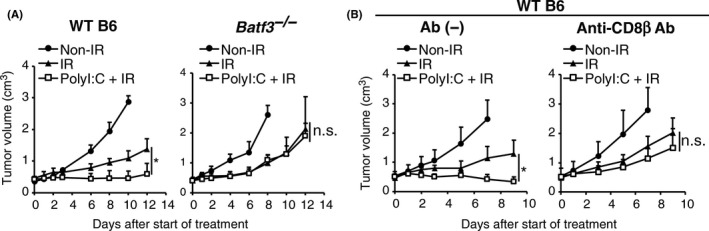
Polyinosinic‐polycytidylic acid (polyI:C) requires basic leucine‐zipper ATF‐like transcription factor 3 (Batf3)‐dependent dendritic cells (DC) and CTL in enhancing tumor growth retardation by ionizing radiation (IR). A, Growth of ovalbumin‐expressing Lewis lung carcinoma (LLC‐OVA) tumors on WT B6 or Batf3 −/− mice in the same treatment as in Figure 1A; n = 3‐4 mice per group. B, Growth of LLC‐OVA tumors on WT B6 mice with or without anti‐CD8β Ab treatment. PolyI:C injection and IR were carried out as shown in Figure 1A. Anti‐CD8β Ab was injected into tumor‐bearing mice 24 hours before and 6 days after giving polyI:C; n = 3‐5 mice per group. Data are shown as average (SD) tumor size. *P < .05. n.s., not significant
Inhibition of LLC‐OVA tumor growth with polyI:C‐radiation therapy was abolished by pretreatment with anti‐CD8β specific Ab (Figure 2B), showing that CD8+ T cells are involved in retardation of tumor growth. Radiation monotherapy poorly induces tumor‐specific CD8+ T cells, which was confirmed in the present study (Figure S2A). This result implies that the main effect of radiation is not the induction of anti‐tumor immunity but rather the direct damage of tumor cells. Indeed, X‐irradiation increased TUNEL‐positive cells in LLC‐OVA tumors within 48 hours after irradiation (Figure S2B). These results suggest that tumor‐effective CD8+ T cells are induced depending upon polyI:C pretreatment.
3.4. PolyI:C and radiation synergistically activate CTL in systemic lymphoid tissue
In lymphoid organs, Batf3‐positive DC are critical for priming CTL by cross‐presentation.27 The activation state of CTL was analyzed in DLN and spleens 6 days after the start of treatment. Separately, PolyI:C and radiation marginally increased the CTL (CD8+ CD3+) population in the spleen. However, a combination of these treatments significantly expanded this cell population (Figure 3A). Although expansion of the CD8+ CD3+ population in DLN was minimal (Figure 3A), the ratio of CTL CD44+ CD62L− cells in DLN and also spleens was most elevated with combination therapy (Figure 3B). CD44+ CD62L− CTL are known to be an effector/memory subset,28, 29 and it appears that polyI:C and radiation synergistically induce CTL‐activation and memory. Similar results were obtained by counting OVA‐tetramer+ CTL; however, the OVA‐tetramer+ population was not necessarily elevated in all mice in the polyI:C‐radiation group (Figure 3C). As CD44+ CD62L− CTL were increased in the polyI:C‐radiation group when compared to the other groups (Figure 3B), tumor antigens other than OVA may act as TAA to be involved in antigen‐cross‐presentation in Batf3‐positive DC. These antigens were not identified, but CTL activity was increased by polyI:C‐radiation therapy in systemic lymphoid organs.
Figure 3.
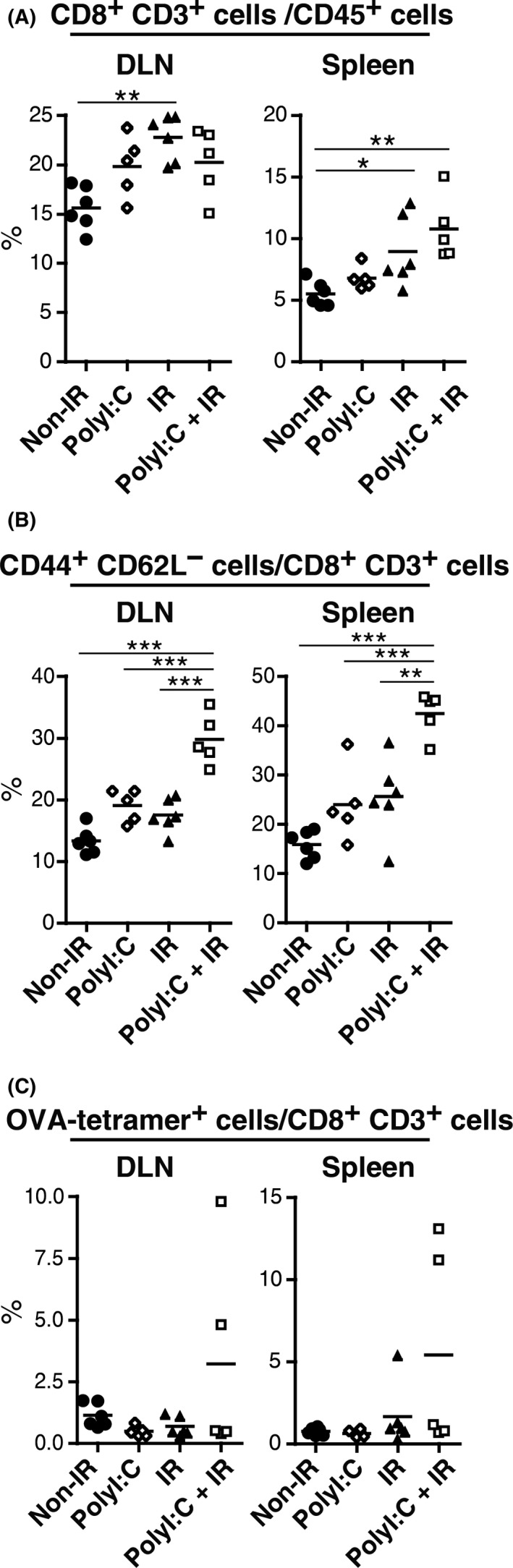
Polyinosinic‐polycytidylic acid (polyI:C) and ionizing radiation (IR) synergistically activate CTL in lymph node and spleen. A‐C, Ovalbumin‐expressing Lewis lung carcinoma (LLC‐OVA)‐implanted WT B6 mice were treated with polyI:C and IR as in Figure 1A. Lymph nodes and spleens were harvested on day 6 after the start of treatment and subjected to flow cytometric analysis; n = 5‐6 mice per group
3.5. CTL tumor infiltration is increased by polyI:C‐radiation therapy
Tumor‐infiltrating CD8+ CD3+ cells were counted in WT B6 mice bearing the LLC‐OVA tumor. Days 8 and 10 after the start of treatment, tumors were harvested and analyzed for tumor‐infiltrating immune cells by FACS. Separately, polyI:C and radiation treatment enhanced CTL infiltration, but an additive effect was observed on both days 8 and 10 when both polyI:C and radiation were given (Figure 4A). The order of polyI:C and radiation delivery did not affect CTL infiltration (data not shown). However, CD11b+ Gr‐1+ cells (MDSC)30, 31 were decreased when radiation was combined with polyI:C pretreatment, but not post‐treatment (Figure S3).
Figure 4.
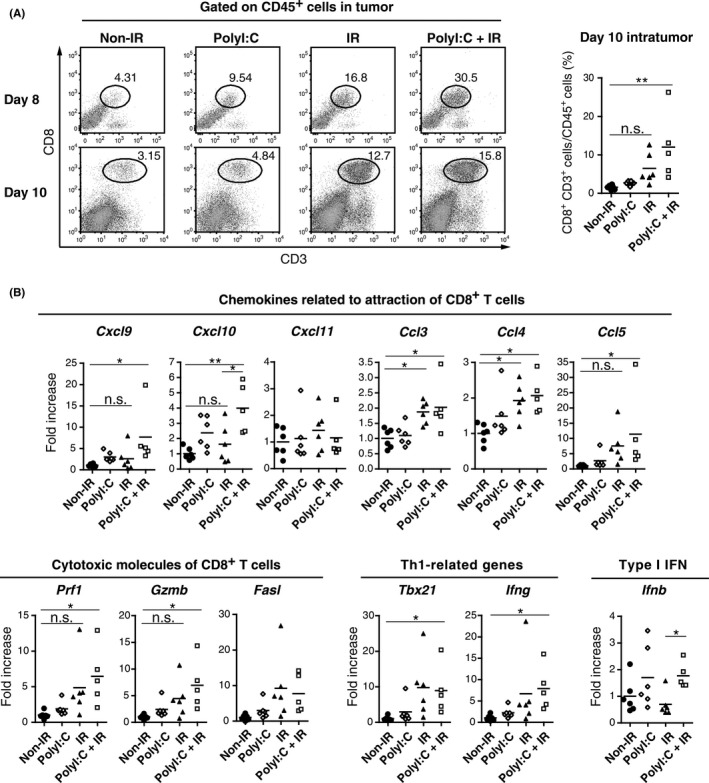
Combination of polyinosinic‐polycytidylic acid (polyI:C) and ionizing radiation (IR) augments CTL infiltration into tumor. A, Ovalbumin‐expressing Lewis lung carcinoma (LLC‐OVA)‐implanted WT B6 mice were treated with polyI:C and IR as in Figure 1A. Tumors were harvested on day 8 or 10 after the start of treatment, and cells were analyzed by FACS Calibur or Aria II. FACS plots of day 8 are representative results of 2 with similar outcomes. For FACS plots of day 10, tumors of 5‐6 mice were mixed into the single group. Dot graph shows frequency of CD8+ CD3+ CD45+ cells in individual tumors before mix. B, Analysis of gene expression levels in tumors by RT‐qPCR. Total RNA was collected from tumors harvested on day 10 after the start of polyI:C‐IR treatment as in Figure 4A; n = 5‐6 mice per group. *P < .05, **P < .01, n.s. not significant
Intratumor mRNA levels for T‐cell‐associated chemotactic and cytotoxic molecules were assessed (Figure 4B). As CD8+ T cells are known to infiltrate into tumor through the signaling of C‐X‐C chemokine receptor 3 (CXCR3) and C‐C chemokine receptor 5 (CCR5),32, 33 expression levels of ligands for these receptors were evaluated. Combination of polyI:C and radiation increased mRNA levels for Cxcl9, Cxcl10, Ccl3, Ccl4, and Ccl5. Likewise, a tendency for increased mRNA levels was observed for cytotoxicity‐related genes such as Prf1, GzmB, and Fasl.34 T helper type 1 (Th1) cells are reportedly crucial to adjust the microenvironment for CD8+ T‐cell activity.35 We also evaluated transcription levels of Th1‐related genes. Enhanced transcription of Tbx21 and Ifng was observed in the polyI:C‐radiation group. Type I interferons assist in CTL immunity36 and Ifnb transcription was increased in the polyI:C‐radiation group in comparison to the radiation‐only group. These results show that polyI:C‐radiation therapy modifies the tumor microenvironment in such a way that CTL infiltration and activation are increased and are more effective than either polyI:C or radiation therapy alone.
3.6. TNF‐α is an effector for LLC‐OVA tumor growth retardation
TNF‐α has been shown to be a pivotal effector for tumor cell death of the Lewis lung carcinoma tumor (3LL)16; hence, the efficacy of polyI:C‐radiation therapy was evaluated in Tnf‐α−/− mice (Figure 5A). Tumor growth retardation by polyI:C‐radiation therapy was abrogated in Tnf‐α−/− mice. TNF‐α was not involved in tumor growth retardation by radiation monotherapy (Figure 5A). TNF‐α was found not only to damage tumor cells but also to sensitize the cells to radiation such that TNF‐α treatment prior to radiation synergistically decreased cell viability of LLC‐OVA cells in vitro (Figure 5B). TNF‐α also decreased viability when given after radiation (Figure S4). Giving further i.p. polyI:C increased TNF‐α levels (Figure 5C) as well as the number of TNF‐α‐producing cells, indicated by intracellular fluorescent staining (Figure 5D). CD11b+ cells also produced TNF‐α in response to polyI:C. CD11c+ DC and CD45− non‐immune cells were essentially TNF‐α negative. CD11b+ cells in tumors were either Gr‐1+ MDSC or F4/80+ macrophages, namely TAM.37 It was found that only F4/80+ cells produce TNF‐α. F4/80+ macrophages constituted approximately 40% of the tumor in both PBS‐ or polyI:C‐treated mice (Figure 5D). Hence, polyI:C‐induced TNF‐α production by TAM enhanced cell death of LLC‐OVA tumors in concert with radiation, which may promote CTL induction secondary to liberated TAA. Thus, TNF‐α is involved in tumor growth retardation during polyI:C‐radiation treatment.
Figure 5.
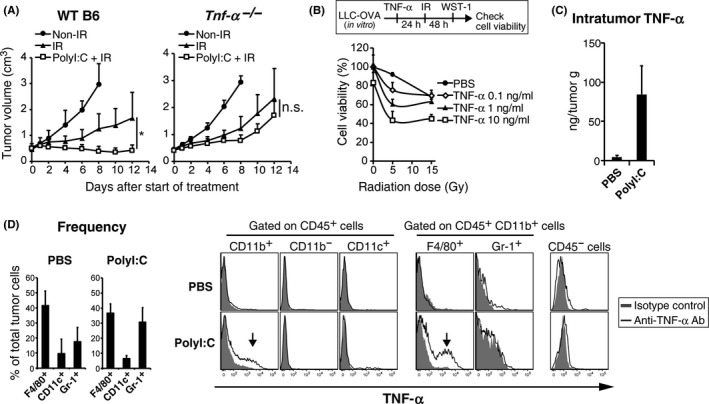
Polyinosinic‐polycytidylic acid (polyI:C) requires tumor necrosis factor alpha (TNF‐α) in augmenting ionizing radiation (IR)‐induced inhibition of tumor growth. A, Growth of ovalbumin‐expressing Lewis lung carcinoma (LLC‐OVA) tumors on WT B6 or Tnf‐α−/− mice. PolyI:C‐radiation combination therapy was carried out as per Figure 1A. B, LLC‐OVA cells were treated with TNF‐α for 24 hours and then with X‐irradiation. After 48 hours culture, cell viability was assessed by WST‐1 assay; n = 3. C, LLC‐OVA tumor‐bearing WT B6 mice were i.p. injected with polyI:C (100 μg/head). After 1 hours, tumors were collected and TNF‐α concentration was determined by cytometric bead assay. D, Tumor‐infiltrating cells from PBS‐ or polyI:C‐treated mice in Figure 5C were cultured with brefeldin A (10 μg/mL) for 5 hours. Frequency of tumor‐infiltrating cells and intracellular TNF‐α expression in cells from PBS or polyI:C‐treated mice were determined by FACS. Representative FACS plots of 4 similar outcomes are shown. Data represent the means (SD)
4. DISCUSSION
The present study shows the importance of the TLR3 signaling pathway in augmentation of radiation‐mediated tumor growth suppression. Intraperitoneal pretreatment of mice with polyI:C before radiation showed higher efficacy against the tumor than post‐treatment. This pretreatment effect did not depend on CTL infiltration nor on TNF‐α (Figure S4), but was possibly a result of the decrease of MDSC (Figure S3). PolyI:C activity was abrogated in Ticam1 −/− but not in Mavs −/− mice, suggesting that the TLR3 pathway rather than the cytoplasmic pathway is predominantly involved in enhancing the therapeutic effect of radiation. Both Batf3‐positive DC and CD8+ T cells were required for tumor growth retardation (Figure 6A). 3LL cells are sensitive to TNF‐α.16 Similarly, TNF‐α increased the susceptibility of LLC‐OVA to radiation and TAM produced TNF‐α in response to polyI:C (Figure 6B).
Figure 6.

Possible mechanisms of elevated therapeutic efficacy of radiation in combination with polyinosinic‐polycytidylic acid (polyI:C). A, PolyI:C‐induced tumor growth suppression with ionizing radiation (IR) depends on basic leucine‐zipper ATF‐like transcription factor 3 (Batf3)‐dependent dendritic cells (DC) and CTL. Toll‐like receptor‐3 (TLR3) stimulation leads to maturation of DC, subsequent CTL activation in lymphoid tissue, and infiltration into tumor. Dead cells arising in IR treatment may supply tumor‐associated antigens (TAA) to DC and contribute to the retardation of tumor growth. B, PolyI:C acts on tumor‐associated macrophages (TAM) and promotes TNF‐α production. As TNF‐α enhances radiosensitivity of LLC‐OVA cells, elevated damage by IR is expected to suppress tumor growth directly and augment anti‐tumor immunity by supplying TAA
LLC‐OVA cells contain OVA and other antigens that can be released from damaged cells. Locally released TAA are cross‐presented by DC with the involvement of Batf3‐positive DC (Figure 6). Tumor‐derived material such as DNA may modulate the tumor microenvironment or DC within DLN. However, this was not the case in that no tumor growth retardation was observed in Tmem173 −/− mice. In contrast, giving i.p. dsRNA localized to DC and then primed the DC,9 whereas tumor cell nucleic acids DAMP did not do so. The reason for this differential effect is unknown. Cytoplasmic delivery of DNA to DC is a complicated process that can be accomplished by cell‐to‐cell contact and may be involved in cross‐priming of CTL that mediate inhibition of tumor growth. These results show that dsRNA differs from DAMP DNA in the context of DC priming in LLC‐OVA tumor‐bearing mice.
Radiotherapy‐induced, locally‐secreted DNA activates the cGAS‐STING pathway, which stimulates the innate immune system. DNA or DNA vaccination induces T‐cell immune responses by way of the TBK1‐interferon regulation factor 3 (IRF3)‐IFN‐α/β pathway. It is likely that DNA from irradiated tumor cells is the mediator of cGAS‐STING signaling in DC. Although the STING pathway is not involved in the therapeutic effect of radiation monotherapy for the LLC‐OVA tumor model, insufficient STING activation can be overcome by giving STING ligands (eg, cGAMP).6 However, cGAMP has been reported to induce a Th2 lymphocyte response38 and Th2 cytokines are known to suppress Th1 immunity in mice39 and patients with cancer.40 Interaction of polyI:C with antigen‐presenting DC would result in Th1 polarization and CTL proliferation.9 Whether polyI:C in combination with tumor‐derived DNA serves as an adjuvant for DC priming in this model is unknown. However, herein, polyI:C has been clearly shown to act as a TLR3 agonist, directly priming Batf3‐positive DC with concomitant tumor growth retardation.
In mouse models, polyI:C induces DC cross‐presentation of antigens to tumor‐specific CTL. TLR3 adjuvant is essential for this form of DC priming.41 However, few reports mention TLR3 ligand‐radiation combined therapy in mice. Why low concentrations of radiation‐released TAA are effective for induction of cross‐priming is of particular interest to this combination study. One explanation is that tumor damage as a result of radiation results in the release of a variety of antigens. With polyI:C‐radiation therapy, both antigen and adjuvant prime DC to stimulate CTL to expand and infiltrate the tumor. Importantly, with this therapy, there is no need for identification or exogenous injection of antigens. Hence, TLR3 ligand is better than previously reported immune adjuvants, such as TLR9 ligand, which required external antigens simultaneously.23 Identification of the precise role of polyI:C in CTL expansion and tumor infiltration in radiotherapy requires further investigation.
The tumor microenvironment is thought to be modified by short‐term free radicals generated by irradiation and multiple integral events subsequent to irradiation (3‐10 days). Radiation affects multiple immune response components including the release of danger signals, recruitment of myeloid cells, modulation of signal transduction, and alteration of innate and adaptive immune responses.42 Herein, a requirement for TLR3 signaling has been shown to be required for effective cross‐priming of anti‐tumor CTL following radiation therapy, at least in some tumor types. It is reasonable to presume that in mice, antigen‐presenting cells express TLR3 and respond to polyI:C with the generation of anti‐tumor immunity.
PolyI:C effectively induces CD4+ T cells, but only poorly induces CD8+ T cells in human patient blood following antigen‐adjuvant therapy.43 Combination therapy with radiation and TLR3 adjuvant has been reported to be effective in patients44, 45 and these studies provide a possible mechanistic basis for that effective therapy. Stereotactic radiation therapy for primary tumors can result in an abscopal effect in patients with metastatic cancer.14 Radiation therapy often exacerbates inflammation, and PolyI:C can also induce undesirable systemic inflammation as a result of cytokines released within 6 hours of administration.46 To minimize cytokine toxicity, TLR3‐specific ligands would be desirable46, 47 instead of polyI:C. TLR3‐specific stimulation can induce CD4+ and CD8+ T cells by acting on DC (Takeda, Yoshida and Matsumoto, unpublished data) and the use of the TLR3 ligand would benefit patients with solid tumors treated with radiation therapy by reducing adverse cytokine effects.
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We are grateful to our laboratory members for their invaluable discussions and technical assistance. We gratefully acknowledge Drs T. Nishimura and H. Kitamura (Hokkaido University) for the gift of the LLC‐OVA cell line and Drs Y. Iwakura (Tokyo University of Science) and S. Akira (Osaka University) for the gift of KO mice. Thanks are also due to Dr J. Inokuchi (Tohoku Medical and Pharmaceutical University) for his valuable discussions. This work was in part supported by the MEXT, AMED and JSPS KAKENHI (Grant Number, 16K08704) from the Japanese Government, and the Uehara Memorial Foundation, GSK Research Fund, and the Takeda Science Foundation. We had non‐profit support from Nobelpharma Co., Ltd through the university control, which we acknowledge gratefully.
Yoshida S, Shime H, Takeda Y, et al. Toll‐like receptor 3 signal augments radiation‐induced tumor growth retardation in a murine model. Cancer Sci. 2018;109:956–965. https://doi.org/10.1111/cas.13543
Funding information
Japan Agency for Medical Research and Development, Uehara Memorial Foundation, Ministry of Education, Culture, Sports, Science and Technology, AMED, JSPS KAKENHI Grant Number JP16K08704, GSK Research Fund, Takeda Science Foundation
Yoshida and Shime equally contributed to this study.
REFERENCES
- 1. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239‐253. [DOI] [PubMed] [Google Scholar]
- 2. Liauw SL, Pitroda SP, Eggener SE, et al. Evaluation of the prostate bed for local recurrence after radical prostatectomy using endorectal magnetic resonance imaging. Int J Radiat Oncol Biol Phys. 2013;85:378‐384. [DOI] [PubMed] [Google Scholar]
- 3. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desmet CJ, Ishii KJ. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol. 2012;12:479‐491. [DOI] [PubMed] [Google Scholar]
- 5. Paludan S, Bowie A. Immune sensing of DNA. Immunity. 2013;38:870‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deng L, Liang H, Xu M, et al. STING‐dependent cytosolic DNA sensing promotes radiation‐induced type I interferon‐dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM‐1, an adaptor molecule that participates in Toll‐like receptor 3–mediated interferon‐β induction. Nat Immunol. 2003;4:161‐167. [DOI] [PubMed] [Google Scholar]
- 8. Jelinek I, Leonard JN, Price GE, et al. TLR3‐specific double‐stranded RNA oligonucleotide adjuvants induce dendritic cell cross‐presentation, CTL responses, and antiviral protection. J Immunol. 2011;186:2422‐2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Azuma M, Ebihara T, Oshiumi H, Matsumoto M, Seya T. Cross‐priming for antitumor CTL induced by soluble Ag + polyI: C depends on the TICAM‐1 pathway in mouse CD11c +/CD8α + dendritic cells. Oncoimmunology. 2012;1:581‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tatematsu M, Nishikawa F, Seya T, Matsumoto M. Toll‐like receptor 3 recognizes incomplete stem structures in single‐stranded viral RNA. Nat Commun. 2013;4:1833. [DOI] [PubMed] [Google Scholar]
- 11. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller JFAP, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015;27:439‐449. [DOI] [PubMed] [Google Scholar]
- 13. Kodama K, Higashiyama M, Okami J, et al. A possible abscopal effect of post‐irradiation immunotherapy in two patients with metastatic lung tumors. Int Cancer Conf J. 2014;3:122‐127. [Google Scholar]
- 14. Adamow M, Ritter E, Sedrak C, et al. Effect in a patient with melanoma. N Engl J Med. 2012;366:925‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azuma M, Takeda Y, Nakajima H, et al. Biphasic function of TLR3 adjuvant on tumor and spleen dendritic cells promotes tumor T cell in fi ltration and regression in a vaccine therapy. Oncoimmunology. 2016;5:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shime H, Matsumoto M, Oshiumi H, et al. Toll‐like receptor 3 signaling converts tumor‐supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA. 2012;109:2066‐2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA‐3) + dendritic cells (DCs) represent a unique myeloid DC subset that cross‐presents necrotic cell antigens. J Exp Med. 2010;207:1247‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akazawa T, Ebihara T, Okuno M, et al. Antitumor NK activation induced by the Toll‐like receptor 3‐TICAM‐1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA. 2007;104:252‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oshiumi H, Okamoto M, Fujii K, et al. The TLR3/TICAM‐1 pathway is mandatory for innate immune responses to poliovirus infection. J Immunol. 2011;187:5320‐5327. [DOI] [PubMed] [Google Scholar]
- 20. Takashima K, Takeda Y, Oshiumi H, et al. STING in tumor and host cells cooperatively work for NK cell‐mediated tumor growth retardation. Biochem Biophys Res Commun. 2016;478:1764‐1771. [DOI] [PubMed] [Google Scholar]
- 21. Yokouchi H, Chamoto K, Wakita D, et al. Tetramer‐blocking assay for defining antigen‐specific cytotoxic T lymphocytes using peptide‐MHC tetramer. Cancer Sci. 2006;97:148‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshida S, Shime H, Funami K, et al. The anti‐oxidant ergothioneine augments the immunomodulatory function of TLR agonists by direct action on macrophages. PLoS ONE. 2017;12:e0169360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chamoto K, Takeshima T, Wakita D, et al. Combination immunotherapy with radiation and CpG‐based tumor vaccination for the eradication of radio‐ and immuno‐resistant lung carcinoma cells. Cancer Sci. 2009;100:934‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takeshima T, Chamoto K, Wakita D, et al. Local radiation therapy inhibits tumor growth through the generation of tumor‐specific CTL: its potentiation by combination with TH1 cell therapy. Cancer Res. 2010;70:2697‐2706. [DOI] [PubMed] [Google Scholar]
- 25. Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature. 2006;441:101‐105. [DOI] [PubMed] [Google Scholar]
- 26. Edelson BT, Kc W, Juang R, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α + conventional dendritic cells. J Exp Med. 2010;207:823‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30:324‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takeda Y, Azuma M, Matsumoto M, Seya T. Tumoricidal efficacy coincides with CD11c up‐regulation in antigen‐specific CD8(+) T cells during vaccine immunotherapy. J Exp Clin Cancer Res. 2016;35:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shime H, Kojima A, Maruyama A, et al. Myeloid‐derived suppressor cells confer tumor‐suppressive functions on natural killer cells via polyinosinic:polycytidylic acid treatment in mouse tumor models. J Innate Immun. 2014;6:293‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. González‐Martín A, Gómez L, Lustgarten J, Mira E, Mañes S. Maximal T cell‐mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res. 2011;71:5455‐5466. [DOI] [PubMed] [Google Scholar]
- 33. Vicari AP, Caux C. Chemokines in cancer. Cytokine Growth Factor Rev. 2014;13:143‐154. [DOI] [PubMed] [Google Scholar]
- 34. Martínez‐Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. 2015;21:5047‐5056. [DOI] [PubMed] [Google Scholar]
- 35. Knutson KL, Disis ML. Tumor antigen‐specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405‐414. [DOI] [PubMed] [Google Scholar]
- 37. Shime H, Misako M, Seya T. The role of innate immune signaling in regulation of tumor‐associated myeloid cells In: Inflammation and Immunity in Cancer. Tokyo: Springer book; 2015:25‐48. [Google Scholar]
- 38. Temizoz B, Kuroda E, Ohata K, et al. TLR9 and STING agonists synergistically induce innate and adaptive type‐II IFN. Eur J Immunol. 2015;45:1159‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)‐12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guenova E, Watanabe R, Teague JE, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T‐cell lymphoma. Clin Cancer Res. 2013;19:3755‐3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419‐426. [DOI] [PubMed] [Google Scholar]
- 42. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2016;17:2‐5. [DOI] [PubMed] [Google Scholar]
- 43. Sabbatini P, Tsuji T, Ferran L, et al. Phase I trial of overlapping long peptides from a tumor self‐antigen and poly‐ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18:6497‐6508. [DOI] [PubMed] [Google Scholar]
- 44. Ostrom QT, Gittleman H, Liao P, et al. A multi‐institution phase II study of poly‐ ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 2014;16:iv1‐iv63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Butowski N, Chang SM, Junck L, et al. A phase II clinical trial of poly‐ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: a North American Brain Tumor Consortium (NABTC01‐05). J Neurooncol. 2009;91:175‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matsumoto M, Tatematsu M, Nishikawa F, et al. Defined TLR3‐specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nat Commun. 2015;6:6280. [DOI] [PubMed] [Google Scholar]
- 47. Takeda Y, Kataoka K, Yamagishi J, et al. A TLR3‐specific adjuvant relieves innate resistance to PD‐L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. 2017;19:1874‐1887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials