

Abstract
T follicular helper (Tfh) cells play an essential role in the formation of germinal center (GC) and generation of high-affinity antibodies. The homing of activated CD4+ T cells into B cell follicles and the involvement of key co-stimulatory/co-inhibitory molecules are critical in controlling both the initiation and the magnitude of GC responses. Meanwhile, studies have shown that a high number of single clone B cells lead to intra-clonal competition, which inhibits the generation of high-affinity antibodies. Our previous work has shown that transcription factor Foxp1 is a critical negative regulator of Tfh cell differentiation. Here we report that the deletion of Foxp1 leads a high proportion of activated CD4+ T cells homing into B cell follicles with a faster kinetics, resulting in earlier GC formation. In addition, we show that Foxp1-deficient Tfh cells restore the generation of high-affinity antibodies when co-transferred with high numbers of single clone B cells. We find that Foxp1 regulates the expression levels of CTLA-4 in activated CD4+ T cells and that Ctla4 is a direct Foxp1 target. Finally, we demonstrate that CTLA-4 expression on conventional CD4+ T cells plays a cell-intrinsic role in Tfh cell differentiation in vivo, and CTLA-4 blockade helps abolish the intra-clonal competition of B cells in generating high-affinity antibodies.
Introduction
T follicular helper (Tfh) cells have been defined as a unique CD4+ T cell subset that provides help to B cells to form germinal centers (GCs) (1-4). GC response is essential for the generation of high-affinity antibodies (5-10), and a faster kinetics that results in the production of high-affinity antibodies can be crucial during infection. The molecular mechanisms that underlie Tfh cell differentiation and Tfh cell help to B cells during GC responses, however, are still not fully understood.
CCR7, a chemokine receptor for T zone chemokines CCL19 and CCL21, is expressed at high levels on naive CD4+ T cells (11-13). T zone fibroblastic reticular cells express CCL19 and CCL21 to attract naive T cells (14, 15). On the other hand, CXCR5, a chemokine receptor for chemokine CXCL13, is involved in activated CD4+ T cell homing to B cell follicles in lymphoid organs (16, 17). The migration of activated CD4+ T cells into B cell follicles and their interaction with B cells are critical, otherwise the Tfh cell responses will be aborted (18, 19). Published studies have shown that CXCR5 induction on activated CD4+ T cells is necessary but not sufficient for T cell homing into B cell follicles (20). CD4+ T cells also require the down-regulation of CCR7 expression for the follicular entry (21). The kinetics of activated CD4+ T cell homing to B cell follicles likely regulates the kinetics of the initiation of GC B cell differentiation and GC formation.
During GC responses, GC B cells up-regulate activation-induced cytidine deaminase (AID) when cycling in the dark zone, undergo somatic hypermutation, and progressively generate high-affinity antibodies via affinity maturation (8-10). Interestingly, studies have shown that a high number of single clone B cells impose intra-clonal competition and result in the inhibition of the generation of high-affinity antibodies (22). The regulation of intra-clonal competition, however, is not clear.
Cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) is well known as a key ‘checkpoint’ in immune tolerance (23-28). Recent studies have shown that CTLA-4 plays important roles in Tfh cell differentiation and Tfh help to B cells (29-31). CTLA-4 deficient mice develop Tfh cells spontaneously, and short-term CTLA-4 blockade using anti-CTLA-4 antibodies in wild-type mice triggers the development of Tfh cells, GC formation and generation of autoantibodies (31). In both the global CTLA-4 deletion and the CTLA-4 blockade model systems, the roles of regulatory T (Treg) cells and T follicular regulatory (Tfr) cells have been emphasized (29, 30). Yet the intrinsic role of CTLA-4 expressed on conventional CD4+ T cells in Tfh cell differentiation is unclear. In addition, whereas studies have shown that CTLA-4 molecules cycle to and from the cell surface (32), the up-regulation of CTLA-4 after T cell activation occurs at the transcriptional level (33-35). Although studies have reported that forkhead proteins bind to the promoter region of the Ctla4 locus and positively regulate CTLA-4 expression (33, 36, 37), to a large extent, the mechanism underlying Ctla4 transcriptional regulation is not well understood.
Previously we have identified transcription factor Foxp1 as a critical negative regulator for the differentiation of Tfh cells (38). Foxp1-deficient CD4+ T cells preferentially differentiate into Tfh cells at the expense of non-Tfh cells, and the constitutive Foxp1A and T cell receptor (TCR)-stimulation induced Foxp1D constitute a ‘double-check’ mechanism limiting Tfh cell differentiation, which greatly affects the subsequent GC and antibody responses (38).
In this study, we demonstrated that Foxp1-deficiency induces a rapid and maintained down-regulation of CCR7 and leads to a high proportion of activated CD4+ T cells homing to B cell follicle at an early stage after antigen challenge. Subsequently, earlier GC formation was observed. We also found that Foxp1 directly controls CTLA-4 expression levels by binding to its promoter and that the CTLA-4 on conventional CD4+ T cells plays a cell-intrinsic and negative regulatory role in Tfh cell differentiation in vivo. In particular, CTLA-4 blockade helps abolish the intra-clonal competition of B cells and restores the generation of high-affinity antibodies.
Materials and Methods
Mice
All mice were maintained in specific pathogen-free barrier facilities and were used in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. B1-8i transgenic mice were purchased from the Jackson Laboratories. CD45.1 SMARTA, Foxp1f/f RosaYFP, Foxp1f/f Cre-ERT2+RosaYFP, OT-IITgFoxp1f/fRosaYFP and OT-IITgFoxp1f/fRosaYFPCre-ERT2+ mice were bred as previously described (38).
Adoptive Transfer, Immunization and CTLA-4 blockade
Eight- to ten-week old mice were treated daily for 4 days with tamoxifen (Sigma-Aldrich) at a dose of 1.5 mg per mouse and rested for 1 day. YFP+CD44loVα2hiCD4+ naive T cells (OT-II Foxp1-cKO) from OT-II Foxp1f/f Cre-ERT2+RosaYFP mice and CD44loVα2hi CD4+ naive T cells (OT-II Foxp1-WT) from OT-II Foxp1f/f RosaYFP mice (or OT-II Foxp1+/+Cre-ERT2+RosaYFP mice) were isolated and sorted with a BD FACS Aria cell sorter (BD Biosciences). Naive Igλ+ B1-8i cells from eight- to ten-week old B1-8i mice were enriched by labeling with phycoerythrin-anti-Igκ (RMK-45; Biolegend) and negative selection with anti-CD43 and anti-PE magnetic beads (both from Miltenyi Biotech) following manufacturer’s protocols. Sorted naive OT-II cells, or together with Igλ+ B1-8i cells in co-transfer experiments, were transferred into age and sex matched SMARTA mice through tail intravenous injection followed by immunization with 100 μg of NP-OVA emulsified in alum adjuvant (Thermo Fisher Scientific) by intraperitoneal injection. For in vivo antibody blocking, recipient mice were treated with 100 μg anti-CTLA-4 (UC10-4F10-1, Bio-X-cell) or 500 μg anti-ICOSL (HK5.3, Bio-X-cell) monoclonal antibodies or PBS by intraperitoneal injection.
Flow cytometry
Flow cytometry was conducted as described (38). Antibodies were as follows: FITC-anti-CD45.2 (104), APC-anti-ICOS (C398.4A; all from eBioscience); APC-anti-CD95 (Jo2; BD Biosciences); PE-anti-CTLA-4 (UC10-4B9), PE-anti-CCR7 (4B12), PE/Cy7-anti-CD38 (90), PE/Cy7-anti-PD1 (29F.1A12), BV421-anti-CXCR5 (11B11), BV510-anti-CD45R (RA3-6B2), APC-e780-anti-CD4 (RM4–5; all from Biolegend). CTLA-4 intracellular staining was performed as previously described (29). Flow cytometry results were analyzed with FlowJo software (Treestar).
Cell migration assays
Transwell chemotaxis assays were performed using 24-well plates with 5-μm pore size inserts (Corning). Navie OT-II Foxp1-WT or OT-II Foxp1-cKO CD4+ T cells were stimulated for 48 h with anti-CD3 (0.5 μg/ml; 145-2C11; eBioscience) and anti-CD28 (1 μg/ml; 37.51; eBioscience) in plates precoated with goat antibody to hamster IgG (0.3 mg/ml; 55397; MP Biomedicals) in complete T cell medium (Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated FCS, 2 mM L-glutamine, penicillin-streptomycin, nonessential amino acids, sodium pyruvate, vitamins, 10 mM HEPES and 50 μM 2-mercaptoethanol), then their populations were expanded for another 24 h in T cell medium containing 100 U/ml recombinant IL-2. OT-II T cells were equilibrated at 37 °C/5% CO2 in T cell medium at 1 × 106 cells/ml for 30 min before use. Total of 500 μl migration medium containing 100 ng/ml CCL19 or CCL21 was applied to the lower chamber and 100 μl cells applied to the upper chamber. After 2 h at 37 °C/5% CO2, percent of migration was determined by flow cytometry as follows: 100 %× ([cell events in lower chamber/input cell events]).
Histology
These procedures were done as described (38). Streptavidin/Biotin Blocking Kit (Vector Labs) was used to block nonspecific binding. Tissue sections were stained in the following three steps: 1) with purified rat anti-mouse CD35 (8c12; BD Biosciences) plus biotin–anti-CD45.2 (104; BD Biosciences) or biotin-anti-PNA (B-1075; Vector Laboratories); 2) with Alexa Fluor 555–conjugated goat polyclonal anti-rat (Invitrogen) plus Alexa Fluor 488–streptavidin (Invitrogen); 3) with Alexa Fluor 647–conjugated rat antibody to mouse IgD (11-26C.2a; Biolegend). Mounted sections were imaged with a 20 × objective on a Nikon A1 confocal microscope.
Real-time RT-PCR
Eight- to ten-week old Foxp1f/f RosaYFP and Foxp1f/f Cre-ERT2+RosaYFP mice were treated with tamoxifen as described above. Naïve CD4+ T cells were activated under Th0, Th1 or Tfh-like polarization conditions, and total RNA was purified as previously described (38). mRNA expression was normalized to that of mRNA encoding the ribosomal protein L32 (Rpl32 mRNA) and is presented relative to that of Foxp1-WT cells. The primers were as follows: Ctla4 -forward (5′-CATGGTGTCGCCAGCTTTC-3′), Ctla4 -reverse (5′-GGTAATCTAGGAAGCCCACTGTA-3′), Rpl32 -forward (5′-CCCAACATCGGTTATGGGAGCA-3′) and Rpl32-reverse (5′-GATGGCCAGCTGTGCTGC-3′).
BCR sequencing
GC B phenotype (B220+CD38loFashiIgλ+) donor B1-8i or endogenous B cells from the spleens of the CD45.1 SMARTA recipient mice were sorted. Genomic DNA of sorted GC B cells was harvested using a DNA/RNA dual isolation kit (Zymo research). VH186.2 sequences were amplified and sequenced as previously described (39). All sequencing results were analyzed with Bioedit V7.2.6 software.
EMSA
Electrophoretic mobility shift assay (EMSA) was performed as previously described with minor modifications (40). Briefly, single-stranded oligonucleotides were annealed with end-labeled with γ-32P-ATP using T4 polynucleotide kinase (New England BioLabs) according to manufacturer’s instructions and were followed by purification with G25 column. Foxp1-ΔN protein was generated using the TNT T7 Quick Coupled Transcription/Translation System (Promega). Binding reactions were performed at room temperature for 20 minutes using either 1μL (Foxp1 A’(A1-A1)) or 3μL (Ctla4 Fkh and Ctla4 Fkh mutant) of in vitro-translated Foxp1-ΔN and approximately 10 pmol of 32P labeled probes in binding buffer (12mM HEPES, 100mM NaCl, 1mM DTT, 1mM EDTA, 12% glycerol, 0.5μg/μL poly(dI-dC) (Thermo Scientific)). Unlabeled Foxp1 A’(A1-A1) probe was added simultaneously for competition where indicated. DNA-protein complexes were visualized by electrophoresis in a 4% polyacrylamide, 0.5 × Tris/Borate/EDTA (TBE) gel. The gel was dried, imaged (PhosphorImager Amersham Biosciences), and quantified using ImageQuant (GE Healthcare). The following oligonucleotide sequences were used in the assay: positive control Foxp1 A’(A1-A1) (5′-CAAGGTAAACAAGACAACGTAAACAA -3′), Ctla4 Fkh (5-ATGGATTTGCTTGTTTTGTTCAGTTTTA-3′) and Ctla4 Fkh mutant (5′- ATGGATTTGCTacggaTGTTCAGTTTTAG-3′, mutated bases in lower case).
ChIP
Foxp1-WT or Foxp1- cKO CD4+ T cells were cultured under Tfh cell-like condition and chromatin immunoprecipitation of Foxp1 was performed as described (38). Immuno-precipitated DNA and input DNA were assessed by Real-time PCR. The primers used were as follows: Ctla4 promoter region forward (5′-CGTACCTTGGATCAAAGCTGTCTA-3′) and reverse (5′-GGAGCAGAGTAAAACCCAACAG-3′); Ctla4 control region forward (5′-GCGAGCATCAGACTTACAATAC-3′) and reverse (5′-CTCAGTTGTAAACACTGCCTGG-3′).
Retroviral transduction
The open reading frame of mouse Ctla4 was amplified and sub-cloned into the retroviral vector MSCV-IRES-hNGFR plasmid. 1 × 106 viral transduced OT-II cells were transferred to the CD45.1 SMARTA recipient mice via intravenous injection followed by immunization with 100 μg of NP-OVA in alum 1 day later.
Statistical Analysis
The two-tailed Student’s t test has been used for all the experiments to calculate P values except the data of W33L+/VH186.2 mutant clone numbers for which the Fisher’s exact test was used.
Results
Foxp1 regulates the kinetics of T cell migration during the initial stage of Tfh cell differentiation
To further understand how Foxp1-deficiency leads to preferential Tfh cell development, we examined some early events involved in Tfh cell differentiation in vivo. Studies have shown that in addition to CXCR5 up-regulation, the down-regulation of CCR7 also plays an important role in the migration of activated CD4+ T cells towards B cell follicles (21). YFP+ Foxp1-deficient naive OT-II (OT-II Foxp1-cKO) or wild-type naive OT-II (OT-II Foxp1-WT) T cells were purified and transferred into SMARTA recipient mice followed by immunization with NP-OVA in alum as previously reported (38). We found that by day 2 after immunization, only about half of the OT-II Foxp1-WT T cells down-regulated CCR7 expression, whereas the majority of the OT-II Foxp1-cKO T cells had uniformly down-regulated CCR7 (Fig. 1A). Most of the CCR7lo donor OT-II T cells also up-regulated CXCR5 (Fig. 1A). By day 3, we found that the majority of donor OT-II T cells in both groups had down-regulated CCR7 and up-regulated CXCR5 (Fig. 1A). Interestingly, by day 4, many of the CCR7loCXCR5+ OT-II Foxp1-WT T cells reversed the expression levels of CCR7 and CXCR5 to become CCR7hiCXCR5−, but the majority of the CCR7loCXCR5+ OT-II Foxp1-cKO T cells maintained their CCR7loCXCR5+ phenotype (Fig. 1A). We also observed a rapid up-regulation of Bcl6 at the protein level in donor OT-II T cells by day 2 as previously reported (41) (Supplemental Fig. 1A). Interestingly, on day 2, the percentage of PD-1+CXCR5+ Tfh cells was higher in Foxp1-deficient OT-II cells than in wild-type OT-II T cells, even though the percentage of Bcl6 expressing cells was the same (Supplemental Fig. 1A). Consistent with the patterns of CCR7 and CXCR5 expression in the beginning 4 days of the response (Fig. 1A), we found that higher percentage of Foxp1-deficient OT-II T cells maintained Bcl6 expression and PD-1+CXCR5+ phenotype (Supplemental Fig. 1A).
Figure 1.
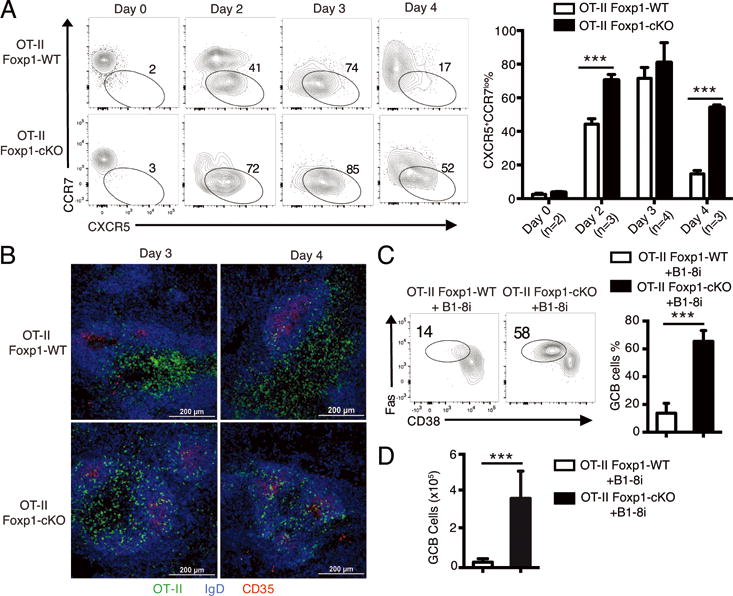
Foxp1 regulates the kinetics of T cell migration during the initial stage of Tfh cell differentiation.
(A) 1 × 106 naive OT-II Foxp1-WT or OT-II Foxp1-cKO T cells were transferred into CD45.1+ SMARTA recipient mice followed by immunization with NP-OVA in alum. CCR7 and CXCR5 expression levels on splenic donor OT-II T cells (left panel) were analyzed 2-4 days later. Percentages of CXCR5+CCR7lo OT-II T cells at different time points are shown in the right panel. Bars represent average ± standard deviation (SD). *** P < 0.001. (B) Confocal microscopy of B cell follicles (IgD+ B cells and CD35+ FDC) and the localization of donor (CD45.2+) OT-II T cells in the recipient mice 2-4 days after immunization as in (A). Scale bars: 200 μm. (C, D) 0.12 × 106 naive OT-II Foxp1-WT or OT-II Foxp1-cKO T cells were co-transferred with 0.6 × 106 B1-8i B cells into CD45.1+ SMARTA recipient mice followed by immunization with NP-OVA in alum. GC B (B220+CD38loFas+) cell differentiation of B1-8i cells (C) and the total numbers of donor GC B cells (D) were analyzed on day 5. Bars in (C, D) represent average ± SD, n=7. *** P < 0.001. Data in (A, B) are representative (or pooled results, A: right panel) of two independent experiments. Data in (C, D) are representative (or pooled results, C: right panel and D) of four independent experiments.
The higher percentage of Foxp1-deficient OT-II T cells that down-regulated CCR7 at day 2 and maintained CCR7loCXCR5+ phenotype to day 4 prompted us to examine whether more activated Foxp1-deficient OT-II T cells migrate into B cell follicles at the early stage of the response, particularly considering that the numbers of total wild-type and Foxp1-deficient donor OT-II T cells were at similar levels (38). We performed immunohistological analysis and found that by day 3 and day 4, the majority of the OT-II Foxp1-WT T cells were in the T cell zone with only some of them in the B cell follicles (Fig. 1B); in contrast, a high proportion of the OT-II Foxp1-cKO T cells left the T cell zone and migrated to the B cell follicles (Fig. 1B). In vitro, we also found that upon activation, CCR7 expression was down-regulated with a faster kinetics in OT-II Foxp1-cKO T cells than in OT-II Foxp1-WT T cells (data not shown); more importantly, transwell experiments showed that a lower percentage of in vitro activated OT-II Foxp1-cKO T cells migrated towards the T zone cytokines CCL19 or CCL21 than OT-II Foxp1-WT T cells (Supplemental Fig. 1B). Thus, both in vitro and in vivo results suggest that in an immune response, newly activated Foxp1-deficient CD4+ T cells leave the T cell zone earlier and move towards the T-B border with a faster kinetics.
The faster kinetics of the migration of Foxp1-deficient OT-II T cells into B cell follicles led us to examine whether the initial GC B cell differentiation and formation also occurs earlier in the recipient mice with OT-II Foxp1-cKO T cells. We sorted naive OT-II Foxp1-WT or OT-II Foxp1-cKO T cells and co-transferred with B1-8i B cell receptor (BCR) transgenic B cells into the SMARTA recipient mice followed by immunization with NP-OVA in alum. B1-8i BCR is derived from the (4-hydroxy-3-nitrophenyl) acetyl (NP)-binding antibody B1-8i (22, 42, 43). In the SMARTA recipient mice, the B1-8i B cells receive help exclusively from the transferred OT-II T cells after NP-OVA challenge.
By day 5 after immunization, whereas only a small percentage of B1-8i B cells differentiated into GC B cells in the recipient mice transferred with OT-II Foxp1-WT T cells, we found that more than half of the B1-8i B cells differentiated into GC B cells in the recipient mice with OT-II Foxp1-cKO T cells (Fig. 1C). The immunohistological analysis showed that by day 5 after immunization, while almost no GC formation was observed in the recipient mice with OT-II Foxp1-WT T cells, the GCs in the recipient mice with OT-II Foxp1-cKO T cells were already formed (Supplemental Fig. 1C). Consistent with the increased percentage of GC B differentiation of B1-8i B cells in the recipient mice with OT-II Foxp1-cKO T cells (Fig. 1C), we found that the total number of B1-8i-derived GC B cells in these recipient mice were also significantly higher than those in the recipient mice with OT-II Foxp1-WT T cells (Fig. 1D). Collectively, these results demonstrate that not only do more Foxp1-deficient OT-II T cells migrate to B cell follicles earlier, they also in turn help induce GC B cell differentiation and GC formation with a faster kinetics. Our study suggests that the Foxp1 pathway-mediated regulation of Tfh cell differentiation and GC responses begins at the early stage of CD4+ T cell response to antigen challenge.
Foxp1-deficient Tfh cells help abolish B cell intra-clonal competition
Single nucleotide mutation of W33L in the BCR VH186.2 region increases NP-binding affinity by 10-fold (42). When only OT-II Foxp1-WT T cells were transferred into the SMARTA recipient mice followed by immunization, NP-specific high-affinity (W33L+) GC B cells derived from polyclonal endogenous B cells were detected on day 12 and 14 post immunization (Fig. 2A and B). Early studies have shown that a high number of B cells with the same specificity will result in intra-clonal competition, which inhibits the production of high-affinity antibodies (22). We confirmed that in the SMARTA recipient mice co-transferred with OT-II Foxp1-WT T cells and a high number (0.6 × 106) of B1-8i B cells, the W33L mutation in both B1-8i and endogenous GC B cells was suppressed (Fig. 2C). Surprisingly, in the recipient mice co-transferred with OT-II Foxp1-cKO T cells and 0.6 × 106 B1-8i B cells, we detected W33L mutation in B1-8i GC B cells (Fig. 2C); the B1-8i GC B cells also accumulated higher levels of somatic hypermutation (Fig. 2D), suggesting that Foxp1-deficient Tfh cells help abolish the B cell intra-clonal competition and restore the generation of high-affinity antibodies.
Figure 2.
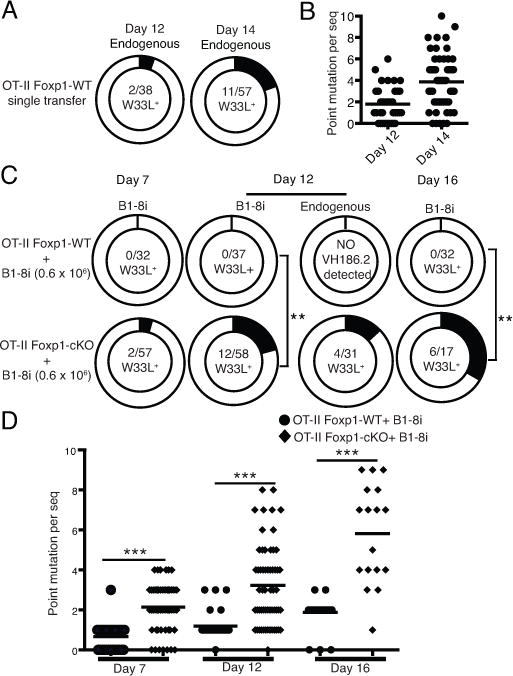
Foxp1-deficient Tfh cells help restore the generation of high-affinity antibodies.
(A) 0.12 × 106 naive OT-II Foxp1-WT T cells were transferred into CD45.1+ SMARTA recipient mice followed by immunization with NP-OVA in alum. On day 12 and 14, endogenous Igλ+ GC B cells were sorted and genomic DNA was isolated. The VH186.2 gene segments were cloned and sequenced. The total number of W33L+ clones and the total number of VH186.2 clones analyzed are shown in the center of each chart. (B) Each dot represents the number of point mutations per sequenced VH186.2 clone as in (A). (C) 0.12 × 106 naive OT-II Foxp1-WT or OT-II Foxp1-cKO T cells were co-transferred with 0.6 × 106 B1-8i B cells into CD45.1+ SMARTA recipient mice followed by immunization with NP-OVA in alum. On day 7, 12 and 16, B1-8i and endogenous GC B cells were sorted, genomic DNA was isolated, and the VH186.2 gene segments were cloned and sequenced. The total number of W33L+ clones and the total number of VH186.2 clones analyzed are shown in the center of each chart. ** P <0.01. (D) Each dot represents the number of point mutations per sequenced VH186.2 clone as in (C). *** P < 0.001. Data in (A, B) are pooled sequences of day 12 (n=4) and day 14 (n=4) from two independent experiments. Data in (C, D) are pooled sequences of day 7, 16 (n=4) from two independent experiments and day12 (n=6) from three independent experiments.
Foxp1 directly controls CTLA-4 expression levels in conventional CD4+ T cells
Recently, CTLA-4 has been shown to play important negative regulatory roles in Tfh cell differentiation (29-31). We examined the CTLA-4 expression in donor OT-II T cells from the SMARTA recipient mice after NP-OVA immunization in alum. We found that the CTLA-4 expression levels were lower in OT-II Foxp1-cKO T cells than in OT-II Foxp1-WT T cells ex vivo (Fig. 3A and Supplemental Fig. 2A). Furthermore, in the in vitro CD4+ T cells activated by anti-CD3/CD28 stimulation under Th0, Th1 or Tfh-like culture conditions (38), we found that the Ctla4 mRNA levels induced after T cell activation were lower in OT-II Foxp1-cKO T cells than in OT-II Foxp1-WT T cells (Fig. 3B). This suggests that Foxp1 may directly regulate CTLA-4 expression levels after T cell activation, although the remaining CTLA-4 expression in activated Foxp1-deficient CD4+ T cells (Fig. 3A) suggests that there are other factors involved in controlling CTLA-4 levels as well. We performed the bioinformatics analysis and identified a forkhead-binding site (Fkh) in the proximal promoter region of the Ctla4 locus (Fig. 3C, left panel), which has been previously reported in the study of Foxo1-mediated regulation of CTLA-4 expression (37). Subsequent EMSA assays showed that in vitro, the Foxp1-ΔN, a truncated Foxp1 protein containing zinc finger, leucine zipper and forkhead domains (40), bound to the oligonucleotides containing the Fkh site (Fig. 3C, right panel). The competition of an unlabeled Foxp1 A’(A1-A1) probe that contains two Foxp1-binding sites (40) effectively reduced the binding of Foxp1-ΔN to Ctla4 Fkh probe in a dosage-dependent manner (Fig. 3C, right panel). Chromatin-immunoprecipitation (ChIP) assay of Foxp1 in activated wild-type CD4+ T cells showed that Foxp1 indeed bound specifically to the Ctla4 promoter region (Fig. 3D). Taken together, these results suggest that Ctla4 is a Foxp1 direct target and Foxp1 regulates CTLA-4 expression levels in CD4+ T cells after activation.
Figure 3.
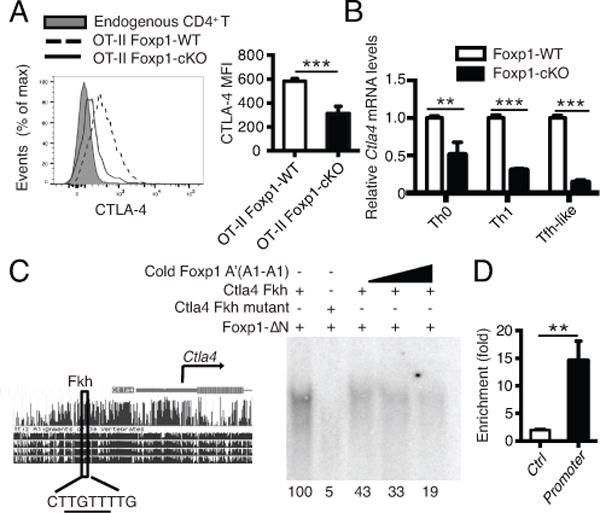
Foxp1 deficiency leads to lower levels of CTLA-4 expression and Ctla4 is a Foxp1 direct target.
(A) Donor OT-II T cells were transferred as in Fig. 1A and CTLA-4 expression levels in splenic donor T cells were analyzed on day 7, n=4. (B) Naive Foxp1-WT or Foxp1-cKO CD4+ T cells were activated under Th0, Th1 or Tfh-like polarization conditions in vitro for 4 days. Ctla4 mRNA levels were analyzed by real-time PCR on day 4. mRNA expression was normalized to mRNA encoding the ribosomal protein L32 (Rpl32 mRNA) and is presented relative to that of Foxp1-WT cells. Bars represent triplicate average ± SD. *** P < 0.001. (C) Predicted forkhead-binding site is indicated in the Ctla4 promoter region (left panel). EMSA of in vitro–translated Foxp1-ΔN protein with Ctla4 Fkh probe (right panel). Foxp1 A’(A1-A1): a positive Foxp1 probe that contains two Foxp1-binding sites; Ctla4 Fkh: a probe that contains the predicted Fkh site in the Ctla4 promoter. Foxp1-ΔN: truncated Foxp1 proteins containing zinc finger, leucine zipper and forkhead domains. Competition assay for Foxp1-ΔN binding was performed with 1x, 5x, 10x cold Foxp1 A’(A1-A1) probes. Quantification values of probe and protein binding were indicated in each corresponding lane. Values were reported relative to the complex formed with Ctla4 Fkh probe and Foxp1-ΔN protein. (D) ChIP analysis of the binding of Foxp1 to a control region (Ctrl) or the Ctla4 promoter region (Promoter), presented as binding in Foxp1-WT T cells relative to binding in the Foxp1-cKO T cells. Bars represent triplicate average ± SD. ** P < 0.01. Data in (A-D) are representative (or pooled results, A: right panel) of two independent experiments.
CTLA-4 on conventional CD4+ T cells inhibits Tfh cell differentiation and GC response in vivo
Published studies on CTLA-4-mediated regulation of Tfh cell differentiation in vivo have either used global CTLA-4 deletion or emphasized T follicular regulatory cells (Tfr) (29, 30). The intrinsic roles of CTLA-4 on conventional CD4+ T cells in Tfh cell differentiation and in Tfh cell help to B cells (29), however, have yet to be tested in vivo. In the OT-II/NP-OVA and SMARTA recipient mouse model system, we found that in wild-type OT-II T cells by day 7 after antigen challenge, the CTLA-4 expression levels were lower in Tfh cells than in non-Tfh cells (Supplemental Fig. 2B), which corresponds with the lower Foxp1 expression levels in Tfh cells than in non-Tfh cells (Supplemental Fig. 2C). Previously we have shown that the Tfh cell differentiation in the OT-II/NP-OVA and SMARTA recipient mouse model system does not involve Treg cells (38). Thus, to further clarify the intrinsic role of CTLA-4 on non-Treg CD4+ T cells in Tfh cell differentiation, we adapted two complementary approaches: 1) Over-expression of CTLA-4 in Foxp1-deficient OT-II T cells; 2) Anti-CTLA-4 antibody blocking with OT-II Foxp1-WT T cell transfer.
Retrovirally infected OT-II Foxp1-cKO T cells were co-transferred with B1-8i B cells into the SMARTA recipient mice followed by NP-OVA immunization in alum. We found that the OT-II Foxp1-cKO T cells infected with CTLA-4-containing retroviruses expressed higher levels of CTLA-4 than those infected with control retroviruses (Fig. 4A), which resulted in a marginal (although statistically significant) decrease of ICOS expression (Supplemental Fig. 3A). The over-expression of CTLA-4 resulted in decreased Tfh cell percentage of OT-II Foxp1-cKO T cells (Fig. 4B and Supplemental Fig. 2D) and B1-8i GC B cells (Fig. 4C), suggesting that reduced CTLA-4 expression levels contribute to the enhanced Tfh cell differentiation of Foxp1-deficient CD4+ T cells and subsequent GC B cell responses.
Figure 4.
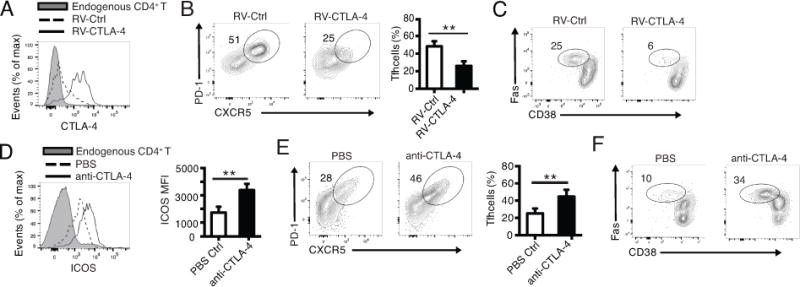
CTLA-4 on conventional CD4+ T cells inhibits Tfh cell differentiation and GC response in vivo
(A, B, C) 1 × 106 OT-II Foxp1-cKO T cells infected with control retrovirus (RV-Ctrl) or retrovirus expressing CTLA-4 (RV-CTLA-4) were co-transferred with B1-8i B cells into CD45.1+ SMARTA recipient mice followed by immunization of the recipient mice 1 day later with NP-OVA in alum. CTLA-4 expression in donor OT-II Foxp1-cKO T cells (A), Tfh cell percent of donor T cells (B) and GC B cell percent of B1-8i cells (C) were analyzed 5 days after immunization. Bars represent average ± SD, n=4. ** P < 0.01. (D, E, F) 1 × 106 naive OT-II Foxp1-WT T cells and 0.6 × 106 B1-8i B cells were co-transferred into the CD45.1+ SMARTA recipient mice followed by immunization with NP-OVA in alum. Recipient mice were treated with PBS or 100 μg anti-CTLA-4 antibodies on day 0, 2 and 4 after immunization. ICOS expression in donor OT-II Foxp1-WT T cells (D), Tfh cell percentage of donor T cells (E) and GC B cell percentage of B1-8i cells (F) were analyzed 5 days after immunization. Bars in (D, E) represent average ± SD, n=4. ** P < 0.01. Data in (A-C) are representative (or pooled results, B: right panel) of two independent experiments. Data in (D-F) are representative (or pooled results, D and E: right panel) of three independent experiments.
In anti-CTLA-4 antibody blocking experiments, the SMARTA recipient mice transferred with both OT-II Foxp1-WT T cells and B1-8i B cells were treated with anti-CTLA-4 antibodies on day 0, 2 and 4, and Tfh cell differentiation of donor T cells was analyzed on day 5. The effectiveness of anti-CTLA-4 blockade was shown by the higher levels of ICOS expression on the donor OT-II T cells from the recipient mice treated with anti-CTLA-4 antibodies (Fig. 4D), which is likely due to the enhanced CD28 co-stimulation as reported (31). By day 5, we found that CTLA-4 blockade resulted in increased percentages of donor OT-II Tfh cells (Fig. 4E) and B1-8i GC B cells (Fig. 4F). Interestingly, in anti-ICOS ligand (ICOSL) blocking experiments, we found that the CTLA-4 expression levels increased slightly (Supplemental Fig. 3B), indicating a negative reciprocal regulation between CTLA-4 and ICOS expression. Collectively, these results suggest that in addition to its role in Treg cell-mediated regulation of Tfh cell differentiation (29, 30), CTLA-4 expression on conventional CD4+ T cells has an intrinsic negative regulatory function in Tfh cell differentiation.
CTLA-4 blockade of Tfh cells enhances high-affinity antibody responses
In our OT-II transfer model, a significant fraction of Tfh cells have formed by day 5 (38). To further examine how CTLA-4 expression on Tfh cells regulates GC B cell responses and verify whether CTLA-4 blocking in vivo also boosts Tfh cell help to GC B cell differentiation as reported in vitro (29), we treated the SMARTA recipient mice co-transferred with OT-II Foxp1-WT T cells and B1-8i B cells with anti-CTLA-4 antibodies on day 5, 7 and 9, and analyzed Tfh and GC B cell differentiation of donor cells on day 12. In the recipient mice treated with anti-CTLA-4 antibodies, the ICOS expression levels on donor OT-II T cells were higher (data not shown), suggesting that the CTLA-4 blocking was effective. On day 12, we found that donor OT-II Tfh cell and B1-8i GC B cell differentiation was maintained at a higher level in the recipient mice treated with anti-CTLA-4 antibodies (Fig. 5A and B). Interestingly, we found that even with a high number (0.6 × 106) of B1-8i cells co-transferred, the CTLA-4 blocking still resulted in the generation of W33L+ high-affinity anti-NP antibodies and increased levels of somatic hypermutation (Fig. 5C and D), suggesting that CTLA-4 blockade also helps abolish the intra-clonal competition in generating high-affinity antibodies.
Figure 5.
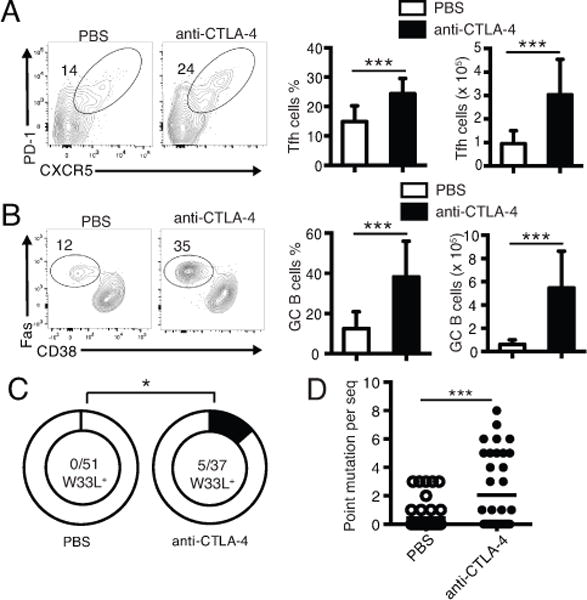
CTLA-4 blockade of conventional CD4+ T cells enhances GC responses and helps abolish intra-clonal competition.
(A, B) Donor OT-II and B1-8i B cells were co-transferred as in Fig. 4D-F. On day 5, 7 and 9, the recipient mice were treated with PBS or 100 μg of anti-CTLA-4 antibodies. The Tfh cell differentiation of donor OT-II T cells (A), the total number of Tfh cells (A), the GC B cell differentiation of Bi-8i B cells (B) and the total number of GC B cells (B) were analyzed on day 12. Bars in (A, B) represent average ± SD, n=10. *** P < 0.001. (C) On day 12, B1-8i GC B cells were sorted and genomic DNA was isolated. The VH186.2 gene segments were cloned and sequenced. The total number of W33L+ mutants and total number of VH186.2 clones analyzed are shown in the center of each chart. * P < 0.05. (D) Each dot represents the number of point mutations per sequenced VH186.2 as in (C). *** P < 0.001. Data in (A, B) are representative (or pooled results, bar figures) of three independent experiments. Data in (C, D) are pooled sequences of 6 mice from two independent experiments.
Discussion
The deletion of Foxp1 in naïve CD4+ T cells leads to a preferential differentiation of Tfh cells with the underlying mechanism incompletely understood (38). The current study demonstrates that Foxp1 deficiency leads a high proportion of activated CD4+ T cells to home into B cell follicles with a faster kinetics, which in turn initiates earlier GC formation. In addition, when co-transferred with a high number of B cells of single clone, Foxp1-deficient Tfh cells help abolish the intra-clonal B cell competition and aid in generating high-affinity antibodies. Finally, we found that Foxp1 directly and positively regulates the expression levels of CTLA-4, and CTLA-4 expression on non-Treg CD4+ T cells has an intrinsic regulatory function in Tfh cell differentiation. The blockade of CTLA-4 signaling also helps abolish the B cell intra-clonal competition.
In the OT-II/NP-OVA model system, it has been reported that CXCR5 is induced by 24 hours, and an early and transient up-regulation of Bcl6 proteins occurs before the first cell division (44). Studies have shown that it is the second wave of the increased Bcl6 expression after day 4 in a subset of activated OT-II T cells that marks the eventual differentiation of Tfh cells (41). In our study, the initial but transient up-regulation of CXCR5 in OT-II Foxp1-WT T cells by day 3 is in accordance with the reported two waves of Bcl6 induction pattern (41); yet, it is intriguing that in Foxp1-deficient OT-II T cells, not only CXCR5+ but also CCR7lo (and Bcl6+) phenotype is maintained, suggesting that Foxp1 deficiency likely facilitates not only the cell migration but also the continuous Tfh cell differentiation. The results of the faster kinetics of activated OT-II Foxp1-cKO T cells down-regulating CCR7 both in vitro (data not shown) and in vivo (by 48 h), and in particular, the lower percentage of them migrating towards the T zone chemokines CCL19 and CCL21 in vitro, suggests that more Foxp1-deficient T cells have been initiated to leave the T cell zone earlier and migrate towards the B cell follicles. Previously we have shown that Foxp1 helps impose T cell quiescence (45, 46). The deletion of Foxp1 may result in faster and stronger T cell activation, subsequently a faster kinetics of downregulation of CCR7 and migration towards T-B border, which would play a critical role in early Tfh cell differentiation. At present it is not clear whether Foxp1 may have some other mechanisms to regulate cell migration. Nevertheless, our study demonstrates that the Foxp1 pathway regulates the kinetics of recently activated CD4+ T cells homing into B cell follicles.
Our results show that ICOS and CTLA-4 signaling negatively and reciprocally regulate the expression levels of these two molecules. Whereas the increase of ICOS expression levels is obvious after anti-CTLA-4 blocking (Fig. 4D), the decrease of ICOS expression levels in CTLA-4 over-expressed T cells is marginal (Supplemental Fig. 3A). We think this is most likely due to the retroviral infection procedure, where T cells are activated in vitro first for the retroviral expression of CTLA-4. Meanwhile, the observation that CTLA-4 over-expression still greatly repressed Tfh cell differentiation suggests an intrinsic inhibitory function of CTLA-4 on Tfh cell differentiation.
Previously we have shown that Foxp1 antagonizes Foxo1 in regulating IL-7α expression by potentially competing for the same forkhead-binding site in the IL-7Rα enhancer (46). It is very intriguing that regarding the regulation of CTLA-4 expression, both Foxp1 and Foxo1 function as positive regulators and it seems that they may also bind to the same forkhead-binding site in the Ctla4 promoter. We have shown that the deletion of Foxp1 in CD4+ T cells results in a slightly reduced amount of Foxo1 protein (38). Thus, in the activated Foxp1-deficient CD4+ T cells, the lower Ctla4 expression levels are possibly due to the reduced amounts of both regulators. How Foxp1 and Foxo1 compete in binding to the same binding site, yet in regulating IL-7α they antagonize each other, whereas in regulating Ctla4 they function the same, is not clear. Considering that these two factors may be involved in different transcription complexes regulating different common target genes, the research in this direction certainly warrants further investigation. Nevertheless, the observation that both Foxp1- and Foxo1-deficient CD4+ T cells express lower levels of CTLA-4 is consistent with their preferential Tfh cell differentiation in vivo.
Studies have suggested that T cell help is a limiting factor for B cell affinity selection in the GC (39), raising the question whether Tfh cell number is a limiting factor or not. Previously we have shown that Foxp1 deletion results in the generation of increased Tfh cell numbers (38). Therefore, it is intriguing that the increased number of OT-II Foxp1-cKO Tfh cells helps abolish the intra-clonal competition between B cells that we observed in our current study. Recent studies have shown that Tfh cells likely have different subpopulations with different B cell help functions (47). At present, how Foxp1 deficiency may affect the differentiation of Tfh cell subsets or the help they provide to GC B cells during affinity maturation is not clear. Nevertheless, our results demonstrate that CTLA-4 expression levels participate in regulating the intra-clonal competition and the generation of high-affinity B cells.
In summary, we have shown that the Foxp1 pathway is involved in regulating the migration kinetics of activated CD4+ T cells towards B cell follicles, indicating that Foxp1 is involved in Tfh cell differentiation from the beginning of a CD4+ T cell response. Our results also demonstrate that CTLA-4 is a direct Foxp1 target and its reduced expression levels in the absence of Foxp1 contributes to the enhanced Tfh cell differentiation of Foxp1-deficient CD4+ T cells. Finally, our study verifies an intrinsic role of CTLA-4 on conventional CD4+ T cells in regulating Tfh cell differentiation in vivo and demonstrates that CTLA-4 blockade helps abolish the intra-clonal competition in generating high-affinity antibodies. The understanding of the fundamental mechanisms in generating faster and enhanced antibody responses will provide knowledge for new strategies to manipulate humoral responses for treatment of infectious diseases and aid in vaccine development.
Supplementary Material
Acknowledgments
We thank Marion Spell for excellent technical help with flow cytometry and Ryan J. McMonigle for scientific discussion and manuscript editing.
This work was supported by Chinese National Science Foundation of Distinguished Young Scholars 31525008 and NSFC 81330072 (to B.L), US National Institutes of Health Grants AI102187 and AI123162 (to S.R.T), AI095439 and AI103162 (to H.H), UAB CFAR Vaccine Concept Grant (to H.H), and UAB Center for AIDS Research (P30AI027767-26).
Abbreviations used in this article
- Tfh
T follicular helper cells
- GC
germinal center
- AID
activation-induced cytidine deaminase
- CTLA-4
cytotoxic T-lymphocyte associated antigen-4
- Treg
regulatory T cells
- Tfr
follicular regulator T cells
- TCR
T cell receptor
- BCR
B cell receptor
- ChIP
chromatin-immunoprecipitation
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 2.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 3.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209:1241–1253. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qi H. T follicular helper cells in space-time. Nat Rev Immunol. 2016;16:612–625. doi: 10.1038/nri.2016.94. [DOI] [PubMed] [Google Scholar]
- 5.Eisen HN, Siskind GW. Variations in Affinities of Antibodies during the Immune Response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- 6.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 7.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 8.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 9.Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity. 2016;45:471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol. 2017;45:21–30. doi: 10.1016/j.coi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 12.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 13.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 14.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nature immunology. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 16.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 17.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 18.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, Laufer TM. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. 2011;187:1091–1095. doi: 10.4049/jimmunol.1100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med. 1999;190:1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 22.Le TV, Kim TH, Chaplin DD. Intraclonal competition inhibits the formation of high-affinity antibody-secreting cells. J Immunol. 2008;181:6027–6037. doi: 10.4049/jimmunol.181.9.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 24.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 26.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 27.Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 28.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 29.Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–1039. doi: 10.1016/j.immuni.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41:1013–1025. doi: 10.1016/j.immuni.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, Kogimtzis A, Kenefeck R, Sansom DM, Walker LS. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U S A. 2015;112:524–529. doi: 10.1073/pnas.1414576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 33.Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, Wang Y, Walunas T, Bluestone J, Listman J, Finn PW. Regulation of CTLA-4 expression during T cell activation. J Immunol. 1996;156:4154–4159. [PubMed] [Google Scholar]
- 34.Gibson HM, Hedgcock CJ, Aufiero BM, Wilson AJ, Hafner MS, Tsokos GC, Wong HK. Induction of the CTLA-4 gene in human lymphocytes is dependent on NFAT binding the proximal promoter. J Immunol. 2007;179:3831–3840. doi: 10.4049/jimmunol.179.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan DV, Gibson HM, Aufiero BM, Wilson AJ, Hafner MS, Mi QS, Wong HK. Differential CTLA-4 expression in human CD4+ versus CD8+ T cells is associated with increased NFAT1 and inhibition of CD4+ proliferation. Genes Immun. 2014;15:25–32. doi: 10.1038/gene.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 37.Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, Katayama CD, Hedrick SM. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Geng J, Wen X, Bi E, Kossenkov AV, Wolf AI, Tas J, Choi YS, Takata H, Day TJ, Chang LY, Sprout SL, Becker EK, Willen J, Tian L, Wang X, Xiao C, Jiang P, Crotty S, Victora GD, Showe LC, Tucker HO, Erikson J, Hu H. The transcription factor Foxp1 is a critical negative regulator of the differentiation of follicular helper T cells. Nature immunology. 2014;15:667–675. doi: 10.1038/ni.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koh KP, Sundrud MS, Rao A. Domain requirements and sequence specificity of DNA binding for the forkhead transcription factor FOXP3. PLoS One. 2009;4:e8109. doi: 10.1371/journal.pone.0008109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baumjohann D, Okada T, Ansel KM. Cutting Edge: Distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. 2011;187:2089–2092. doi: 10.4049/jimmunol.1101393. [DOI] [PubMed] [Google Scholar]
- 42.Allen D, Simon T, Sablitzky F, Rajewsky K, Cumano A. Antibody engineering for the analysis of affinity maturation of an anti-hapten response. EMBO J. 1988;7:1995–2001. doi: 10.1002/j.1460-2075.1988.tb03038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, Rajewsky K. B cell development under the condition of allelic inclusion. Immunity. 1997;6:225–233. doi: 10.1016/s1074-7613(00)80325-8. [DOI] [PubMed] [Google Scholar]
- 44.Chen X, Ma W, Zhang T, Wu L, Qi H. Phenotypic Tfh development promoted by CXCR5-controlled re-localization and IL-6 from radiation-resistant cells. Protein Cell. 2015;6:825–832. doi: 10.1007/s13238-015-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng X, Ippolito GC, Tian L, Wiehagen K, Oh S, Sambandam A, Willen J, Bunte RM, Maika SD, Harriss JV, Caton AJ, Bhandoola A, Tucker PW, Hu H. Foxp1 is an essential transcriptional regulator for the generation of quiescent naive T cells during thymocyte development. Blood. 2010;115:510–518. doi: 10.1182/blood-2009-07-232694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nature immunology. 2011;12:544–550. doi: 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, Craft J. TFH cells progressively differentiate to regulate the germinal center response. Nature immunology. 2016;17:1197–1205. doi: 10.1038/ni.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.