

Abstract
There are currently no approved vaccines or small molecule therapeutics available for the prophylaxis or treatment of Middle East Respiratory Syndrome coronavirus (MERS-CoV) infections. MERS-CoV 3CL protease is essential for viral replication; consequently, it is an attractive target that provides a potentially effective means of developing small molecule therapeutics for combatting MERS-CoV. We describe herein the structure-guided design and evaluation of a novel class of inhibitors of MERS-CoV 3CL protease that embody a piperidine moiety as a design element that is well-suited to exploiting favorable subsite binding interactions to attain optimal pharmacological activity and PK properties. The mechanism of action of the compounds and the structural determinants associated with binding were illuminated using X-ray crystallography.
Keywords: MERS-CoV, 3CL protease, Piperidine moiety, Antiviral, Peptidomimetic inhibitors
Graphical abstract

Highlights
-
•
The structure-guided design of a new class of Middle East Respiratory Syndrome Coronavirus (MERS-CoV) 3CL protease inhibitors was performed.
-
•
A piperidine moiety was used as a design element in the synthesis of peptidomimetic inhibitors that exploit interactions with the enzyme S3-S4 subsites.
-
•
The inhibitor design rationale was validated by X-ray crystallographic studies.
-
•
X-ray crystallography confirmed the mechanism of action of the inhibitors.
1. Introduction
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses of the family Coronaviridae [1]. Human and animal coronaviruses are classified into at least 25 species in four genera, α to δ coronaviruses [1]. Mouse hepatitis virus (MHV) is the most prominently studied coronavirus both in vitro and in vivo, serving as the prototype coronavirus. Human coronaviruses, including 229E, NL63, OC43, and HKU1 strains, have been implicated in respiratory infections, otitis media, exacerbations of asthma, diarrhea, myocarditis, and neurological disease [[1], [2], [3], [4]]. In contrast, severe acute respiratory distress syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) pose a significant threat to public health worldwide due to their ability to cause serious human disease with high mortality rates [[2], [3], [4]]. MERS-CoV has become a global threat due to continuous outbreaks in countries on the Arabian peninsula and the potential of spread to other countries with a high mortality rate [[5], [6], [7], [8]]. Coronaviruses are also important pathogens in animals and are associated with respiratory and enteric illnesses, including transmissible gastroenteritis virus (TGEV), porcine epidemic diarrhea virus (PEDV), feline infectious peritonitis virus (FIPV), and bovine coronavirus [1]. There are currently no effective vaccines or small molecule therapeutics for the treatment of MERS (or SARS) coronavirus infection [[9], [10], [11]].
The highly conserved genome of coronaviruses contains two open reading frames, ORF1a and ORF1b, on the 5′ end that encode nonstructural proteins. Genes encoding the coronavirus structural proteins, S (spike glycoprotein), E (envelope protein), M (membrane glycoprotein), and N (nucleocapsid protein), which play a critical role in virion-cell receptor binding, replication and virion assembly, are located at the 3′ end of the genome [1,12]. Coronavirus entry is initiated by the binding of the spike protein (S) to cell receptors, specifically, dipeptidyl peptidase 4 (DDP4) and angiotensin converting enzyme 2 (ACE2) for MERS-CoV and SARS-CoV, respectively [[1], [2], [3], [4], [5]]. Entry into cells requires host proteases for cleavage at two sites in the S protein, in the case of most CoV [13,14] Translation of the genomic mRNA of ORF1a yields polyprotein pp1a, while a second polyprotein (pp1b) is the product of a ribosomal frame shift that joins ORF1a together with ORF1b. ORF1a encodes a papain-like cysteine protease (PLpro) and a 3C-like cysteine protease (3CLpro). Polyproteins pp1a and pp1b are processed by 3CLpro (11 cleavage sites) and PLpro (3 cleavage sites) resulting in sixteen mature nonstructural proteins, including RNA-dependent RNA polymerase (RdRp) and helicase, which play important roles in the transcription and replication of coronaviruses (Fig. 1 ). Both proteases are essential for viral replication, making them attractive targets for drug development [9,10,[15], [16], [17]].
Fig. 1.

Overview of the organization and proteolytic processing of MERS-CoV polyproteins, pp1a and pp1b. Scissor images indicate cleavages by 3CLpro, and black arrowheads by PLpro.
MERS-CoV 3CLpro is a chymotrypsin-like cysteine protease having a catalytic Cys148-His41 dyad and an extended binding site [[18], [19], [20], [21]]. The protease displays a stringent primary substrate specificity for a P1 Gln residue [22] and has a strong preference for a P2 Leu residue. The P3 residue side chain is oriented toward the solvent while the S4 subsite is shallow, preferring a small hydrophobic P4 residue (Ala). Functional and structural studies have delineated the similarities between the 3CLpro of coronaviruses that can be exploited in the design of broad-spectrum inhibitors [23].
We have recently reported the first demonstration of clinical efficacy of a coronavirus protease inhibitor (a dipeptidyl aldehyde bisulfite adduct inhibitor designated GC376) [24,25]. Specifically, administration of GC376 to cats infected with FIPV, a coronavirus that is 100% fatal in cats, reversed the progression of fatal FIP and resulted in clinical remission in a majority of animals (>90%). Since FIP disease progression is quite rapid and its pathogenesis primarily immune-mediated, features shared by MERS-CoV, we hypothesized that a viral protease inhibitor could reverse the pathogenesis of MERS-CoV in affected hosts. Interrogation of this hypothesis entailed, as a first step, the design of a new and versatile class of peptidomimetic inhibitors of MERS-CoV 3CL protease. We describe herein the structure-guided design of inhibitors of MERS-CoV 3CLpro that embody a piperidine moiety as a novel design element, as well as pertinent structural and biochemical studies. These inhibitors were also examined against other coronaviruses, including SARS-CoV, FIPV and MHV to evaluate the spectrum of activity against multiple coronaviruses.
2. Results and discussion
2.1. Inhibitor design rationale
The structure-guided design of inhibitor (I) encompassed the following steps: (a) we first determined a high resolution X-ray crystal structure of MERS-CoV 3CLpro in complex with GC376 (Fig. 2 /Panel A). Examination of the active site of the complex revealed that the aldehyde bisulfite adduct had reverted to the precursor peptidyl aldehyde, which subsequently formed a tetrahedral hemi-thioacetal upon reaction with the active site Cys148. Notably, the electron density at this stereocenter was consistent with the formation of both R and S enantiomers at the covalent binding site (also observed for the other structures described in the following sections). The structure reveals a network of backbone hydrogen bonds which ensure correct positioning of the inhibitor to the active site, as well as two critical hydrogen bonds with the P1 Gln surrogate [26] side chain. The inhibitor P2 Leu side chain is ensconced in the hydrophobic S2 subsite of the enzyme. Importantly, the structure shows a hydrophobic-driven interaction between the benzyl group of the inhibitor and the γ-lactam ring of the Gln surrogate side chain; (b) based on the forgoing, we reasoned that extending the “cap” would allow the inhibitor to assume an extended conformation and orient the phenyl ring toward the hydrophobic S4 pocket of the enzyme. Validation of this idea was obtained by synthesizing extended inhibitor GC813 and determining a high resolution X-ray crystal structure of the MERS-CoV 3CLpro:GC813 complex (Fig. 2/Panel B). The m-Cl phenethyl side chain is clearly shown to occupy the hydrophobic S4 subsite. In addition to an array of H-bonds with Gln192, Glu169, and Gln167 and the backbone of the inhibitor, which serve to correctly position the inhibitor at the active site, the inhibitor interacts with the S1, S2 and S4 subsites, but not the S3 subsite; (c) we hypothesized that the attachment of a piperidine ring to the peptidyl component would yield a structurally novel peptidomimetic (I) capable of (1) orienting recognition elements R3 and R4 in a correct vector relationship for optimal interactions with the S3 and S4 subsites, (2) rendering a dipeptidyl inhibitor equivalent to a tetrapeptidyl inhibitor with potentially diminished PK liabilities and, (3) providing a flexible means for the structure-guided parallel optimization of ADMET/PK and physicochemical properties using diversity sites R3 and R4 in inhibitor (I) (Fig. 3 ). In summary, the piperidine-based design strategy is a hitherto unrecognized effective means of rendering a dipeptidyl inhibitor equivalent to a tetrapeptidyl inhibitor capable of engaging in optimal binding interactions with all four S1-S4 subsites but which, however, is anticipated to display diminished PK liabilities due to its reduced peptidyl character. Furthermore, the aforementioned piperidine-based design strategy has wide applicability and can be extended to any protease with an extended binding site. Preliminary evidence in support of this approach is provided by the results of enzyme and cell-based screening of derivatives of (I) (Table 1, Table 2 ), as well as the results of structural studies (vide infra) (see Table 3).
Fig. 2.

X-ray crystal structures of A) MERS-CoV 3CLpro:GC376 and B) MERS-CoV 3CLpro:GC813.
Fig. 3.

Evolution of design and general structure of inhibitor (I).The EC50 values of inhibitors GC376 and GC813 were determined against MERS-CoV in cell culture.
Table 1.
Anti-coronavirus activity of compounds 9a-f and 10a-f in the FRET enzyme assay.
| Compound | R3 | R4 | X | IC50 (μM) |
||
|---|---|---|---|---|---|---|
| MERS-CoV | SARS-CoV | FIPV | ||||
| 9a | H | Boc | CHO | 0.6 | 2.1 | 0.8 |
| 10a | CH(OH)SO3Na | 0.4 | 5.1 | 2.4 | ||
| 9b | CH3SO2 | CHO | 0.7 | 28.8 | 3.5 | |
| 10b | CH(OH)SO3Na | 0.6 | 42.1 | 2.3 | ||
| 9c | (C6H5)CH2 | Boc | CHO | 0.8 | 5.2 | 1.6 |
| 10c | CH(OH)SO3Na | 0.7 | 6.3 | 2.1 | ||
| 9d | CH3SO2 | CHO | 0.7 | 3.9 | 0.8 | |
| 10d | CH(OH)SO3Na | 0.9 | 4.3 | 1.1 | ||
| 9e | CH3CH2 | Boc | CHO | 6.1 | 5.5 | 5.5 |
| 10e | CH(OH)SO3Na | 7.5 | 4.1 | 6.7 | ||
| 9f | H | CH3CH2O(CO) | CHO | 0.6 | 3.2 | 1.3 |
| 10f | CH(OH)SO3Na | 0.5 | 8.8 | 1.1 | ||
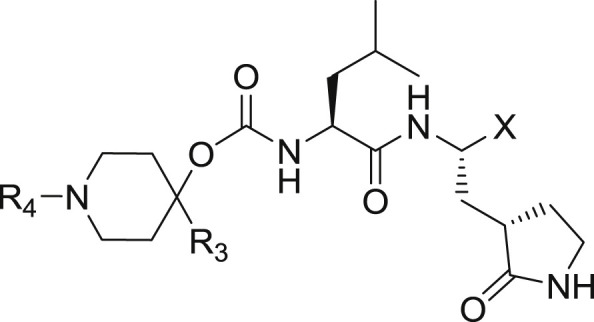
Table 2.
Anti-coronavirus activity (EC50) of compounds 10a and 10c in the cell based assay and cytotoxicity (CC50).
| EC50 (μM) |
CC50 (μM) | |||
|---|---|---|---|---|
| MERS-CoV | MHV | FIPV | ||
| 10a | 0.5 | 0.6 | 1.5 | >100 |
| 10c | 0.8 | 1.0 | 0.1 | >100 |
Table 3.
Crystallographic data for MERS-CoV 3CLpro in complex with compounds GC376, GC813, 10c and 10e.
| Data Collection | MERS-CoV 3CLpro: GC376 | MERS-CoV 3CLpro: GC813 | MERS-CoV 3CLpro: Compound 10c | MERS-CoV 3CLpro: Compound 10e |
|---|---|---|---|---|
| Unit-cell parameters (Å,o) | a = 101.81 | a = 101.58 | a = 101.25 | a = 100.63 |
| b = 58.29 | b = 58.00 | b = 58.10 | b = 58.11 | |
| c = 49.78 | c = 49.73 | c = 49.73 | c = 49.86 | |
| β = 112.3 | β = 112.2 | β = 112.0 | β = 112.1 | |
| Space group | C2 | C2 | C2 | C2 |
| Resolution (Å)a | 49.57–2.05 (2.11–2.05) | 49.03–1.55 (1.58–1.55) | 49.94–1.85 (1.89–1.85) | 46.63–2.25 (2.32–2.25) |
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Observed reflections | 56,558 | 130,081 | 75,599 | 42,870 |
| Unique reflections | 16,912 | 37,535 | 22,772 | 12,726 |
| <I/σ(I)>a | 9.3 (1.8) | 9.4 (1.9) | 11.3 (2.0) | 12.2 (2.0) |
| Completeness (%)a | 99.2 (99.6) | 96.7 (96.7) | 99.1 (99.0) | 99.7 (99.8) |
| Multiplicitya | 3.3 (3.4) | 3.5 (3.4) | 3.3 (3.2) | 3.4 (3.3) |
| Rmerge (%)a,b | 9.9 (75.9) | 7.5 (86.8) | 7.8 (68.5) | 8.7 (62.4) |
| Rmeas (%)a,d | 11.8 (90.2) | 8.8 (103.2)) | 9.3 (82.4) | 10.4 (74.8) |
| Rpim (%)a,b | 6.4 (48.2) | 4.7 (55.3) | 5.0 (45.2) | 5.6 (40.7) |
| CC1/2a,e | 0.996 (0.673) | 0.997 (0.569) | 0.997 (0.774) | 0.997 (0.776) |
| Refinement | ||||
| Resolution (Å)a | 41.79–2.05 | 30.77–1.55 | 46.94–1.85 | 46.63–2.25 |
| Reflections (working/test)a | 16,036/865 | 35,666/1853 | 21,634/1127 | 12,039/683 |
| Rfactor/Rfree (%)a,c | 16.0/23.4 | 15.6/18.5 | 16.3/21.2 | 16.1/22.8 |
| No. of atoms (Protein/Ligand/Water) | 2260/58/179 | 2286/62/272 | 2255/60/128 | 2239/46/69 |
| Model Quality | ||||
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.009 | 0.009 | 0.010 | 0.012 |
| Bond angles (o) | 1.011 | 1.017 | 1.072 | 1.193 |
| Average B-factor (Å2) | ||||
| All Atoms | 31.4 | 19.5 | 28.4 | 37.1 |
| Protein | 31.0 | 18.4 | 28.2 | 37.1 |
| Ligand | 28.7 | 17.3 | 27.4 | 34.7 |
| Water | 37.4 | 29.0 | 32.9 | 37.0 |
| Coordinate error(maximum likelihood) (Å) | 0.30 | 0.14 | 0.18 | 0.24 |
| Ramachandran Plot | ||||
| Most favored (%) | 99.0 | 99.7 | 99.0 | 98.0 |
| Additionally allowed (%) | 1.0 | 0.3 | 1.0 | 2.0 |
Values in parenthesis are for the highest resolution shell.
Rmerge = ƩhklƩi |Ii(hkl) - <I(hkl>|/ƩhklƩiIi(hkl), where Ii(hkl) is the intensity measured for the ith reflection and <I(hkl)> is the average intensity of all reflections with indices hkl.
Rfactor = Ʃhkl ||Fobs (hkl) | - |Fcalc (hkl) ||/Ʃhkl |Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
2.2. Chemistry
The synthesis of final compounds 9(a-f) and 10(a-f) is outlined in Scheme 1 . 1-Boc-4-piperidinone was reacted with different Grignard reagents to yield the corresponding 1-Boc-4-piperidinol derivatives (1c and 1e). Refluxing (L) leucine methyl ester hydrochloride with trichloromethyl chloroformate yielded the isocyanate which was reacted with (1c and 1e, or commercially-available N-substituted 4-piperidinol 1a) to form the corresponding carbamate adducts (4a, 4c and 4e) that were hydrolyzed to the corresponding acids (5a, 5c and 5e) with lithium hydroxide in aqueous THF. Subsequent coupling with glutamine surrogate methyl ester hydrochloride 11 afforded the desired dipeptidyl esters (6a, 6c and 6e) which were either treated with lithium borohydride directly or were first treated with dry HCl in dioxane followed by reaction with an alkyl sulfonyl chloride or alkyl chloroformate, to yield esters (7b, 7d and 7f) prior to reduction with lithium borohydride, to yield alcohols 8 (a-f). Dess-Martin oxidation, followed by flash chromatography, yielded pure aldehydes 9(a-f). The enantiomeric purity of the aldehydes was consistently high, with the amount of epimerized aldehyde ranging between 0 and 10%. The corresponding bisulfite adducts 10(a-f) were readily obtained as white solids by stirring the aldehydes with sodium bisulfite in an ethyl acetate/water mixture. The synthesized compounds are listed in Table 1.
Scheme 1.

Synthesis of inhibitors 9a-f and 10a-f.
2.3. Biochemical studies
The inhibitory activity of the synthesized compounds against 3CLpro of MERS-CoV, SARS-CoV or FIPV, and the antiviral activity of two representative compounds (compounds 10a and 10c) in a cell-based system including MERS-CoV, FIPV and MHV were evaluated as described in the experimental section. The IC50, EC50, and CC50 values, are listed in Table 1, Table 2 These are the average of at least two determinations.
It is evident that derivatives of (I) function as highly potent inhibitors of all tested coronaviruses in enzyme (Table 1) and cell based assays (Table 2 and Fig. 4 ). More importantly, representative aldehyde bisulfite adduct compounds 10a and 10c display potent inhibition toward MERS-CoV in both enzyme and cell-based systems, with low cytotoxicity (CC50 > 100 μM) (Table 2 and Fig. 4). For example, compound 10a has a selectivity index (SI = CC50/EC50) of >250. With the exception of compounds 9e-10e, the aldehyde and aldehyde bisulfite adducts were found to have comparable in vitro potency toward MERS-CoV 3CLpro. Furthermore, pharmacological activity was found to be dependent on the nature of the R3 group (compounds 9e-10e are 10-fold less active toward MERS-CoV 3CLpro than compounds 9a-d, 10a-d and 9f-10f).
Fig. 4.

Effects of compounds 10a and 10c on the replication of MERS-CoV in cell culture. Virus titers by various drug concentrations are shown as % to the control (no compound).
In order to establish the mechanism of action of (I), as well as obtain structural information that can be used to guide the optimization of pharmacological activity, the high resolution X-ray crystal structures of several derivatives of (I) bound to MERS-CoV 3CLpro were determined, including the cocrystal structure of the MERS-CoV 3CLpro:inhibitor 10c complex (Fig. 5 A). The formation of a tetrahedral adduct via the reaction of the aldehyde, generated from aldehyde bisulfite adduct 10c under the crystallization conditions used [27,28], with the active site cysteine (Cys148) is clearly evident, confirming the mechanism of action of (I). Inspection of the structure reveals the presence of prominent electron density consistent with the structure of inhibitor 10c; however, the N-Boc-piperidinyl moiety was disordered. The position and orientation of the benzyl group suggest that the piperidine ring is likely projecting toward the S4 subsite. Inhibitor 10c is bound to the active site of the enzyme via a network of backbone H-bonds with Gln192, Gln167, and Glu169 (Fig. 5B). Additionally, a H-bond with His41 serves to stabilize the hemi-thioacetal tetrahedral adduct. Also clearly evident are three critical H-bonds involving the P1 Gln surrogate ring oxygen and nitrogen with Glu169, His166 and Phe143. The H-bonding interactions are near identical to those of inhibitor GC813 (Fig. 2/Panel B). The structural complementarity of inhibitors 10c and GC813 is also evident in the electrostatic surface representation of the enzyme with the two inhibitors nestled in the active site (Fig. 6 ).
Fig. 5.

Binding of compound 10c (gray) in the active site of MERS-CoV 3CLpro (magenta). A) Fo-Fc omit map (green mesh) and 2Fo-Fc map (blue mesh) contoured at 3σ and 1σ respectively. B) Chemical structure of compound 10c. The atoms colored red were disordered and could not be fit to the electron density maps. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 6.

Overlay of X-ray crystal structures of GC813 and compound 10c.
The cocrystal structure of the MERS-CoV 3CLpro:aldehyde bisulfite adduct 10e complex also showed that, under the crystallization conditions used, the aldehyde bisulfite adduct reverted to the precursor aldehyde, which subsequently formed a tetrahedral adduct with the active site cysteine (Cys148) (Fig. 7 A). The piperidinyl moiety was disordered and consequently its precise location could not be discerned. However, inhibitor 10e is engaged in the same H-bonding interactions as inhibitor 10c (Fig. 7B).
Fig. 7.

Binding of compound 10e (gray) in the active site of MERS-CoV 3CLpro (magenta). A) Fo-Fc omit map (green mesh) and 2Fo-Fc map (blue mesh) contoured at 3σ and 1σ respectively. B) Chemical structure of compound 10e. Atoms colored red were disordered and could not be modeled. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Conclusion
MERS-CoV constitutes a global public health concern. There are currently no licensed vaccines or antiviral drugs for the prevention and treatment of coronavirus infections. We disclose herein for the first time the design and utilization of a general class of piperidine-based peptidomimetic inhibitors of coronavirus 3CL proteases. Attachment of the piperidine moiety to a dipeptidyl component permits the resultant hybrid inhibitor to engage in favorable binding interactions with the S3 and S4 subsites of the enzyme. More importantly, the approach disclosed herein can be extended to other proteases of medical relevance. Finally, the disclosed compounds potently inhibit MERS-CoV, and their mechanism of action and mode of binding to MERS-CoV 3CL protease have been illuminated using X-ray crystallography.
4. Experimental section
4.1. General
Reagents and dry solvents were purchased from various chemical suppliers (Aldrich, Acros Organics, Chem-Impex, TCI America, and Bachem) and were used as obtained. Silica gel (230–450 mesh) used for flash chromatography was purchased from Sorbent Technologies (Atlanta, GA). Thin layer chromatography was performed using Analtech silica gel plates. Visualization was accomplished using UV light and/or iodine. NMR spectra were recorded in CDCl3 or DMSO-d6 using a Varian XL-400 spectrometer. Melting points were recorded on a Mel-Temp apparatus and are uncorrected. High resolution mass spectrometry (HRMS) was performed at the University of Kansas Mass Spectrometry lab using an LCT Premier mass spectrometer (Waters, Milford, MA) equipped with a time of flight mass analyzer and an electrospray ion source. The purity of the compounds was determined by high-performance liquid chromatography (HPLC) using a Waters Alliance HPLC system with a reverse phase column (Symmetry® C18 3.5 μm, 4.6 × 75 mm) and a Varian Pro-star HPLC system with a normal phase column (Kinetex 2.6u HILIC 100A, 75 × 4.6 mm) at 254 nm. Analysis was conducted using two different methods. Method A: Revered phase system with gradient elution. Gradient was started with water 98%: methanol 2% to water 60%: methanol 40% over a period of 15 min, mobile phase flow rate 1.0 mL/min. Method B: Normal phase system with isocratic with 20% acetonitrile and 80% dichloromethane, mobile phase flow rate 1.0 mL/min. All final compounds had a purity of ≥95% by both methods.
4.1.1. Synthesis of Boc-piperidinol derivatives 1c and 1e. General procedure
An appropriate Grignard reagent (11 mmol/1.1 eq) was added dropwise under a N2 atmosphere to a solution of 1-Boc-4-piperidinone (10 mmol) in dry THF (15 mL) in an ice bath kept at 0 °C. The reaction mixture was stirred for 3 h at room temperature under a N2 atmosphere while monitoring completion of the reaction by TLC. The reaction mixture was diluted with water (25 mL) and the solution was acidified to pH ∼3 using 5% hydrochloric acid. The solvent was removed on the rotary evaporator and the residue was extracted with ethyl acetate (75 mL) and the layers separated. The organic layer then washed with brine (40 mL) and dried over anhydrous sodium sulfate, filtered and concentrated to yield a colorless oily product which was purified by flash chromatography to yield 1c and 1e.
4.1.2. Synthesis of (L) leucine methyl ester isocyanate 3
(L) Leucine methyl ester hydrochloride 2 (100 mmol) was placed in a dry 500-mL RB flask and then dried overnight on the vacuum pump. The flask was flushed with nitrogen and dry dioxane (200 mL) was added followed by trichloromethyl chloroformate (29.67 g, 150 mmol), and the reaction mixture was refluxed for 10 h. The solvent was removed on the rotary evaporator and the residue was vacuum distilled to yield pure isocyanate 3 as a colorless oil [27,28].
4.1.3. Synthesis of substituted piperidine-derived carbamates 4a, 4c and 4e. General procedure
A solution of substituted or unsubstituted 1-Boc-4-piperidinol (1c, 1e or 1a) (20 mmol) in dry acetonitrile (15 mL) was treated with triethylamine (4.05 g, 40 mmol) followed by the amino acid methyl ester isocyanate 3 (20 mmol). The resulting solution was refluxed for 2 h and then allowed to cool to room temperature. The solution was concentrated and the residue was taken up in ethyl acetate (75 mL). The organic layer was washed with 5% HCl (2 × 20 mL) and brine (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, leaving compounds 4a, 4c and 4e as colorless oils.
4.1.4. Synthesis of acids 5a, 5c and 5e. General procedure
A solution of ester (4a, 4c or 4e) (20 mmol) in tetrahydrofuran (30 mL) was treated with 1 M LiOH (40 mL). The reaction mixture was stirred for 3 h at room temperature and the disappearance of the ester was monitored by TLC. Most of the solvent was evaporated off and the residue was diluted with water (25 mL). The solution was acidified to pH ∼3 using 5% hydrochloride acid (20 mL) and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated to yield the corresponding compounds 5a, 5c and 5e as colorless oils.
4.1.5. Synthesis of compounds 6a, 6c and 6e. General procedure
EDCI (2.40 g, 12.5 mmol, 1.25 eq) and HOBt (1.92 g, 12.5 mmol, 1.25 eq) were added to a solution of compound (5a, 5c or 5e ) (10 mmol) in dry DMF (20 mL) and the mixture was stirred for 30 min at room temperature. In a separate flask, a solution of deprotected glutamine surrogate 11 (2.23 g, 10 mmol) in DMF (15 mL) cooled to 0–5 °C was treated with diisopropylethylamine (DIEA) (9.5 g, 40 mmol, 4 eq), stirred for 30 min, and then added to the reaction mixture containing the acid. The reaction mixture was stirred for 12 h while monitoring the reaction by TLC. The solvent was removed and the residue was partitioned between ethyl acetate (100 mL) and 10% citric acid (2 × 40 mL). The layers were separated and the ethyl acetate layer was further washed with saturated aqueous NaHCO3 (40 mL), followed by saturated NaCl (50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to yield a yellow-colored oily product. Purification by flash chromatography yielded esters 6a, 6c and 6e as white solids.
4.1.6. Synthesis of compounds 7b, 7d and 7f. General procedure
4 M HCl in dioxane (8 mL) was added to a solution of compound (6a, 6c and 6e) (10 mmol) in dry DCM (5 mL) and the mixture was stirred for 1 h at room temperature. The solvent was removed and the residue was dried under high vacuum for 2 h before the product was dissolved in dry THF (20 mL). An appropriate alkyl sulfonyl chloride or alkyl chloroformate derivative (11 mmol/1.1 eq) was added to the solution with stirring. The reaction mixture was stirred for 12 h at room temperature and the residue was dissolved in ethyl acetate (50 mL) and washed with 5% HCl (2 × 20 mL). The ethyl acetate layer was further washed with saturated NaCl (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to yield a crude product. Purification by flash chromatography yielded the corresponding esters 7b, 7d and 7f as white solids.
4.1.7. Synthesis of alcohols 8 (a–f). General procedure
Lithium borohydride (2 M in THF, 7.5 mL, 15 mmol) was added dropwise to a solution of ester (6 or 7) (5 mmol) in anhydrous THF (30 mL), followed by absolute ethyl alcohol (15 mL) and the reaction mixture was stirred at room temperature overnight. The reaction mixture was then acidified by adding 5% HCl and the pH adjusted to ∼2. Removal of the solvent left a residue which was taken up in ethyl acetate (100 mL). The organic layer was washed with brine (25 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to yield compounds 8 (a-f) as white solids.
4.1.8. Synthesis of aldehydes 9a-f. General procedure
Compound 8 (a-f) (5 mmol) was dissolved in anhydrous dichloromethane (50 mL) under a nitrogen atmosphere and cooled to 0 °C. Dess-Martin periodinane reagent (3.18 g, 7.5 mmol, 1.5 eq) was added to the reaction mixture with stirring. The ice bath was removed and the reaction mixture was stirred at room temperature for 3 h (monitoring by TLC indicated complete disappearance of the starting material). A solution of 10% aqueous sodium thiosulfate (20 mL) was added and the solution was stirred for another 15 min. The aqueous layer was removed and the organic layer was washed with 10% aqueous sodium thiosulfate (20 mL), followed by saturated aqueous sodium bicarbonate (2 × 20 mL), water (2 × 20 mL) and brine (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The yellow residue was purified by flash chromatography (silica gel/methylene chloride/ethyl acetate/methanol) to yield a white solid 9 (a-f).
4.1.8.1. tert-Butyl 4-({[(S)-4-methyl-1-oxo-1-({(S)-1-oxo-3-[(S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)pentan-2-yl]carbamoyl}oxy)piperidine-1-carboxylate (9a)
White solid (yield 73%), M.p 62–64 °C. 1H NMR (400 MHz, CDCl3-d): δ ppm 0.88–1.03 (m, 6H), 1.46 (s, 9H), 1.54–1.74 (m, 3H), 1.76–1.90 (m, 2H), 1.92–2.04 (m, 1H), 2.43 (d, J = 8.89 Hz, 2H), 3.14–3.26 (m, 2H), 3.32–3.43 (m, 2H), 3.60 (br. s., 1H), 3.65–3.77 (m, 3H), 3.94–4.02 (m, 1H), 4.17–4.26 (m, 1H), 4.28–4.37 (m, 1H), 4.75–4.84 (m, 1H), 5.22–5.29 (m, 1H), 6.12 (s, 1H), 7.81–7.86 (m, 1H), 8.37–8.42 (m, 1H), 9.49 (s, 1H). 13C NMR (400 MHz, DMSO-d6): δ ppm 200.84, 178.90, 172.33, 155.39, 153.90, 78.71, 69.23, 59.74, 53.29, 52.92, 40.78, 40.14, 37.49, 31.90, 30.68, 29.29, 28.04, 24.23, 22.95. HRMS (ESI) calcd for C24H40N4O7: [M+H]: 497.2975. Found: 497.2865. HPLC 96.2% (Method A).
4.1.8.2. 1-(Methylsulfonyl)piperidin-4-yl {(S)-4-methyl-1-oxo-1-[{(S)-1-oxo-3-[(S)-2-oxopyr rolidin-3-yl]propan-2-yl}amino]pentan-2-yl}carbamate (9b)
White solid (yield 70%), M.p 53–55 °C. 1H NMR (400 MHz, CDCl3-d): δ ppm 0.92–1.02 (m, 6H), 1.50–1.74 (m, 5H), 1.78–1.89 (m, 2H), 1.91–2.06 (m, 4H), 2.26–2.53 (m, 1H), 2.80 (s, 3H), 3.14–3.25 (m, 2H), 3.38 (dd, J = 7.62, 4.74 Hz, 4H), 4.24–4.34 (m, 1H), 4.84 (d, J = 3.47 Hz, 1H), 5.20–5.26 (m, 1H), 5.77–5.83 (m, 1H), 7.32–7.34 (m, 1H), 8.51–8.56 (m, 1H), 9.48 (s, 1H). HRMS (ESI) calcd for C20H34N4O7S: [M+H]: 475.2226. Found: 475.2230. HPLC 98.8% (Method A).
4.1.8.3. tert-Butyl 4-benzyl-4-([{(S)-4-methyl-1-oxo-1-[{(S)-1-oxo-3-[(S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino]pentan-2-yl}carbamoyl]oxy)piperidine-1-carboxylate (9c)
White solid (yield 76%), M.p 57–59 °C. 1H NMR (400 MHz, CDCl3-d): δ ppm 0.78–0.91 (m, 6H), 1.33 (s, 9H), 1.35–1.47 (m, 2H), 1.51–1.72 (m, 6H), 1.79–1.88 (m, 2H), 2.14–2.40 (m, 3H), 2.74–2.96 (m, 1H), 3.12–3.27 (m, 2H), 3.42–3.60 (m, 1H), 3.78–3.94 (m, 1H), 4.07–4.18 (m, 1H), 4.22 (d, J = 4.78 Hz, 1H), 4.93–5.03 (m, 2H), 5.94 (d, J = 5.52 Hz, 1H), 6.99–7.08 (m, 1H), 7.19–7.29 (m, 4H), 7.64–7.70 (m, 1H), 8.21–8.26 (m, 1H), 9.37 (s, 1H). HRMS (ESI) calcd for C31H46N4O7: [M+H]: 587.3445. Found: 587.3452. HPLC 100% (Method A).
4.1.8.4. 4-Benzyl-1-(methylsulfonyl)piperidin-4-yl {(S)-4-methyl-1-oxo-1-[{(S)-1-oxo-3-[(S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino]pentan-2-yl}carbamate (9d)
White solid (yield 69%), M.p 56–58 °C. 1H NMR (400 MHz, CDCl3-d): δ ppm 0.89–1.04 (m, 6H), 1.52 (td, J = 8.69, 4.52 Hz, 2H), 1.60–1.76 (m, 5H), 1.80–1.89 (m, 1H), 1.95 (dd, J = 6.53, 3.53 Hz, 2H), 2.26–2.50 (m, 2H), 2.80 (s, 3H), 3.25–3.37 (m, 2H), 3.44–3.52 (m, 1H), 3.54–3.64 (m, 2H), 3.91–4.00 (m, 1H), 4.19–4.28 (m, 1H), 4.29–4.37 (m, 1H), 5.10 (d, J = 2.10 Hz, 2H), 5.90 (br. s., 1H), 7.13 (d, J = 7.08 Hz, 1H), 7.30–7.38 (m, 4H), 7.77–7.82 (m, 1H), 8.33–8.37 (m, 1H), 9.49 (s, 1H). 13C NMR (400 MHz, DMSO-d6): δ ppm 200.84, 178.94, 172. 68, 155.90, 137.16, 130.44, 128.27, 127.86, 77.93, 65.20, 53.38, 48.57, 42. 38, 41.25, 40.83, 37.49, 37.24, 32.85, 32.44, 27.67, 24.20, 22.96. HRMS (ESI) calcd for C27H40N4O7S: [M+H]: 565.2696. Found: 565.2678. HPLC 100% (Method A).
4.1.8.5. tert-Butyl 4-ethyl-4-({[(S)-4-methyl-1-oxo-1-{[(S)-1-oxo-3-{(S)-2-oxopyrrolidin-3-yl}propan-2-yl]amino}pentan-2-yl]carbamoyl}oxy)piperidine-1-carboxylate (9e)
White solid (yield 75%), M.p 58–60 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.77 (t, J = 7.30 Hz, 3H), 0.86 (dd, J = 6.80, 6.70 Hz, 6H), 1.39 (s, 9H), 1.50 (q, J = 5.80 Hz, 2H), 1.57–1.67 (m, 2H), 1.72–1.87 (m, 4H), 1.96–2.06 (m, 1H), 2.07–2.18 (m, 2H), 2.19–2.30 (m, 1H), 2.97–3.20 (m, 2H), 3.67–3.80 (m, 2H), 3.85–3.94 (m, 1H), 3.96–4.08 (m, 1H), 4.16–4.25 (m, 1H), 4.35–4.44 (m, 1H), 4.47–4.55 (m, 1H), 4.60–4.74 (m, 1H), 7.60–7.73 (m, 1H), 8.41–8.43 (m, 1H), 8.56–8.59 (m, 1H), 9.41 (s, 1H). HRMS (ESI) calcd for C26H44N4O7: [M+H]: 525.3288. Found: 525.3276. HPLC 100% (Method A).
4.1.8.6. Ethyl 4-([{(S)-4-methyl-1-oxo-1-[{(S)-1-oxo-3-[(S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino]pentan-2-yl}carbamoyl]oxy)piperidine-1-carboxylate (9f)
White solid (yield 82%), mp 60–61 °C. 1H NMR (400 MHz, CDCl3-d): δ ppm 0.96 (dd, J = 9.07, 5.87 Hz, 6H), 1.26 (t, J = 7.10 Hz, 3H), 1.48–1.73 (m, 4H), 1.80–1.90 (m, 2H), 1.94–2.03 (m, 1H), 2.35–2.52 (m, 2H), 3.19–3.30 (m, 2H), 3.33–3.46 (m, 2H), 3.67–3.80 (m, 2H), 4.08–4.20 (m, 5H), 4.33 (d, J = 8.71 Hz, 1H), 4.78–4.87 (m, 2H), 5.30 (s, 1H), 6.14–6.25 (m, 1H), 8.38 (s, 1H), 9.49 (s, 1H). HRMS (ESI) calcd for C22H36N4O7: [M+H]: 469.2662. Found: 469.2643. HPLC 100% (Method A).
4.1.9. Synthesis of bisulfite adducts 10a-f. General procedure
Absolute ethanol (12 mL) was added to a solution of aldehyde 9 (a-f) (5 mmol) in dry ethyl acetate (20 mL) with stirring, followed by a solution of sodium bisulfite (540 mg; 5 mmol) in water (5 mL) and the reaction mixture was stirred for 3 h at 50 °C. The reaction mixture was allowed to cool to room temperature and then vacuum filtered. The solid was thoroughly washed with absolute ethanol and the filtrate was dried over anhydrous sodium sulfate, filtered, and concentrated to yield a yellowish oil. The oily product was treated with ethyl ether (2 × 50 mL) to form a white solid. The white solid was stirred with ethyl ether (30 mL) and ethyl acetate (15 mL) for 5 min. Careful removal of the solvent using a pipette left the corresponding aldehyde bisulfite adducts 10 (a-f) as white solids.
4.1.9.1. Sodium (2S)-2-{(S)-2-[({[1-(tert-butoxycarbonyl)piperidin-4-yl]oxy} carbonyl)amino]-4-methylpentanamido}-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate (10a)
White solid (yield 72%), M.p 87–89 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.80–0.90 (m, 6H), 1.04–1.13 (m, 4H), 1.20 (d, J = 6.35 Hz, 1H), 1.39 (s, 9H), 1.51–1.67 (m, 2H), 1.69–1.84 (m, 4H), 1.99–2.31 (m, 2H), 3.10 (dt, J = 15.85, 7.87 Hz, 2H), 3.55–3.66 (m, 2H), 3.71–3.80 (m, 1H), 3.83–3.90 (m, 1H), 3.93–4.04 (m, 2H), 4.59–4.74 (m, 2H), 7.20 (s, 1H), 7.54 (s, 1H), 8.44–8.48 (m, 1H). HRMS (ESI) calcd for C24H41N4O10SNa: [M+H]: 601.2519. Found: 601.25. HPLC 97.6% (Method B).
4.1.9.2. Sodium (2S)-1-hydroxy-2-{(S)-4-methyl-2-[({[1-(methylsulfonyl)piperidin-4-yl]oxy}carbonyl)amino]pentanamido}-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate (10b)
White solid (yield 76%), M.p 77–80 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.79–0.95 (m, 6H), 1.14–1.23 (m, 2H), 1.31–1.47 (m, 2H), 1.61 (br. s., 4H), 1.90 (d, J = 6.54 Hz, 2H), 2.03–2.22 (m, 2H), 2.26–2.36 (m, 1H), 2.87 (s, 3H), 3.02–3.20 (m, 4H), 3.83–3.97 (m, 2H), 4.20 (d, J = 7.66 Hz, 1H), 4.33 (dd, J = 14.72, 4.56 Hz, 1H), 4.64 (d, J = 3.61 Hz, 2H), 7.27 (d, J = 8.20 Hz, 1H), 7.63–7.69 (m, 1H), 8.38–8.47 (m, 1H). HRMS (ESI) calcd for C20H35N4O10S2Na: [M+H]: 579.1771. Found: 579.18. HPLC 98.7% (Method B).
4.1.9.3. Sodium (2S)-2-{(S)-2-[({[4-benzyl-1-(tert-butoxycarbonyl)piperidin-4-yl]oxy} carbonyl)amino]-4-methylpentanamido}-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl] propane-1-sulfonate (10c)
White solid (yield 71%), M.p 65–67 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.87 (d, J = 7.03 Hz, 6H), 1.03–1.15 (m, 2H), 1.38 (s, 9H), 1.58–1.69 (m, 4H), 1.79 (d, J = 10.91 Hz, 2H), 2.01–2.32 (m, 4H), 2.96–3.17 (m, 4H), 3.70–3.81 (m, 3H), 3.93–4.12 (m, 2H), 4.62–4.76 (m, 1H), 4.95–5.08 (m, 2H), 7.12–7.26 (m, 1H), 7.34 (br. s., 4H), 7.52 (s, 1H), 7.62–7.72 (m, 1H), 8.49 (d, J = 6.86 Hz, 1H). HRMS (ESI) calcd for C31H47N4O10SNa: [M+H]: 691.2989. Found: 691.2954. HPLC 100% (Method B).
4.1.9.4. Sodium (2S)-2-{(S)-2-[({[4-benzyl-1-(methylsulfonyl)piperidin-4-yl]oxy}carbonyl)amino]-4-methylpentanamido}-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate (10d)
White solid (yield 71%), M.p 68–70 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.80–0.97 (m, 6H), 1.04–1.14 (m, 2H), 1.32–1.53 (m, 4H), 1.57–1.69 (m, 2H), 1.74–1.82 (m, 1H), 2.06–2.34 (m, 2H), 2.85 (s, 3H), 3.00–3.19 (m, 2H), 3.31–3.46 (m, 2H), 3.50–3.64 (m, 2H), 3.72–3.86 (m, 3H), 3.93–4.10 (m, 2H), 4.95–5.09 (m, 2H), 7.16 (d, J = 6.25 Hz, 1H), 7.34 (d, J = 2.29 Hz, 4H), 7.47–7.56 (m, 1H), 7.62–7.70 (m, 1H), 8.49 (d, J = 7.23 Hz, 1H). 13C NMR (400 MHz, DMSO-d6): δ ppm 179.40, 172. 68, 156.33, 137.56, 130.86, 128.70, 127.96, 126.78, 65.63, 64.31, 61.62, 53. 81, 49.00, 42.38, 41.25, 37.92, 34.73, 32.85, 32.16, 28.08, 24.63, 22.30. HRMS (ESI) calcd for C27H41N4O10S2Na: [M+H]: 669.2240. Found: 669.22. HPLC 100% (Method B).
4.1.9.5. Sodium (2S)-2-{(S)-2-[({[1-(tert-butoxycarbonyl)-4-ethylpiperidin-4-yl]oxy}carbonyl)amino]-4-methylpentanamido}-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate (10e)
White solid (yield 71%), M.p 61–63 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.81–0.97 (m, 6H), 1.08 (dq, J = 13.44, 6.83 Hz, 3H), 1.39 (s, 9H), 1.47–1.65 (m, 4H), 1.79 (d, J = 11.18 Hz, 4H), 1.99–2.33 (m, 4H), 3.00–3.20 (m, 2H), 3.62 (d, J = 4.98 Hz, 2H), 3.74 (dd, J = 14.18, 7.05 Hz, 2H), 3.94–4.09 (m, 2H), 4.16–4.24 (m, 1H), 4.42 (d, J = 10.74 Hz, 2H), 4.60–4.75 (m, 1H), 7.53 (d, J = 6.83 Hz, 1H), 7.63–7.72 (m, 1H), 8.37 (d, J = 7.96 Hz, 1H). HRMS (ESI) calcd for C26H45N4O10SNa: [M+H]: 629.2832. Found: 629.28. HPLC 95.9% (Method B).
4.1.9.6. Sodium (2S)-2-{(S)-2-[({[1-(ethoxycarbonyl)piperidin-4-yl]oxy}carbonyl)amino]-4-methylpentanamido}-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate (10f)
White solid (yield 76%), M.p 86–88 °C. 1H NMR (400 MHz, DMSO-d6): δ ppm 0.80–0.90 (m, 6H), 1.03–1.12 (m, 1H), 1.18 (t, J = 7.05 Hz, 3H), 1.38–1.50 (m, 4H), 1.60 (d, J = 6.88Hz, 2H), 1.75–1.85 (m, 2H), 2.04–2.23 (m, 2H), 3.06 (dd, J = 12.40, 7.96Hz, 2H), 3.16 (d, J = 6.93 Hz, 2H), 3.61–3.75 (m, 2H), 3.80–3.88 (m, 1H), 3.95 (dd, J = 10.42, 5.98 Hz, 1H), 4.00–4.07 (m, 2H), 4.18–4.25 (m, 1H), 4.33–4.41 (m, 1H), 4.62–4.74 (m, 2H), 7.22 (s, 1H), 7.45 (s, 1H), 7.60 (d, J = 6.10 Hz, 1H). HRMS (ESI) calcd for C22H37N4O10SNa: [M+H]: 573.2206. Found: 573.22. HPLC 94.8% (Method B).
4.2. Enzyme assays and inhibition studies. FRET protease assays
The FRET protease assay was performed by preparing stock solutions of the substrate (Dabcyl-KTSAVLQ/SGFRKME-Edans derived from the cleavage sites on the viral polyproteins of SARS-CoV) and inhibitor in DMSO and diluting into assay buffer which was comprised of 20 mM HEPES buffer, pH 8, containing NaCl (200 mM), EDTA (0.4 mM), glycerol (60%), and 6 mM dithiothreitol (DTT). The expression and purification of the 3CLpro of MERS-CoV, SARS-CoV or FIPV was performed by a standard method described previously by our lab [24,29]. The protease (3CLpro of MERS-CoV, SARS-CoV or FIPV) was mixed with serial dilutions of each compound or with DMSO in 25 μL of assay buffer and incubated at 37 °C for 30 min, followed by the addition of 25 μL of assay buffer containing substrate. Fluorescence readings were obtained using an excitation wavelength of 360 nm and an emission wavelength of 460 nm on a fluorescence microplate reader (FLx800; Biotec, Winoosk, VT) 1 h following the addition of substrate. Relative fluorescence units (RFU) were determined by subtracting background values (substrate-containing well without protease) from the raw fluorescence values, as described previously [29]. The dose-dependent FRET inhibition curves were fitted with a variable slope by using GraphPad Prism software (GraphPad, La Jolla, CA) in order to determine the IC50 values of the inhibitors.
4.3. Antiviral assays/cell-based inhibition assays
The effects of compounds 10a and 10c on the replication of MERS-CoV, FIPV or MHV-A59 were examined in Vero81, CRFK or CCL9.1 cells, respectively [30]. Briefly, confluent and semi-confluent cells were infected at an MOI of 0.01 PFU/cell. Following adsorption, cells were incubated with medium containing DMSO (<0.1%) or each compound (up to 100 μM) for 48 h. After incubation, viral titers were determined with a TCID50 (FIPV or MHV) or plaque assay (MERS-CoV). EC50 values were determined using GraphPadPrism software [31].
4.4. Nonspecific cytotoxic effects
The cytotoxic dose for 50% cell death (CC50) for compounds 10a and 10c was determined in Vero81, CRFK or CCL9.1 cells. Confluent cells grown in 96-well plates were treated with various concentrations (1–100 μM) of each compound for 72 h. Cell cytotoxicity was measured by a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI). The in vitro therapeutic index was calculated by dividing the CC50 by the EC50.
4.5. X-ray crystallographic studies. Crystallization and data collection
Purified MERS-CoV 3CLpro, in 100 mM NaCl, 20 mM Tris pH 8.0, was concentrated to 8 mg/mL (0.5 mM). Stock solutions of 100 mM GC376, GC813, compound 10c or compound 10e were prepared in DMSO and the complex with MERS 3CLpro was prepared by mixing the concentrated protein supplemented with 3 mM compound and incubating overnight at 4 °C. All crystallization experiments were conducted using Compact 300 (Rigaku Reagents) sitting drop vapor diffusion plates at 20 °C using equal volumes of protein and crystallization solution equilibrated against 75 μL of the latter. Crystals of MERS 3CLpro in complex with GC813, compound 10c and compound 10e that displayed a prismatic morphology were obtained from the Index HT screen (Hampton Research) condition G10 (25% (w/v) PEG 3350, 100 mM Bis-Tris pH 5.5, 200 mM MgCl2) in 1–2 days. Crystals of the GC376 complex were obtained from the Index HT screen (Hampton Research) condition E6 (30% (v/v) PEG 550 MME, 100 mM Bis-Tris pH 6.5, 50 mM CaCl2). Samples were transferred to a fresh drop containing 80% crystallant and 20% (v/v) PEG 200 before storing in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source beamline 17-ID using a Dectris Pilatus 6 M pixel array detector.
4.6. Structure solution and refinement
Intensities were integrated using XDS [32,33] using Autoproc [34] and the Laue class analysis and data scaling were performed with Aimless [35], which suggested that the highest probability Laue class was 2/m and space group C2. Structure solution was conducted by molecular replacement with Phaser [36] using a previously determined isomorphous structure of MERS 3CLpro (PDB: 4RSP [37]) as the search model. Structure refinement and manual model building were conducted with Phenix [38] and Coot [39], respectively. Disordered side chains were truncated to the point for which electron density could be observed. Structure validation was conducted with MolProbity [40] and figures were prepared using the CCP4MG package [41].
4.7. Accession codes
Coordinates and structure factors for the MERS 3CLpro inhibitor complexes were deposited to the Worldwide Protein Data Bank (wwPDB) with the accession codes: 5WKJ (GC376), 5WKK (GC813), 5WKL (inhibitor 10c) and 5WKM (inhibitor 10e).
Acknowledgments
Use of the University of Kansas Protein Structure Laboratory was supported by a grant from the National Institute of General Medical Sciences (P30GM110761) of the National Institutes of Health. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-06CH11357. We are grateful to Dr. Terry L. Bowlin, Microbiotix, Inc. for assistance in determining the cell-based values of compounds GC376 and GC813. The research was supported in part by grants from the National Institutes of Health (R01 AI109039 to K.O.C. and P01 AI060699 and R01 AI129269 to S. P.) and a faculty development KU Endowment Dolph Simons Award in Biomedical Sciences (W.C.G.).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2018.03.004.
Contributor Information
Kyeong-Ok Chang, Email: kchang@vet.ksu.edu.
William C. Groutas, Email: bill.groutas@wichita.edu.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- 1.Masters P.S., Perlman S. Coronaviridae in Field's virology. In: Knipe D.M., Howley P.M., editors. vol. 1. Williams & Wilkins; Philadelphia: 2013. pp. 825–858. (Lippincott). [Google Scholar]
- 2.Coleman C.M., Frieman M.B. Coronaviruses: important emerging human pathogens. J. Virol. 2014;88:5209–5212. doi: 10.1128/JVI.03488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg S.B. Update on human rhinovirus and coronavirus infections. Semin. Respir. Crit. Care Med. 2016;37:555–571. doi: 10.1055/s-0036-1584797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vijay R., Perlman S. Middle East respiratory syndrome and severe acute respiratory syndrome. Curr. Opin. Virol. 2016;16:70–76. doi: 10.1016/j.coviro.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van den Brand J.M.A., Smits S.L., Haagmans B.L. Pathogenesis of Middle East respiratory syndrome coronavirus. J. Pathol. 2015;235:175–184. doi: 10.1002/path.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan J.F.W., Lau S.K.P., To K.K.W., Cheng V.C.C., Woo P.C.Y., Yuen K.-Y. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015;28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hui D.S., Peiris M. Middle East respiratory syndrome. Am. J. Respir. Crit. Care Med. 2015;192:278–279. doi: 10.1164/rccm.201506-1221ED. [DOI] [PubMed] [Google Scholar]
- 9.Zumla A., Chan J.F., Azhar E., Hui D.S., Yuen K.-Y. Coronaviruses: drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adedeji A.O., Sarafianos S.G. Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 2014;8:45–53. doi: 10.1016/j.coviro.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Modjarrad K. MERS-CoV vaccine candidates in development: the current landscape. Vaccine. 2016;34:2982–2987. doi: 10.1016/j.vaccine.2016.03.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian Z., Dominguez S.R., Holmes K.V. Role of the spike glycoprotein of human Middle East syndrome coronavirus (MERS-CoV) in virus and syncytia formation. PLoS One. 2013;8 doi: 10.1371/journal.pone.0076469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirato K., Kawase M., Matsuyama S. Middle East respiratory syndrome infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013;87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller J.K., Whittaker G.R. Host cell entry of Middle East respiratory distress syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. U.S.A. 2014;111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of severe acute respiratory distress syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baez-Santos Y.M., St. John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar V., Tan K.-P., Wang Y.-M., Lin S.-W., Liang P.-H. Identification, synthesis, and evaluation of SARS-CoV and SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016;24:3035–3042. doi: 10.1016/j.bmc.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Needle D., Lountos G.T., Waugh D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. 2015;D71:1102–1111. doi: 10.1107/S1399004715003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu A., Wang Y., Zeng C., Huang X., Xu S., Su C., Wang M., Chen Y., Guo D. Prediction and biochemical analysis of putative cleavage sites of the 3C-like protease of Middle East respiratory syndrome coronavirus. Virus Res. 2015;208:56–65. doi: 10.1016/j.virusres.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H., Lei H., Santarsiero B.D., Gatuz J.L., Cao S., Rice A.J., Patel K., Szypulinski M., Ojeda I., Ghosh A.K., Johnson M.E. Inhibitor recognition specificity for MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 2015;10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nomenclature used is that of L. Schechter, A. Berger. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. where S1, S2, S3, …. Sn and S1’, S2’, S3’, …. Sn’ correspond to the enzyme subsites on the N-terminus and C-terminus side, respectively, of the scissile bond. Each subsite accommodates a corresponding amino acid residue side chain designated P1, P2, P3,…..Pn and P1’, P2’, P3’,…..Pn’ of a substrate or inhibitor. P1 is the primary substrate specificity residue and P1-P1’ is the scissile bond. [DOI] [PubMed] [Google Scholar]
- 23.Wang F., Chen C., Tan W., Yang K., Yang H. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016;6:22677. doi: 10.1038/srep22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y., Liu H., Galasiti Kankanamalage A.C., Weerasekara S., Hua D.H., Groutas W.C., Chang K.O., Pedersen N.C. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen N.C., Kim Y., Liu H., Galasiti Kankanamalage A.C., Eckstrand C., Groutas W.C., Bannasch M., Meadows J.M., Chang K.O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2017 doi: 10.1177/1098612X17729626. 17 Sep 1:1098612X17729626. ([Epub ahead of print]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webster S.E., Okano K., Little T.L., Reich S.H., Xin Y., Fuhrman S.A., Matthews D.A., Love R.A., Hendrickson T.F., Patick A.K., Meador J.W., Ferre R.A., Brown E.L., Ford C.E., Binford S.L., Worland S.T. Tripeptide aldehyde inhibitors of human rhinovirus 3C protease: design, synthesis, biological evaluation, and cocrystal structure solution of P1 glutamine isosteric replacements. J. Med. Chem. 1998;41:2786–2805. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- 27.Galasiti Kankanamalage A.C., Kim Y., Weerawarna P.W., Uy R.A.Z., Damalanka V.C., Mndadapu S.R., Alliston K.R., Mehzabeen N., Battaille K.P., Lovell S., Chang K.-O., Groutas W.C. Structure-guided design and optimization of dipeptidyl inhibitors of norovirus 3CL protease. Structure-activity relationships and biochemical, X-ray crystallographic, cell-based, and in vivo studies. J. Med. Chem. 2015;58:3144–3155. doi: 10.1021/jm5019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandadapu S.R., Gunnam M.R., Tiew K.-C., Uy R.A.Z., Prior A.M., Alliston K.R., Hua D.H., Kim Y., Chang K.-O., Groutas W.C. Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg. Med. Chem. Lett. 2013;23:62–65. doi: 10.1016/j.bmcl.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim Y., Lovell S., Tiew K.C., Mandadapu S.R., Alliston K.R., Battaille K.P., Groutas W.C., Chang K.-O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses and coronaviruses. J. Virol. 2012;6:1754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Wilde A.H., Falzarano D., Zevenhoven-Dobbe J.C., Beugeling C., Fett C., Martellaro C., Posthuma C.C., Feldmann H., Perlman S., Snijder E.J. Alisporivir inhibits MERS- and SARS-coronavirus replication in cell culture, but not SARS-coronavirus infection in a mouse model. Virus Res. 2017;228:7–13. doi: 10.1016/j.virusres.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.http://www.graphpad.com/quickcalcs/ConfInterval1.cfm (accessed April 2017).
- 32.Kabsch W. Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 1988;21:67–72. [Google Scholar]
- 33.Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011;67(Pt4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans P.R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston T.S., St. John S.E., Osswald H.L., Nyalapatla P.R., Paul L.N., Ghosh A.K., Denison M.R., Mesecar A.D. Ligand-induced dimerization of Middle East Respiratory Syndrome (MERS) coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J. Biol. Chem. 2015;290:19403–19422. doi: 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., David I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potterton L., McNicholas S., Krissinel E., Gruber J., Cowlan K., Emlsey P., Murshudov G.N., Cohen S., Perrakis A., Noble M. Developments in the CCP4 molecular graphics project. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 42.Evans P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 43.Diederichs K., Karplus P.A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 44.Weiss M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001;34:130–135. [Google Scholar]
- 45.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans P. Biochemistry. Resolving some old problems in protein crystallography. Science. 2012;336:986–987. doi: 10.1126/science.1222162. [DOI] [PubMed] [Google Scholar]
- 47.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov N., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. 2012;60:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.