

Abstract
Vascular cognitive impairment is a major cause of dementia caused by chronic hypoxia, producing progressive damage to white matter (WM) secondary to blood-brain barrier (BBB) opening and vascular dysfunction. Tight junction proteins (TJPs), which maintain BBB integrity, are lost in acute ischemia. Although angiogenesis is critical for neurovascular remodeling, less is known about its role in chronic hypoxia. To study the impact of TJP degradation and angiogenesis during pathological progression of WM damage, we used the spontaneously hypertensive/stroke prone rats with unilateral carotid artery occlusion and Japanese permissive diet to model WM damage. MRI and IgG immunostaining showed regions with BBB damage, which corresponded with decreased endothelial TJPs, claudin-5, occludin, and ZO-1. Affected WM had increased expression of angiogenic factors, Ki67, NG2, VEGF-A, and MMP-3 in vascular endothelial cells and pericytes. To facilitate the study of angiogenesis, we treated rats with minocycline to block BBB disruption, reduce WM lesion size, and extend survival. Minocycline-treated rats showed increased VEGF-A protein, TJP formation, and oligodendrocyte proliferation. We propose that chronic hypoxia disrupts TJPs, increasing vascular permeability, and initiating angiogenesis in WM. Minocycline facilitated WM repair by reducing BBB damage and enhancing expression of TJPs and angiogenesis, ultimately preserving oligodendrocytes.
Keywords: magnetic resonance imaging, chronic hypoxia, BBB permeability, tight junction proteins, angiogenesis, vascular cognitive impairment and dementia, white matter, and spontaneously hypertensive/stroke prone rat
Introduction
Vascular cognitive impairment and dementia (VCID) is the second most common form of dementia (Gorelick et al., 2011; Toth et al., 2017). The major form of VCID is subcortical ischemic vascular disease (SIVD), which is progressive damage to white matter secondary to small blood vessel pathology (Faraco and Iadecola, 2013; Hachinski et al., 2006; O’Brien et al., 2003; Pantoni, 2010; Taheri et al., 2011). We showed that the pathophysiology of SIVD involves inflammatory disruption of the blood-brain barrier (BBB) associated with increased expression of matrix metalloproteinases (MMPs) in the brain (Rosenberg et al., 2001) and the cerebrospinal fluid (CSF) (Adair et al., 2004; Candelario-Jalil, 2011). Hypoxia initiates a neuroinflammatory response with the production of free radicals and proteases that disrupts the BBB, leading to vasogenic edema and myelin damage (Iadecola, 2013).
Spontaneously hypertensive/stroke prone rats (SHR/SP) show many of the pathologic features of patients with SIVD, including fibrosis of small blood vessels, injury to the WM, and behavioral deficits (Sironi et al., 2004). In SHR/SP fed a diet consisting of low protein and high salt (the Japanese Permissive Diet, JPD), along with unilateral carotid artery occlusion (UCAO), we used an oxygen sensitive crystal with electron paramagnetic resonance to show an increase in tissue oxygen in the WM at 12 weeks of life with a drastic decrease in oxygen from the 13th to the 16th week when death occurs (Weaver et al., 2014). We showed that the UCAO/JPD SHR/SPs underwent an accelerated injury to the WM that is associated with BBB disruption (Jalal et al., 2012). This BBB disruption occurred secondary to increased hypoxia-inducible factor-1α (HIF-1α), which induced a MMP-9-mediated infiltration of leukocytes (Jalal et al., 2015).
The integrity of the neurovascular unit is maintained by multiple components, including tight junctions (TJ) that seal capillary endothelial cells, astrocyte end feet, pericytes, and the extracellular matrix (ECM)(Neuwelt et al., 2011). The limited permeability of the BBB is mainly due to the existence of tight junction proteins (TJPs), which are integral transmembrane proteins that form the TJ strands between endothelial cells. Prominent BBB structural changes occur after cerebral ischemic stroke, including TJP degradation and redistribution (Krueger et al., 2015; Neuwelt et al., 2011). However, the role of chronic hypoxia in TJP degradation and redistribution that leads to BBB opening and WM injury secondary to chronic hypertension is unclear.
Angiogenesis provides critical neurovascular substrates for remodeling after stroke and other neurodegenerative diseases by generating new vessels necessary for neurogenesis and synaptogenesis (Beck and Plate, 2009; Hermann and Zechariah, 2009; Yang et al., 2013). Growing evidence suggests that angiogenesis might represent a new pathogenic mechanism involved in the progression of aging and neurodegeneration. A study suggested that decreased serum levels of the angiogenic factors VEGF and TGF-1β in Alzheimer’s disease (AD) and amnestic mild cognitive impairment (MCI) might be related to the severity of cognitive impairment (Huang et al., 2013). A decline in cerebrovascular angiogenesis may inhibit recovery from hypoxia-induced capillary loss (Brown and Thore, 2011). In spite of the fact that hypoxia initiates inflammation and BBB disruption, it also promotes vessel growth by up-regulating multiple pro-angiogenic pathways that mediate key aspects of endothelial, stromal, and vascular support cell biology (Krock et al., 2011). We previously demonstrated that hypoxia induced MMP-mediated inflammation produced neuroinflammatory white matter (WM) damage in SHR/SP. Angiogenesis can occur when VEGF is induced in response to hypoxia via the HIF-1 transcription factor, which plays a major role in angiogenesis. We showed that one-week after UCAO/JPD, there was a significant increase in HIF-1α in WM, which increased further by 3 weeks (Weaver et al., 2014). In addition, the early vascular endothelial injuries and dysfunction observed in the sham SHR/SPs (Schreiber et al., 2013) and UCAO/JPD SHR/SPs may induce the endothelial cells to release factors triggering neural stem cell proliferation and angiogenesis, contributing to recovery (Shen et al., 2004). Thus, there is evidence to suggest that UCAO/JPD induces angiogenesis in injured WM related to BBB disruption. However, whether angiogenesis and vascular remodeling are involved in the pathological progression of chronic hypoxia in SIVD is unclear.
In this study, we tested the hypothesis that chronic hypoxia induces BBB disruption by degrading TJPs resulting in WM damage, and triggers angiogenesis in response to WM injury in the UCAO/JPD SHR/SPs. Using multimodal MRI, we studied the correlation between the extent of WM injury in different regions and the increase in BBB permeability and serum IgG leakage. In addition, we investigated the association of chronic hypoxia with disruption of TJPs, increase in BBB permeability and endothelial dysfunction. In order to investigate hypoxia-induced angiogenesis, we characterized the expression of angiogenic factors in vascular cells in lesion WM. However, UCAO/JPD caused death of rats after 4 weeks (Jalal et al., 2012). Minocycline is a tetracycline derivative that has shown neuroprotective effects by anti-inflammatory and inhibition of MMPs in models of acute cerebral ischemia and neurodegenerative disease (Fagan et al., 2011; Yang et al., 2015; Yenari et al., 2006). We showed before that minocycline significantly reduced WM damage, improved behavior, and prolonged life in UCAO/JPD SHR/SPs (Jalal et al., 2015). In this study, we used long-term treatment of minocycline to protect the BBB, unmask angiogenesis and oligodendrocyte repair.
Methods
Animal groups and surgery
All animal studies were reviewed and approved by the Institutional Committee on Animal Use and Care at the University of New Mexico in accordance with institutional guidelines.
Male SHR/SPs (Charles River Laboratories) were divided into the following groups: 1) carotid sham-operated with normal diet; 2) UCAO/JPD. Rats in these groups were sacrificed at week 4 following UCAO/JPD. Naïve SHR-SP and sham Wistar–Kyoto rats at age of 16 weeks were employed as normal controls. 3) Carotid sham-operated with normal diet; 4) UCAO/JPD with vehicle (DMSO)-treatment; 5) UCAO/JPD with minocycline-treatment (50 mg/kg in DMSO). Rats in these groups were sacrificed at week 9 following UCAO/JPD for drug treatment study. The methods for the unilateral carotid artery occlusion (UCAO) surgery, Japanese permissive diet (JPD), and blood-pressure measures have been reported previously and will be described briefly (Jalal et al., 2012). Animals were purchased at 6 weeks of age and monitored with weekly weight measurements and systolic blood pressure by tail-cuff non-invasive blood pressure measurements using the CODA system (Kent Scientific Inc.). At 12 weeks of age the blood pressure reached a maximum, where it remains unless they are fed the JPD with added salt, which causes the blood pressure to continue to rise (Guerrini et al., 2002). The diet was given and UCAO performed at 12-week of age in SHR/SP. In the UCAO groups, the right carotid artery was isolated and double-ligated permanently with 4-0 silk sutures under deep anesthesia with 2.0 % isoflurane. Following UCAO, rats were placed on the JPD (16% protein, 0.75% potassium, 4% sodium; Ziegler Bros, Inc.) with 1 % sodium chloride added to drinking water. In the sham-operated group the right carotid artery was isolated and rats were fed with regular rodent diet with tap water following this procedure. The vehicle for the minocycline study consisted of 30% DMSO with 25% solutol in normal saline. DMSO was needed to dissolve the drug; both drug or vehicle injections were delivered every other day intraperitoneally for 9 weeks beginning in the 12th week.
Patient
The UNM Human Research Review Committee approved the study. All patients gave informed consent to undergo a lumbar puncture and to imaging studies. Permission to perform autopsies was provided by the patients’ families. Patients were seen in the Neurology Clinics at either the University of New Mexico Hospital or the Albuquerque Veterans Administration Hospital.
The patient had mixed SIVD and AD that was confirmed at autopsy; he died at age 86 after a 5-year illness characterized by personality change and cognitive impairment. The patient was seen multiple times over the course of his illnesses and had routine clinical MRI as well a proton magnetic resonance spectroscopy (1H-MRS) with N-acetylasparate (NAA) measurements. He had evidence of white matter hyperintensities and white matter lesions as shown by reduced NAA that were more pronounced on one side, but were present in both hemispheres on MRI (FLAIR). Pathological examination showed evidence of gliosis in the white matter along with amyloid plaques and neurofibrillary tangles that were restricted to limbic cortex with an overall distribution pattern most consistent with Braak stage III. Clinical diagnosis in this patient was mixed dementia, which was made according to published criteria and was confirmed at autopsy (Gorelick et al., 2011; Hachinski et al., 2006). Brain tissue was obtained at autopsy and fixed in formalin. Antigen retrieval methods were used to allow for immunohistochemistry.
Magnetic Resonance Imaging (MRI)
Multimodal MRI of the rat, including relaxation time imaging, diffusion imaging, perfusion imaging and dynamic permeability imaging, was conducted as described before (Yang et al., 2015). The rat was placed in a dedicated holder, and positioned in the isocenter of a 4.7-Tesla MRI scanner (Bruker Biospin), which was equipped with a 40-cm bore, a 660mT/m (rise time within 120μs) gradient and shim systems. To obtain good signal-to-noise ratio, a small-bore linear RF coil (Inner Diameter = 72mm) and a single tuned surface coil (RAPID Biomedical) were employed for signal excitation and detection, respectively (Sood et al., 2009; Taheri and Sood, 2007; Yang et al., 2013). During MRI, rats were anaesthetized with 2.5% isoflurane by mechanical ventilation. Respiration and heart rate were monitored during MRI measurements, and body temperature was maintained at 37.0±0.5 °C.
T2-weighted images were acquired with a fast spin-echo sequence (RARE) (TR/TE = 5000ms/56ms, FOV=4cm×4cm, slice thickness=1mm, interslice distance = 1.1mm, number of slice = 12, matrix = 256×256, number of average = 3). Infarct area and peri-infarct area were manually delineated from T2 images. The delineated areas were used as the reference for all the other parametric images. The same slice location was prescribed for all the subsequent MR protocols.
Multi-slice, multi-shot, diffusion-weighted echo-planar imaging (EPI) (TR/TE=3800ms/38ms; b-values=600 and 1900 s/mm2 in 30 directions; FOV = 4cm×4cm, slice thickness=1mm, matrix = 256 × 256) was performed to assess tissue architecture. Quantitative apparent diffusion coefficient (ADC) maps were calculated on a voxel-wise basis with a linear least square fit on the logarithm of the signal intensity versus the b-value for each diffusion direction. Based on the ADC maps, the eigenvalues of the diffusion tensor, quantitative mean diffusivity (ADC) and fractional anisotropy (FA) maps were generated using ParaVision 5.1 (Bruker Biospin MRI).
Cerebral blood flow (CBF) was measured using the arterial spin labeling (ASL) method. The sequence: Flow-sensitive Alternating Inversion Recovery Rapid Acquisition with Relaxation Enhancement (FAIR-RARE) was used to implement ASL with parameters: TE/TR=46ms/16000ms, FOV=4cmx4cm, slice thickness=1mm, number of slice=1, matrix=128×128. Perfusion map was calculated using ASL Perfusion Processing macro in ParaVision 5.1. The principle is as follows: Inversion recovery data from the imaging slice are acquired after selective inversion of the slice and after inversion of the slice including the surrounding tissue, containing the supplying arteries. The difference of the inverse of the apparent T1 images then yields a measure of the cerebral blood flow.
To non-invasively evaluate BBB permeability, we applied dynamic contrast-enhanced MRI (DCEMRI) and graphical analysis of the resultant image data (Patlak et al., 1983). The contrast agent Gd-DTPA at dose of 0.1mM/kg was injected into the femoral vein. DCEMRI was performed using a transverse fast T1 mapping that consisted of obtaining precontrast (3 sequences) and post contrast (16 sequences) images up to 45 minutes after the contrast injection. The details of pulse sequence T1_EPI for T1 mapping are: FOV=4cm×4cm, slice thickness=1.5mm, slice gap=0, matrix size = 128×128, TR/TE =10000ms/8.3 ms, number of segment = 4, number of average = 1, total scan time = 2m40s0ms. T1 map was reconstructed with the t1epia fitting function in Bruker Paravision Image Sequence Analysis (ISA) Tool. Previous research (Ewing et al., 2003) has demonstrated that the blood-to-tissue transfer or influx constant, Ki and tissue volume, Vp, could be obtained by graphical analysis of a timed series of tissue and arterial concentrations of contrast agent. Since the contrast agent concentration is proportional to changes of 1/T1(Δ(1/T1(t))), map of Ki was constructed from repeated estimates of Δ(1/T1(t)). An in-house computer program in MATLAB (Mathworks), which implemented the above principle, was used to generate the Ki map.
Immunohistochemistry (IHC)
After the MRI scans, animals were sacrificed and rat brains were removed for IHC analysis. Ten μm sections from rat brain fixed with 2% paraformaldehyde, 0.1 M sodium periodate, 0.075 M lysine in 100 mM phosphate buffer, pH 7.3 (PLP) were used for immunohistochemical analysis. Autopsy tissue of lesion WM from the patient who died with vascular dementia underwent routine autopsy with fixation of the brain in 10% formalin for neuropathological examination. Brain blocks were dissected and prepared for paraffin embedding and sectioning. Primary antibodies and dilutions used in IHC were claudin-5 (1:500) (Invitrogen, Grand Island, NY, USA), occludin (1:500) (Invitrogen), zonula occludens-1 (ZO-1) (1:200, Invitrogen), MMP-3 (1:1500, Chemicon), Ki67 (1:1000, Abcam), RECA-1 (1:300, Abcam), glial fibrillary acidic protein (GFAP) (1:400, Sigma-Aldrich; 1:100, Abcam), NG2 (1:300, Millipore), PDGFR-β (1:100, Cell Signaling). Cy™3-conjugated AffiniPure Goat anti-rat IgG (1:50, Jackson ImmunoReaserch) was used to stain serum IgG extravasation.
For immunofluorescence, 10 μm sections of frozen rat brain were treated with acetone and blocked with 5% normal serum. For human brain sample, paraffin embedded 10 μm sections were prepared as described before (Rosenberg et al., 2001). Primary antibodies were incubated for 24 h at 4°C. Sections were incubated for 90 min at 25°C with secondary antibodies conjugated with FITC or Cy-3 (Invitrogen) or Cy5 (Santa Cruz). 4’-6-diamidino-2-phenylinidole (DAPI) (Molecular Probes) was used to label cell nuclei. Immunohistochemistry (IHC) negative controls were incubated without the primary antibody or with normal (non-immune) IgGs and no specific immunolabeling was detected.
All IHC slides were viewed on an Olympus BX-51 bright field and fluorescence microscope (Olympus America Inc.). Dual or triple immunofluorescence slides were also imaged with Nikon ECLPSE TiS Inverted Microscope capable of 3D (motorized XY stage and Z focus) Imaging Stitching for both Bright-field and Single Wave-length/filter cube Epi-Fluor and software (Nikon Instruments Inc.).
Quantification of fluorescent intensity for MMP-3 and IgG
Rats were killed and perfused for IHC of MMP-3 and IgG at 4 and 8 weeks after UCAO/JPD. For analysis and quantification of MMP-3 and IgG, 8 brain sections were used from each animal with an interval of 100 μm covered a span of 800 μm in the peri-Hippocampal Plane (~Bregma −3.20 to −4.2 mm) (Yang et al., 2013). Left and right corpora callosa (CC) were measured in images captured with a 20 X objective lens by using Image J (NIH). Indicators of animal identity on slides were blinded to the investigators. Integrated density of fluorescence was calculated as the mean of the intensity obtained from the imaged sections.
Western blots
Western blots were performed to determine protein levels in affected white matter. Frozen coronal sections from another set of the 5 designed groups were cut at 400 micrometer thickness and micropunched biopsies were collected from both brain hemispheres in WM areas of external capsule (EC), corpus callosum (CC), and internal capsule (IC) at −25°C (Jalal et al., 2015). WM proteins were extracted in RIPA buffer and 50 μg total protein were separated on 4% to 20% gradient gels (Bio-Rad). The proteins were transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were then incubated with primary antibodies: claudin-5 (1:500, Invitrogen), occludin (1:500, Invitrogen), ZO-1 (1:500, Invitrogen), VEGF-A (1:2,000, Abcam). The membranes were incubated with the respective secondary antibodies and blots were developed using the West Pico Detection System (Thermo Fisher Scientific). Protein bands were visualized on X-ray film. Semiquantitation of target protein intensities was done with the use of ImageJ (NIH), and actin (1:7500, Sigma-Aldrich) immunoblots on the same PVDF membranes were used to normalize protein loading and transfer. The results are reported as normalized band intensity with actin for quantifying relative protein expression.
Statistics
Unpaired t-tests or one-way ANOVA were performed for two groups or for multiple group comparisons (with a post-hoc Student-Newman-Keuls test), respectively. Two-way (-factors) ANOVA were performed for group’s comparisons with time course analysis. In all statistical tests, differences were considered significant when p <0.05. Data are presented as means ± SE. Statistical analyses were performed using Prism, version 6.0 (GraphPad Software Incorporated).
Results
UCAO/JPD induced WM damage at 4 weeks demonstrated by MRI
We examined the WM injury in various regions including infarct volumes in corpus callosum (CC), external capsule (EC), and internal capsule (IC) in left (L) and right (R) hemispheres. Compared with the sham group, T2-weighted images showed significant WM injury in LCC, LEC, LIC, RCC, and REC in the UCAO/JPD group (Fig. 1A). When we compared the damage between left and right WM, significantly smaller infarct volumes were seen in REC and RIC than LEC and LIC, respectively. IC presented minimum injury in both hemispheres. A unilateral infarct was located in the left cortex. Consistent with the anatomical T2 images, the major WM tissue loss as shown by increased apparent diffusion coefficients (ADC, Fig. 1B) and white matter fiber tract degeneration by fractional anisotropy (FA, Fig. 1C) was seen in LCC, LEC, and RCC (Yang et al., 2015; Yang et al., 2013). We measured the cerebral blood flow (CBF) in WM with ASL (Fig. 1D). Compared with sham group, CBF maps showed hypoperfusion in all WM regions measured in both hemispheres, specifically in the LCC, LEC, RCC, and REC in UCAO/JPD group, which correlates to the higher tissue damage regions measured by T2, ADC, and FA in WM.
Figure 1.
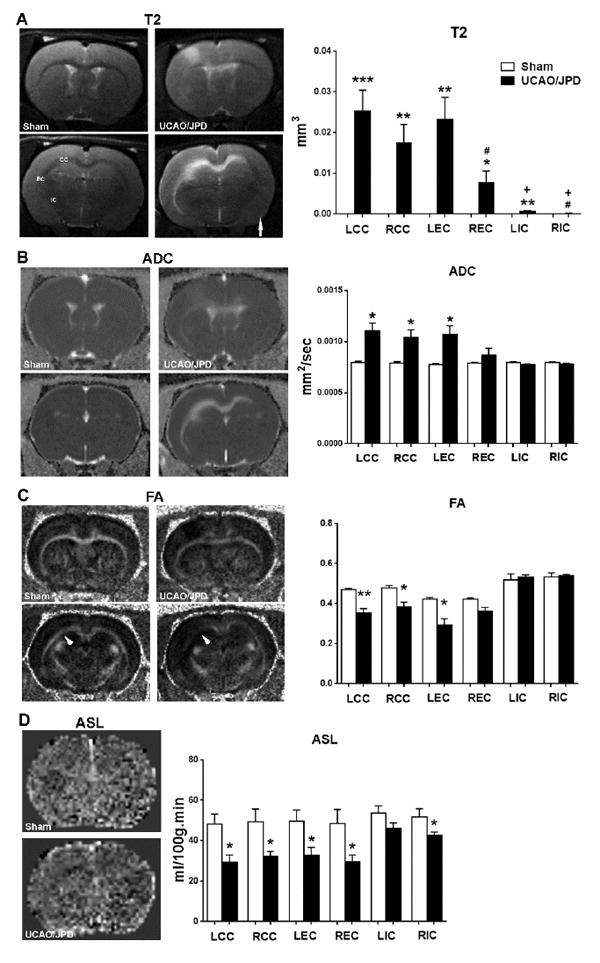
White matter (WM) damage and blood flow monitored by MRI in rats at 4 weeks after UCAO/JPD. (A). Anatomical T2-weighted (T2) images. Arrow indicates right hemispheres. Graph demonstrates quantification of infarct volumes in corpus callosum (CC), external capsule (EC), and internal capsule (IC) in left (L) and right (R) hemispheres. Major WM lesions were seen in left hemisphere. Arrow indicates the right hemisphere. (B). Apparent diffusion coefficient (ADC) for diffusion. Graph demonstrates quantification of tissue loss in WM. (C). Fractional anisotropy (FA) maps. Arrowheads indicate WM loss in UCAO/JPD rat compared with sham rat. Graph demonstrates quantification of WM lesion in WM. (D). Arterial spin labeling (ASL) maps. Graph demonstrates quantification of blood flow in WM. *p<0.05, **p<0.01, ***p<0.001 UCAO/JPD vs. Sham. #p<0.05 REC vs. LEC and LIC. +p<0.01 LIC and RIC vs. LCC, LEC, RCC, and LEC, respectively. n=8 in sham group, n=9 in UCAO/JPD group.
Increased BBB permeability and WM injury associated with abnormal TJPs
The BBB transfer rate (Ki) maps measured with DCEMRI showed significantly increased BBB permeability in rat brain subjected to UCAO/JPD compared with sham group (Fig. 2A). Increased BBB permeability was seen in both WM and gray matter (GM), and overlapped with the lesion regions shown by images of T2, ADC, and FA (Fig.1A, B, and C). Notably, the Ki maps and quantification of BBB transfer rate in WM show significantly higher BBB leakage in the left hemisphere WM versus the right, which is in line with the extent of WM injury in these regions. The MRI observations corresponded with histology of serum IgG extravasation in lesion WM and GM (Fig. 2B). Our data indicated that the UCAO/JDP induced WM damage reflected the extent of injury corresponding to the increase of BBB permeability.
Figure 2.
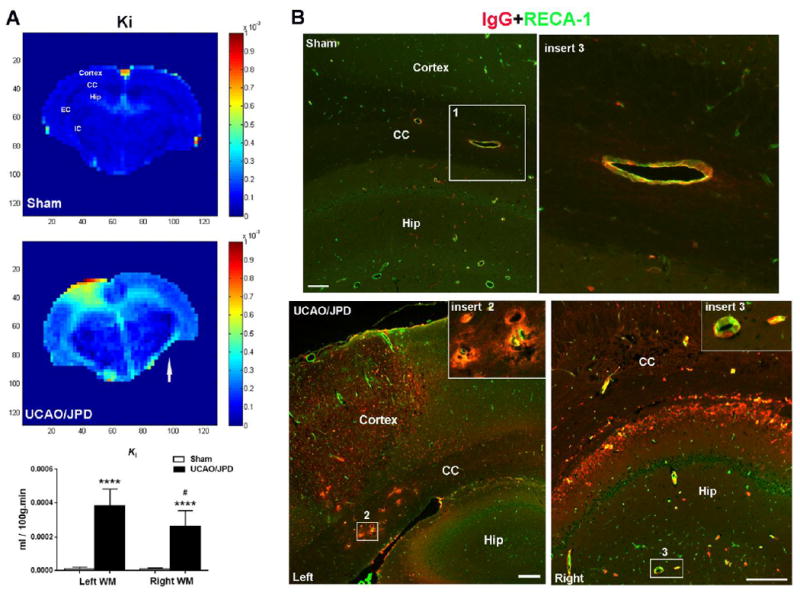
BBB permeability monitored by MRI and IHC in rats at 4 weeks after UCAO/JPD. (A). Parametric image Ki map represents BBB transfer rate for BBB permeability. Color-coded permeability coefficient maps reconstructed from contrast-enhanced MRI data demonstrate the regions of high (red) and low (blue) permeability. Arrow indicates the right hemisphere. ****p<0.0001 UCAO/JPD vs. Sham. #p<0.05 vs. left WM, n=8 in sham group, n=9 in UCAO/JPD group. CC: corpus callosum, EC: external capsule, Hip: hippocampus. (B). BBB leakage by double-immunostaining for serum IgG (red) extravasation and RECA-1 (green) in sham and UCAO/JPD SHR/SPs. RECA-1 is a cellular marker for vascular endothelial cells. Inserts show higher magnification images of leaking vessels in corpus callosum and hippocampus. Scale bars = 50 μm.
In addition, we also observed small but definite leakage of IgG in naïve and sham-operated SHR/SP brains, including WM, at age of 16 weeks (Supplementary Fig.1 and Fig. 2A). Double immunostaining of IgG with RECA-1, a cellular marker of endothelial cells, presented serum IgG leakage from the microvessels in WM and GM including the hippocampus (Fig. 2B). This is consistent with other studies (Lippoldt et al., 2000; Schreiber et al., 2013), indicating SHR/SPs have disruption of the BBB with endothelial cell dysfunction at 13 to 15 weeks of age.
To further determine the effect of UCAO/JPD on BBB disruption, we used double-immunohistochemistry (IHC) to examine the expression of TJPs in vessels in WM. Unexpectedly, we rarely detected the typical linear appearance of TJP strands, such as claudin-5, in WM microvessels of sham-operated SHR/SPs, which were evident in sham Wistar–Kyoto rats (Fig. 3A) (Liebner et al., 2000b; Sood et al., 2009; Yang et al., 2007). Double-immunostaining showed co-localizations of ZO-1, occludin, and claudin-5 with endothelial cells (RECA-1 and CD31, markers of endothelial cell) in a few microvessels (Fig. 3B). The abnormal TJ formation in sham SHR/SPs may correspond to the small amount of BBB leakage detected by MRI and IgG staining.
Figure 3.
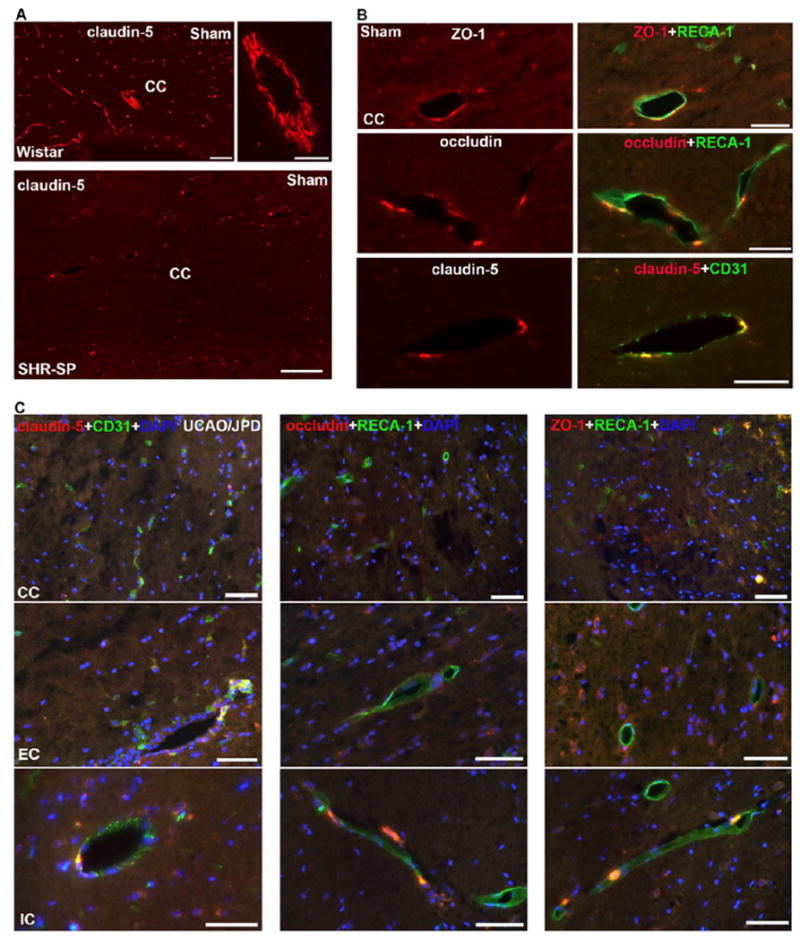
Photomicrographs represent double immunostaining of TJPs with RECA-1 in WM of sham Wistar, sham-operated SHR/SP, and UCAO/JPD SHR/SP rats at 4 weeks. (A). Photomicrographs represent expression of claudin-5 in CC of sham Wistar and sham-operated SHR/SP rats. Scale bars = 100 μm. (B). High magnification photomicrographs represent expression of TJPs in vascular endothelial cells in CC of sham-operated SHR/SP rats. Scale bars = 50 μm. (C). Photomicrographs represent expression of TJPs in vascular endothelial cells in WM of UCAO/JPD SHR/SP rats. Scale bars = 50 μm. DAPI was used to show nuclei.
At 4 weeks after UCAO/JPD, very low signal of TJPs was detected in the endothelial cells of vessels in lesion WM, specifically, no claudin-5 and occludin were seen in the vessels of lesion CC (Fig. 3C). We also found that the degree of TJP loss in vessels related to the degree of WM injury, such as the CC regions presented highest degree of TJP loss. In addition, in peri-lesion areas of WM, reactive astrocytes and proliferating vascular pericytes also expressed increased TJPs (Supplementary Fig.2), particularly ZO-1 and occludin in the reactive astrocytes that are significantly induced inside and surrounding the lesion WM areas (Jalal et al., 2012; Jalal et al., 2015). Next, we examined the protein levels of these three TJPs in lesion WM (Fig. 4). As expected, claudin-5 level was significantly decreased in both LCC and RCC, in UCAO/JPD group compared with sham rats. The reduction of occludin in LCC and claudin-5 in LCC, LIC, and RIC was also detected in the UCAO/JPD group. However, a significant increase of ZO-1 in LEC and REC, occludin in LEC, RCC, and REC, claudin-5 in REC was also detected, which could be due to increased expression of TJPs in active glial cells in injured WM.
Figure 4.
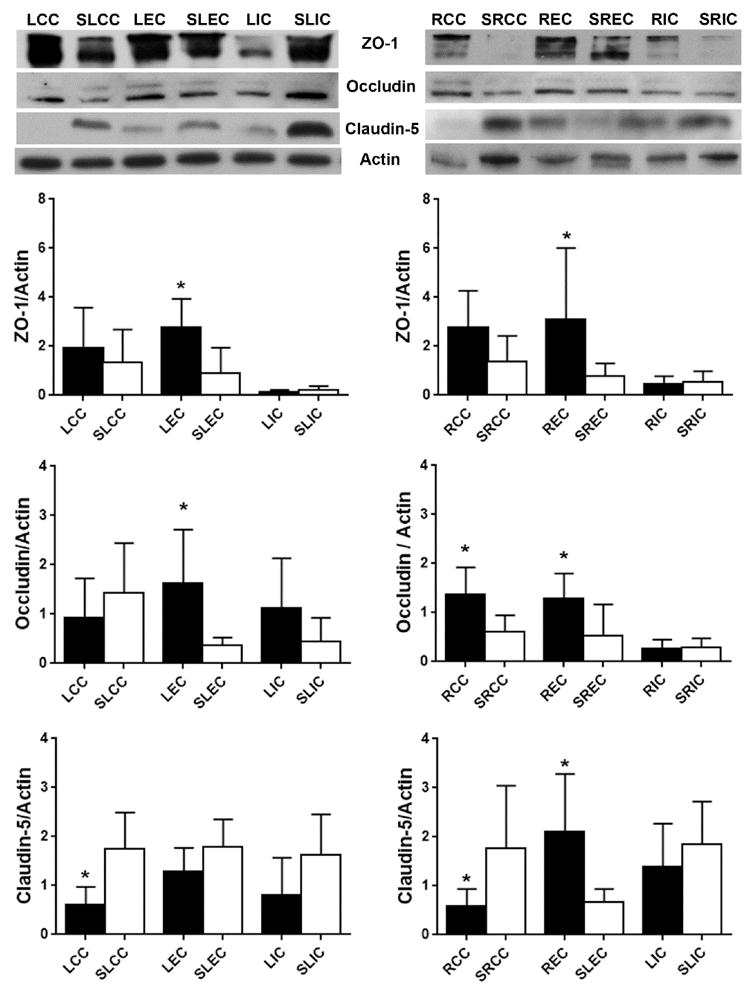
Western blot analysis of protein levels for TJPs in WM of left and right hemispheres from sham-operated rat and UCAO/JPD SHR/SP rat at 4 weeks. *p<0.05 UCAO/JPD vs. Sham. n=6 in each group. WM regions in sham group were labeled as SLCC, SLEC, SLIC, SRCC, SREC, and SRIC. WM regions in sham group were labeled as SLCC, SLEC, SLIC, SRCC, SREC, and SRIC. This labeling was also referred to all bar graphs of Western blot analysis.
Increased expression of angiogenic factors in neurovascular cells in lesion WM
RECA-1-positive vessels were seen in WM both in UCAO/JPD and sham rats at 4 weeks, while fewer blood vessels were detected in lesion CC of UCAO/JPD group compared with sham group (Fig. 5A). Double-immunostaining showed that the vascular endothelial cells contained Ki67 in the nuclei in the peri-lesion area in UCAO/JPD WM (Fig. 5B), suggesting formation of new vessels (Li et al., 2010; Yang et al., 2013). The transmembrane proteoglycan, NG2, identifies pericytes in angiogenic microvasculature (Virgintino et al., 2007; Yang et al., 2013). Co-labeling showed increased co-localization of NG2 with PDGFR-β (Fig. 5C), an established early marker of activated pericytes, in UCAO/JPD WM, particularly in lesion EC and IC where lower injury severity was seen compared with CC area. Expression of NG2 in pericytes closely surrounding highly vascularized structures confirmed the proliferating/angiogenic state of pericytes in lesion WM in UCAO/JPD rat brains(Virgintino et al., 2007; Yang et al., 2013).
Figure 5.
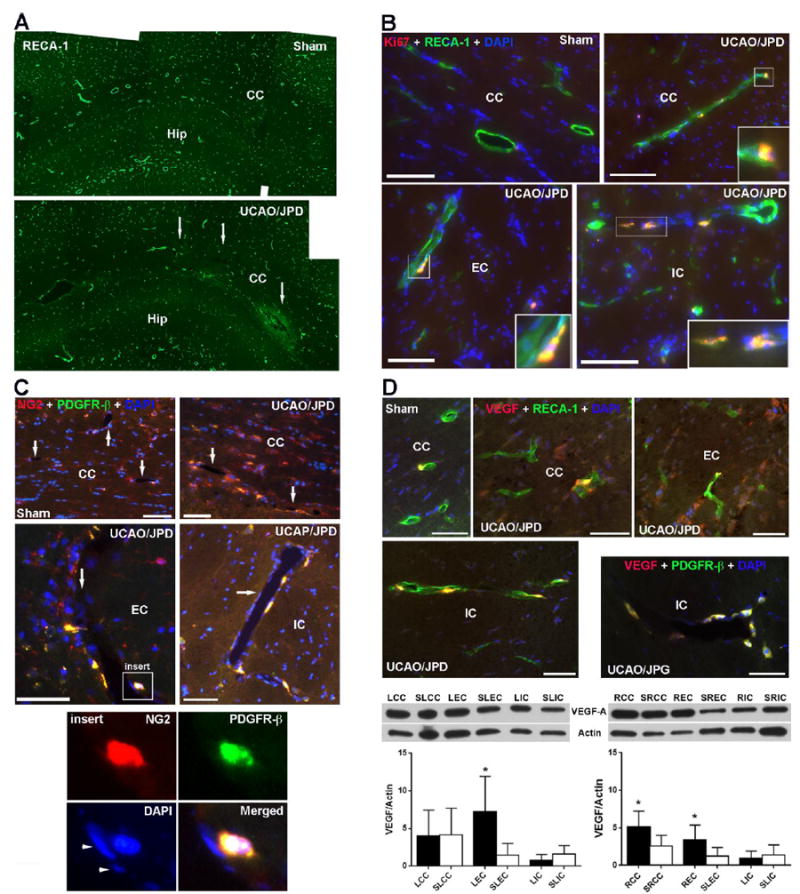
Expression of angiogenitic factors in lesion WM detected by IHC and WB at 4 weeks. (A). Photomicrographs represent vessels with RECA-1 in left hemispheres of sham- operated and UCAO/JPD SHR/SPs. Arrows indicate lesion regions in CC. (B). Double immunostaining shows expression of Ki67, a marker of cell proliferation, vascular endothelial cells (RECA-1) in WM of sham-operated and UCAO/JPD SHR-SP rats. Inserts show Ki67 in nuclei (DAPI) of endothelial cells in injured WM. (C). Double immunostaining of NG2 and with PDGFR-β, a marker of pericytes, in WM of sham- and UCAO/JPD SHR/SPs. Arrows indicate vessels in WM. Insert shows a vessel that has PDGFR-β-positive pericytes expressing NG2. Insert images show NG2 in PDGFR-β positive pericyte surrounding a vessel. Arrowheads indicate the nuclei of endothelial cells. Scale bars = 50 or 100 μm. (D). Expression of VEGF-A in WM of sham-operated and UCAO/JPD SHR/SP rats by IHC and Western blot. Double immunostaining of VEGF-A with RECA-1 shows increased VEGF-A in endothelial cells, as well in non-endothelial cells in lesion CC and EC. The expression of VEGF-A in vessel in lesion IC was co-localized with PDGFR-β-positive pericytes. Scale bars = 50 μm. Western blot analysis of protein levels for VEGF-A in WM of left and right hemispheres from sham-operated rat and UCAO/JPD SHR/SP rat at 4 weeks. *p<0.05 UCAO/JPD vs. Sham. n=6 in each group.
VEGF is a critical regulator of angiogenesis and barriergenesis (Lee et al., 2009). At 4 weeks, there was increased expression of VEGF-A, an isoform of VEGF associated with angiogenesis, in the injured WM compared with sham-operated rats (Fig. 5D). In the peri- and lesion regions, double-immunostaining showed VEGF-A co-localized with endothelial cells and pericytes surrounding vessels. Western blot analysis demonstrated that the significantly increased levels of VEGF-A were seen in damaged LEC, RCC, and REC compared with the sham group. Though increased VEGF-A was detected in UCAO/JPD LCC by IHC, there is no difference of VEGF-A protein level between LCCs of UCAO/JPD and sham animals. This may due to the fact that lesion LCCs have the most extensive injury and tissue loss. NG2 and VEGF-A were also detected in the WM of sham-operated rats, which may reflect the development of high blood pressure, chronic hypoxia, BBB leakage, and growth factor expression in SHR/SPs.
We previously showed that MMP-3, expressed by proliferating pericytes that are located next to newly formed vessels, is involved in angiogenesis during ischemia-induced brain repair (Yang et al., 2013). As expected the UCAO/JPD induced significant increase of MMP-3 expression in WM and GM at 4 weeks (Fig.6A). In affected WM, increased MMP-3 was seen in reactive astrocytes surrounding the border of infarct regions in LCC and in oligodendrocyte-like cells (Jalal et al., 2012) in peri-lesion areas (Fig. 6B). Within the peri-lesion regions, primarily in EC and IC, increased MMP-3 expression, which was not co-localized with GFAP-positive astrocytes, was seen in cells closely surrounding vessels. In the rat model of UCAO/JPD, the cells expressing MMP-3 surrounding vessels were pericytes (Fig. 6C). This observation of MMP-3 expression was also seen in human brains with SIVD. MMP-3 was detected in the cells closely surrounding endothelial cells without co-localization with GFAP-positive astrocytes within the injured WM in brain tissue from a patient with mixed dementia (SIVD and Alzheimer’s disease) (Fig. 6D).
Figure 6.
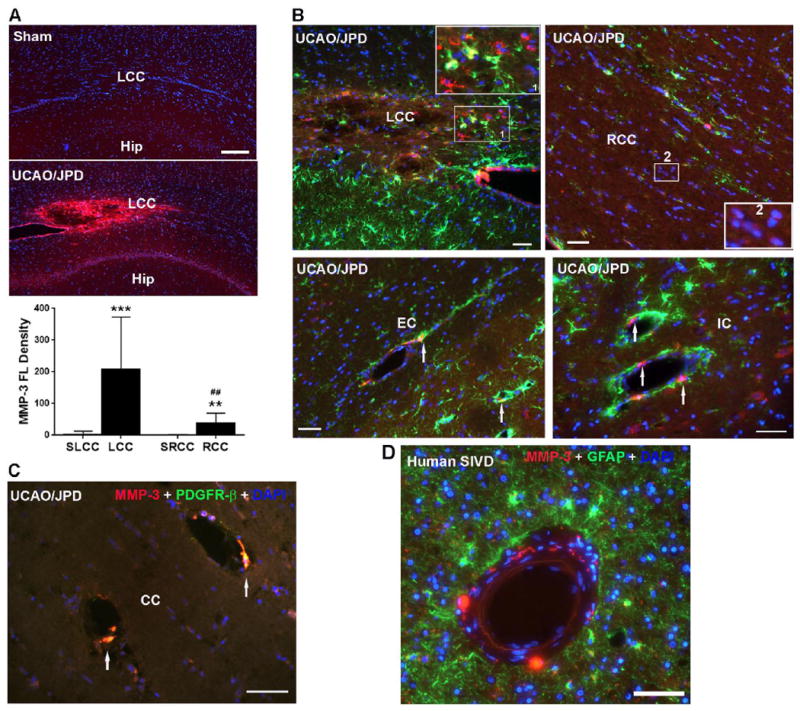
Expression of MMP-3 in WM of sham- and UCAO/JPD SHRs and human brain with subcortical ischemic vascular disease (hSIVD). (A) Increase of MMP-3 expression in lesion WM by IHC at 4 weeks. DAPI was used to show nuclei. Scale bar =100 μm. Graph shows the quantification of MMP-3 fluorescent (FL) intensity in LCC and RCC. **p<0.01, ***p<0.001 UCAO/JPD vs. Sham, ##p<0.01 RCC vs. LCC. n=8 in each group. (B). Double immunostaining shows expression of MMP-3 in astrocytes (GFAP) in WM of sham-operated and UCAO/JPD SHR-SP rats at 4 weeks. Insert 1 shows co-localization of MMP-3 and astrocytes. Insert 2 shows expression of MMP-3 in oligodendrocyte-like cells. Arrows indicate MMP-3-positivel cells surrounding vessels. (C). Double immunostaining shows expression of MMP-3 in pericytes (PDGFR-β) in WM of sham-operated and UCAO/JPD SHR/SP rats at 4 weeks. Scale bars = 50 or 100 μm. (D). Double immunostaining shows expression of MMP-3 in astrocytes in WM of human brain with SIVD. GFAP+ cells are seen surrounding a blood vessel, and the MMP-3+ cells are distinct from the astrocytes.
Minocycline treatment enhanced expression of Ki67, NG2, and MMP-3 in proliferating vascular cells, and significantly increased VEGF-A protein level in WM at 9 weeks
Our results suggest that UCAO/JPD triggers angiogenesis in response to WM injury. However, UCAO/JPD caused death of rats after 4 weeks. We previously reported that both vehicle (DMSO) and minocycline extended rat survival to 9 weeks after UCAO/JPD, and minocycline significantly attenuated WM damage (Jalal et al., 2015). In this study, MRI T2-weighted images and the quantification of infarct volumes show that the WM lesion in CC, EC, and IC in both hemispheres are significantly protected with minocycline treatment at 9 weeks after UCAO/JPD (Fig. 7A). In addition, significantly larger WM infarct volumes (Table 1) were detected in regions of CC and EC in the DMSO/UCAO/JPD group at 9 weeks than the ones in the UCAO/JPD group at 4 weeks (Figs. 7A and 1A). However, the two groups at different time points show a similar pattern of asymmetric WM damage with less injury to the right hemisphere, as well as a unilateral infarct in left cortex. Compared with the serum IgG extravasation in lesion WM at 4 weeks (Fig. 2B), significant increase of IgG extravasation was seen in lesion WMs of both left and right hemispheres in vehicle-treated rats at 9 weeks (Fig. 7B), which correlated with the significantly increased of infarct volumes (Fig. 7A). Minocycline almost entirely blocked the BBB leakage. Thus, administration of DMSO and minocycline allow us to extend our investigation on angiogenesis in response to WM injury induced by UCAO/JPD.
Figure 7.
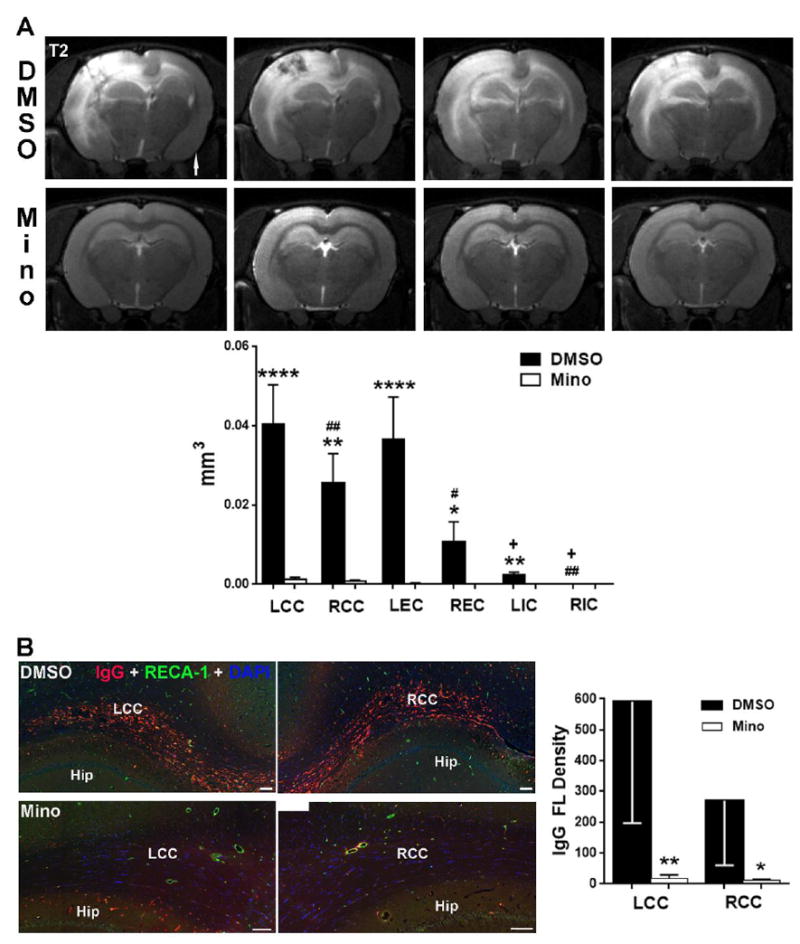
MRI studies of WM injury from the minocycline treated- and vehicle-groups following UCAO/JPD at 9 weeks. (A). Representative T2-weighted images from 4 rats out of 8 rats in minocycline (Mino) treated, and DMSO (vehicle) groups, respectively, at 9 weeks following UCAO/JPD. Arrow indicates the right hemisphere. Graph shows that the changes in infarct volumes in CC, EC, and IC in both hemispheres are significantly protected with minocycline treatment. *p<0.05, **p<0.01, ****p<0.0001 vehicle vs. MINO group; #p<0.05 REC vs. LEC, ##p<0.01 RCC and RIC vs. LCC and LIC. +p<0.01 LIC and RIC vs. LCC/LEC and RCC/LEC, respectively. n =8 in each group. (B). BBB leakage by double-immunostaining for IgG and RECA-1 in lesion WM from the minocycline treated- and vehicle-groups following UCAO/JPD at 9 weeks. Scale bars = 100 μm. DAPI was used to show nuclei. Graph shows the quantification of IgG fluorescent (FL) intensity in LCC and RCC. *p<0.05, **p<0.01 UCAO/JPD vs. Sham, ##p<0.01 RCC vs. LCC, n=8 in each group.
Table 1.
WM lesion volumes quantified by MRI T2-weighted images.
| Regions of lesion WM | LCC | RCC | ||
|---|---|---|---|---|
| 4W | 9W+DMSO | 4W | 9W+DMSO | |
| Lesion (cm3) Mean ± SE | 0.0284 ± 0.0046 | 0.0530 ± 0.0074 | 0.0197 ± 0.0044 | 0.0355 ± 0.0057 |
| p values | 0.0212 * | 0.0459 * | ||
| Regions of lesion WM | LEC | REC | ||
| 4W | 9W+DMSO | 4W | 9W+DMSO | |
| Lesion (cm3) Mean ± SE | 0.0261 ± 0.0051 | 0.0488 ± 0.0097 | 0.0261 ± 0.0051 | 0.0488 ± 0.0097 |
| p values | 0.0475 * | 0.4912 | ||
| Regions of lesion WM | LIC | RIC | ||
| 4W | 9W+DMSO | 4W | 9W+DMSO | |
| Lesion (cm3) Mean ± SE | 0.0007 ± 0.0002 | 0.0031 ± 0.0006 | 0.0007 ± 0.0002 | 0.0031 ± 0.0006 |
| p values | 0.0018 * | 0.4082 | ||
4W: Rats were scanned by MRI at 4 weeks after UCAO/JPD. 9W+DMSO: Rats were scanned by MRI at 9 weeks after UCAO/JPD with DMSO administration.
p<0.05. n=8 for each group.
We first examined the impact of the administration with vehicle (DMSO) and minocycline on expression of angiogenic factors in injured WM at 9 weeks. Due to the much larger infarct areas and tissue loss in vehicle-treated WM (Fig. 7), all IHC images of vehicle group were captured from the peri-infarct areas. Higher fluorescence signal of Ki67 was detected in minocycline-treated WM compared with vehicle (Fig. 8A). Furthermore, the co-localization of Ki67 with endothelial nuclei were seen in all of minocycline-treated LCC, LEC, and LIC, while vehicle-treated CC showed no co-localization of Ki67 with endothelial cells. IHC showed that higher fluorescence signal of MMP-3 was detected in WM of SHR/SPs-treated with minocycline, while more reactive astrocytes were seen in vehicle-treated WM (Fig. 8B). Minocycline treatment reduced NG2 expression in lesion LCC compared with the one treated with vehicle, however, co-localization of NG2 and PDGFR-β could be detected in lesion LCC (Fig. 8C). Western blot analysis was used to determine the changes of VEGF-A in WM from sham-operated, vehicle-treated, and minocycline-treated rats. The levels of VEGF-A protein in LCC and LEC were significantly augmented by minocycline compared with the sham-operated group (Fig. 8D). Interestingly, at 9 weeks after UCAO/JPD, a significant increase of VEGF-A was detected in both vehicle- and minocycline-treated ICs, compared with sham-operated one. This observation is different from the one observed in UCAO/JPD-treated IC at 4 weeks that has no changes compared with the sham-operated group (Fig. 5D). Factors released by endothelial cells in vitro trigger neural stem cell proliferation and angiogenesis, contributing to functional recovery (Lee et al., 2009). UCAO/JPD induced lowest injury in IC compared with in CC and EC (Figs. 1 and 7), which could provide a better cellular basis to trigger angiogenesis, in IC areas (Hermann and Zechariah, 2009).
Figure 8.
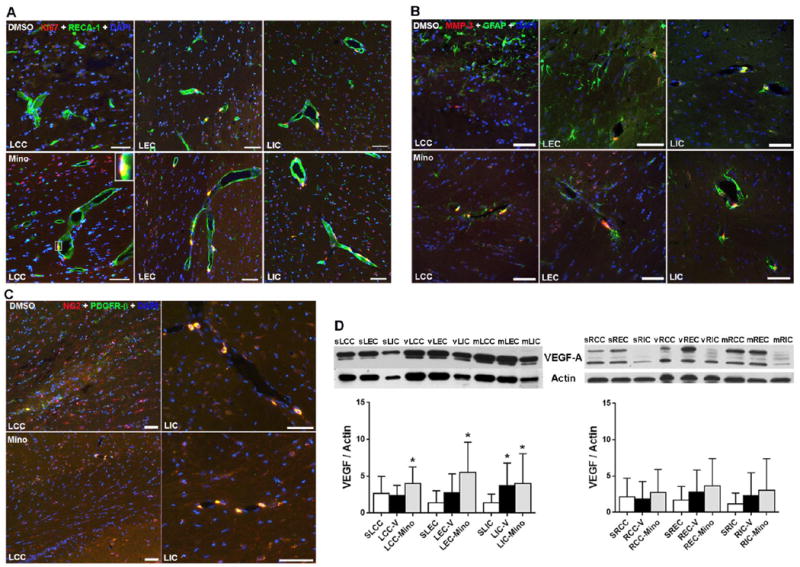
Minocycline enhanced the expression of angiogenitic factors in vascular cells in lesion WM at 9 weeks by IHC and Western blot. (A). Representative IHC images show higher expression of Ki67 in CC and EC treated with minocycline compared to vehicle. Insert presents RECA-1-positive vessel contains nuclei (DAPI) expressing Ki67 in minocycline treated LCC. (B). Double-immunostaining shows less reactive astrocytes (GFAP) and higher expression of MMP-3 surrounding vessels in CC and EC treated with minocycline compared to vehicle. (C). Representative IHC images show higher expression of NG2 in WM treated with minocycline compared to vehicle. However, higher co-localization of NG2 and pericytes surrounding vessels is seen in WM treated with minocycline. Scale bars = 50 μm. DAPI was used to show nuclei. (D). Western blot analysis of protein level for VEGF-A in WM of left and right hemispheres from sham-operated rats, and DMSO-treated and minocycline-treated SHR-SP rats following UCAO/JPD at 9 weeks. *p<0.05 UCAO/JPD or minocycline vs. Sham. n=6 in sham, n=8 in vehicle and minocycline groups.
Minocycline treatment enhanced the protein levels of TJPs and GST-π in WM at 9 weeks
Western blot analysis was used to examine the protein levels of TJPs in WM from sham-operated, vehicle-treated, and minocycline-treated rats (Fig. 9A). In vehicle-treated rats, significantly increased protein levels of ZO-1 in REC and RIC, claudin-5 in LIC, REC and RIC were detected compared with sham-operated rats. Significantly higher levels of ZO-1 and claudin-5 were seen in both left and right WM of minocycline-treated rats compared with the sham-operated ones, while increased occludin was detected only in left CC and EC of minocycline-treated rats. Similar with VEGF-A, significantly increased ZO-1 and claudin-5 were detected in both vehicle- and minocycline-treated ICs at 9 weeks after UCAO/JPD, compared with sham-operated rats. Importantly, we found that claudin-5 in the LCC, LEC, RCC, and REC of minocycline-treated rats was significantly enhanced compared with the vehicle-treated group. Since study showed that claudin-5 is expressed only by vascular endothelial cells in brain (Morita et al., 1999b), the results indicate that the endothelial cells in the angiogenic status in these damaged WM are expressing claudin-5, and minocycline enhanced the TJ strand formation in angiogenic vessels.
Figure 9.

Western blot analysis of protein levels for TJPs, ZO-1, occludin, and claudin-5, in WM of left and right hemispheres from sham-operated rats, and DMSO-treated and minocycline-treated SHR-SP rats following UCAO/JPD at 9 weeks. Graphs show that minocycline enhanced the protein levels of TJPs *p<0.05, **p<0.01 minocycline vs. vehicle; #p<0.05, ##p<0.01 UCAO/JPD or minocycline vs. sham.
Since we observed increased NG2 expression in injured WMs associated with linear rows of DAPI-staining nuclei formed by mature oligodendrocyte-lineage cells (Yang et al., 2011) (Figs. 5C and 8C), we studied the impact of the minocycline on expression of NG2 in oligodendrocytes at 9 weeks to analyze the repair of WM from injury (Fig. 10A and Supplementary Fig.3). Compared with minocycline-treated group, less co-localization of NG2 with CC1-positive oligodendrocytes was seen in vehicle-treated CC. In minocycline-treated group, most of NG2 was expressed by CC1-positive oligodendrocyte-lineage cells. Furthermore, we observed that the morphological shape of CC1-positive oligodendrocytes in vehicle-treated LCC is different from that in minocycline-treated LCC that are presented in intact linear rows of DAPI-staining nuclei formed by CC1-positive oligodendrocyte-lineage cells (Takeda et al., 2016). In addition, CC1 and DAPI staining also demonstrated reduction of oligodendrocytes in vehicle-treated LCC compared to minocycline-treated LCC.
Figure 10.
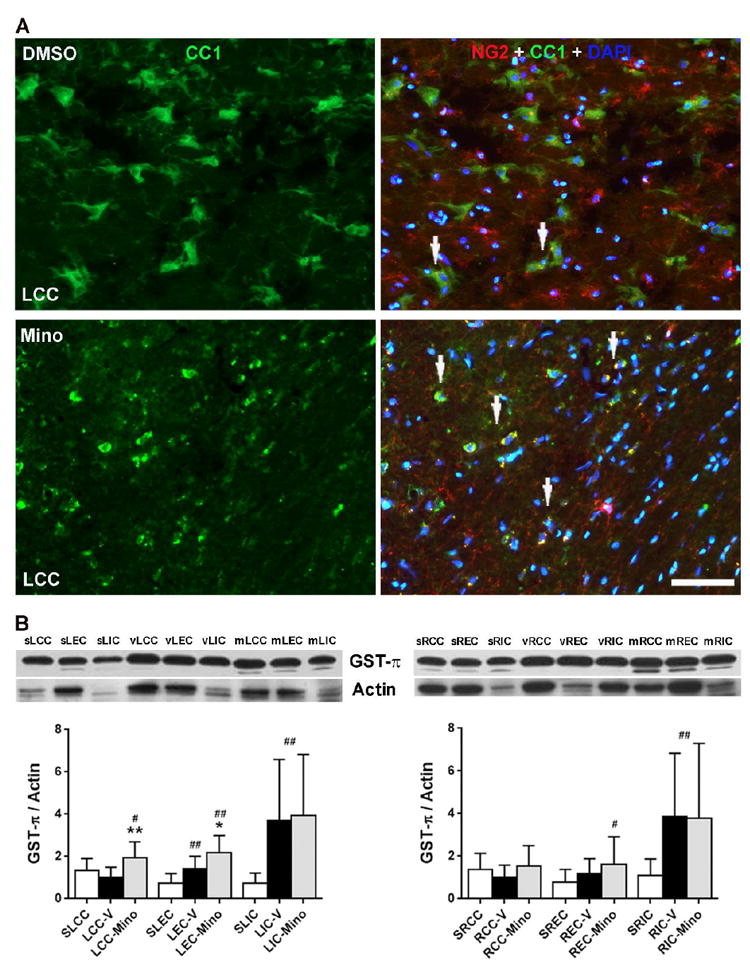
Minocycline preserved oligodendrocytes and enhanced protein level of GST-π in WM SHR/SP rats following UCAO/JPD at 9 weeks. (A). Representative IHC images show the expression of NG2 in oligodendrocytes (CC1) in lesion WM. Arrows indicates co-localization of NG2 and CC1. DAPI was used to show nuclei. Scale bar = 50 μm. (C). Western blot for GST-π protein levels in WM of left and right hemispheres from sham-operated, and DMSO-treated and minocycline-treated SHR-SP rats. Graphs show that minocycline enhanced the protein levels of GST-π. *p<0.05, **p<0.01 vs. vehicle; #p<0.05, ##p<0.01 vs. sham. n=6 in sham, n=8 in vehicle and minocycline groups.
We next examined the protein level of GST-π, a cellular marker of mature oligodendrocytes, in WM from sham-operated, vehicle-treated, and minocycline-treated rats (Fig. 10B). Western blot analysis showed significantly increased levels of GST-π in vehicle-treated LEC, LIC, and RIC compared with sham-operated rats. In minocycline-treated rats, significantly increased levels of GST-π in LCC, LEC, LIC, REC, and RIC were detected compared with sham-operated rats. There were significantly enhanced GST-π levels in LCC and LEC with minocycline treatment compared to those with vehicle treatment. Furthermore, significantly increased GST-π levels were detected in both vehicle- and minocycline-treated ICs at 9 weeks after UCAO/JPD, compared with sham-operated animals.
Discussion
Our results show that hypoxic hypoperfusion-induced WM damage is associated with increased BBB permeability on MRI and serum albumin IgG leakage at 4 weeks after starting UCAO/JPD. Although sham-operated SHR/SPs showed abnormalities in TJPs in vessels by 16 weeks of age compared with Wistar–Kyoto rats, and a minimal increase of BBB permeability, major changes were found in rats subjected to 4-weeks of UCAO/JPD, accompanied by significantly decreased TJPs in the vascular endothelial cells in injured WM, which indicated that the increased BBB permeability in lesion WM was associated with TJP disruption in injured microvessels. The increased expression of Ki67, NG2, MMP-3, and VEGF-A in proliferating neurovascular cells in the injured WM provides the first evidence that angiogenesis and vascular remodeling occurs secondary to chronic hypoxia in a rat SIVD model. We propose that hypoxia-induced angiogenesis contributes to formation of new vessels in damaged WM, and that minocycline reduced BBB disruption, promoting reformation of TJ between endothelial cells in the angiogenic vessels involved in WM repair.
Unilateral right carotid occlusion produced asymmetric damage with smaller injury to the right hemisphere as observed in our previous studies (Jalal et al., 2012; Jalal et al., 2015), presumably due to the reduced pressure on the right side after surgery, which was confirmed by the multimodal MRI studies, using anatomical T2, ADC, FA and ASL. Although right carotid occlusion with JPD induced white matter lesions in both hemispheres, our data demonstrated significantly larger WM lesion area and volume in left hemisphere than the right with a unilateral cortical infarct seen with greater frequency in the left hemisphere. In both hemispheres, the maximum lesion volume appeared in CCs, while ICs showed the least damage. This regional specific asymmetric WM damage induced by UCAO/JPD presents multiple stages in the injury process in SHR/SP brain that appear to be occurring simultaneously, suggesting that the SHR/SP/UCAO/JPD is an optimal animal model to investigate the pathobiological mechanisms, including angiogenesis and TJ reformation in BBB, for SIVD, a progressive WM damage disease in human patients. However, it is still not clear what induced greater damage in the non-occluded hemisphere than that seen in the occluded hemisphere. We speculate that higher blood pressure was induced in the left hemisphere due to occlusion of the right common carotid artery which resulted in greater damage in the left hemisphere. Further study will be needed to clarify the mechanism of apparent protection on the occluded side.
Using dynamic contrast-enhanced MRI (DCEMRI) and gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA), we measured the BBB permeability in WM for the same rats that were subjected to MRI analysis for T2, ADC, FA, and ASL (Yang et al., 2015). We observed a significantly increased BBB transfer rate (Ki) in both left and right hemispheres of rats at 4 weeks after UCAO/JPD compared to sham rats. The Ki maps and quantification of BBB transfer rate in WM showed significantly higher BBB leakage in the left WM than the right one, with highest BBB permeability in left CC areas. These observations by MRI were supported by histologic analysis of serum IgG extravasation in lesion WM and GM. The present findings are also compatible with earlier reports that increased BBB permeability in WM of SHRSPs started at week 2 and continued in week 3 after UCAO/JPD, which followed a significant increase of HIF-1α at one week (Jalal et al., 2015).
In this study, we examined the expression of TJPs, including ZO-1, occludin, and claudin-5, in the WM. Unexpectantly, there was an abnormal expression of the three TJPs in WM microvessels in sham SHR/SP at age of 16 weeks compared with either WKY or SHR (Sood et al., 2009; Yang and Rosenberg, 2011). An earlier MRI-histological study shows no vasculopathy in deep penetrating arterioles, and no WM hyperintensities in SHR/SP aged up to 10 months (Brittain et al., 2013). The sham SHR/SPs presented lower expression of TJPs with a lack of linear appearance in vessels that is typical in normal BBB (Tsukita and Furuse, 1998; Tsukita and Furuse, 1999; Tsukita and Furuse, 2000; Yang et al., 2007), which was accompanied with a minimal serum IgG leakage from the microvessels in WM and GM, indicating abnormal TJ function with increased BBB permeability in the SHR/SPs appearing by 16 weeks of age. Our finding is in agreement with a previous study that showed a disturbed polarity of the TJs in SHR/SP endothelial cells at 13 weeks of age (Lippoldt et al., 2000), In addition, studies on vascular pathology in SHR/SP suggest the early BBB breakdown following erythrocyte accumulation as the starting point of cerebral small vessel disease in SHR/SPs (Mencl et al., 2013; Schreiber et al., 2013; Schreiber et al., 2012). Our results indicate an early abnormality in TJPs in WM, suggesting for the first time a potential new molecular mechanism for the WM injury in the SHR/SPs.
TJPs change expression, subcellular location, post-translational modification, and protein-protein interactions under both physiological and pathological conditions (Bolton et al., 1998; Steed et al., 2010; Yang et al., 2007). ZO-1, occludin, and claudin-5 are the main structural barrier proteins and are considered sensitive indicators of normal and disturbed functional state of the BBB (McCaffrey et al., 2009; Willis et al., 2004). By 4 weeks after UCAO/JPD, using IHC, we observed significant degradations of ZO-1, occludin, and claudin-5 in affected WM, with a complete absence of claudin-5 and occludin in microvessels of lesioned CC areas. Due to an increase in reactive astrocytes and other glial cells in the injured WM, which also expressed ZO-1 and occludin, we failed to detect significant decreases in protein levels for ZO-1 and occludin in injured CCs. However, significantly decreased claudin-5 protein in affected CCs shown by Western blot confirmed the loss of TJPs in WM. In the brain, the only claudins to be found are claudin-1, -5 and -11, while claudin-11/OSP was identified in oligodendrocytes (Morita et al., 1999a), Claudin-5 is an EC specific component of TJ strands (Morita et al., 1999b), so that claudins-1 and -5, besides occludin in tight junctions, appear to be the most important structural components of BBB TJs (Liebner et al., 2000a). Furthermore, both IHC and Western blot demonstrated that the degree of TJP degradations in WM was similarly region-dependent as was seen for WM damage and BBB leakage in the MRI analysis. We speculate that UCAO/JPD induced chronic hypoxia in WM, accelerating the degradation of TJPs in the BBB endothelial cells in SHR/SPs. The vessel injury reduced tissue oxygen content, resulting in the progressive WM damage. Our results suggest that chronic hypoxia degrades TJPs. One possibility is that HIF-1α which has been identified as an important mediator in the signaling pathways of BBB disruption after hypoxic insult (Ogunshola and Al-Ahmad, 2012), could mediate TJP alteration, possibly by inducing the expression and activation of VEGF, a strong inducer of vascular permeability. This could induce the redistribution of ZO-1 and occludin and also actin cytoskeleton (Yan et al., 2012; Yeh et al., 2007).
Angiogenesis, the growth of new blood vessels, is the physiological process through which new blood vessels form from pre-existing vessels, which is an important process that occurs both during health and disease (Lee et al., 2009). HIF-1α regulates the transcription of various angiogenic factors, such as vascular VEGF, platelet derived growth factor (PDGF), angiopoietin, and transforming growth factor-β1 (TGF-β1), and plays an important role in vascular development and remodeling (Ahn et al., 2008; Lee et al., 2009). There is a question whether chronic hypoxia could induce angiogenesis in lesion WM under conditions similar to those occurring in patients with SIVD. Increased vascular densities and angiogenic vessels can be seen in animal models of vascular disease from chronic hypoxia, indicating that angiogenesis might play a role in the pathophysiology of SIVD (Boero et al., 1999; Brown and Thore, 2011; Chakraborty et al., 2017; Yang et al., 2017). In the present study, we found that some vessels stained with endothelial cell markers remained in the lesion areas of WM at 4 weeks after UCAO/JPD, which could provide the cellular basis to trigger angiogenesis (Hermann and Zechariah, 2009; Shen et al., 2004). Some of these vessels expressed Ki67 in nuclei of the endothelial cells, while pericytes that closely surrounded these vessels were NG2 positive. NG2 is expressed by microvascular pericytes in angiogenic vessels and plays a key role in promoting endothelial cell migration and morphogenesis during the early stages of neo-vascularization (Fukushi et al., 2004; Jungner et al., 2013). The expression of Ki67 in endothelial nuclei and NG2 in the vascular associated pericytes suggests that they are newly formed and contribute to new vessel proliferation in the lesion WM areas induced by UCAO/JPD (Ozerdem and Stallcup, 2004; Virgintino et al., 2007; Yang et al., 2013).
We observed significantly increased VEGF-A levels in lesion WM at 4 weeks after UCAO/JPD, which was expressed by endothelial cells, vascular associated pericytes, and astrocytes. During hypoxia, HIF-1α plays an important role in expression of VEGF which is a critical regulator of angiogenesis and barriergenesis (Chakraborty et al., 2017; Krock et al., 2011; Lee et al., 2009). The VEGF-A in endothelial cells, and vascular-associated astrocytes and pericytes may provide a microenvironment to orchestrate angiogenesis that favors this process during neurovascular remodeling (Yang et al., 2013). Pericytes and astrocytes, which expressed VEGF, expressed the MMP -3 in the lesion WM at 4 weeks after UCAO/JPD. Interestingly, the expression of MMP-3 closely surrounding vessels in lesion WM was also observed in human brains with SIVD. In angiogenesis, loss of vascular integrity and degradation of the extracellular matrix are crucial initiating steps. MMPs degrade the extracellular matrix preparing the stage for growth factors and guidance molecules, and facilitate vessel migration in injured tissues. This endothelial migration is aided by growth factors, including VEGF (Dore-Duffy and LaManna, 2007).
By taking advantage of DMSO to prolong survival of SHR/SPs subjected to UCAO/JPD (Jalal et al., 2015), we extended our investigation on angiogenesis and tested the impact of minocycline on the UCAO/JPD induced vascular remodeling and oligodendrocyte repair in lesion WM. At 9 weeks, UCAO/JPD induced significantly greater WM damage and BBB leakage in most of regions examined compared with the ones at 4 weeks. The IgG extravasation correlated significantly with increased infarct volumes. Minocycline treatment almost entirely blocked the BBB leakage, significantly attenuating the WM damage. Notably, more healthy vessels were seen in the WM of animals treated with minocycline as well as higher signals of angiogenic factors, including Ki67, NG2, and MMP-3, in endothelial cells, pericytes, or astrocytes, respectively. Treatment with minocycline enhanced the level of VEGF-A in all regions of left hemisphere WM, while only LIC showed significant increases of VEGF-A protein, compared to sham SHR/SPs. These findings suggest that minocycline protected the WM from damage by blocking the BBB disruption, promoting angiogenesis. VEGF-induced angiogenesis leads to immature and dysfunctional vessels, impairing the BBB, causing more harm than benefit (Chakraborty et al., 2017), while chronic vascular remodeling in SHR/SPs may contribute to reduced cerebral perfusion and development of stroke. We found that minocycline treatment promoted TJP expression in lesion WM; claudin-5 was significantly increased in the LCC, LEC, RCC, and REC of minocycline-treated rats compared both with the vehicle-treated and sham groups. Considering that claudin-5 is only expressed by endothelial cells in brain (Morita et al., 1999b), these results suggest that new vessels are forming in these areas of damaged WM with increased expression of TJPs, in particular claudin-5, in response to UCAO/JPD at 9 weeks, and minocycline enhanced the TJ strand formation in angiogenic vessels.
We finally investigated the impact of the minocycline on expression of NG2 in oligodendrocytes and the protein level of GST-π in lesion WM at 9 weeks in order to analyze the repair of WM. In the adult brain, oligodendrocyte progenitor cells (OPCs) have been identified as NG2-containing cells (Nishiyama et al., 1997) (Tamura et al., 2007). OPCs lose NG2 immunoreactivity to differentiate into immature oligodendrocytes, resulting in the expression of GST-π exclusively in the cytoplasm (Li et al., 2009). Our data demonstrate a morphological change of oligodendrocytes in CC of vehicle-treated, compared with the ones in minocycline-treated CC that are present in intact linear rows of nuclei formed by normal oligodendrocyte-lineage cells (Takeda et al., 2016). There was a reduction of oligodendrocytes in vehicle-treated CC with breakdown of myelin-like structure. Although minocycline treatment reduced the total expression of NG2 in lesion WM, the increased NG2 in vehicle-CC partly co-localized with oligodendrocytes, but was mostly expressed in oligodendrocyte-lineage cells after minocycline treatment, and in the cells surrounding vessels. GST-π has been used as an marker of mature oligodendrocyte-lineage cells in the adult rodent brain (Tansey and Cammer, 1991). Localization of GST-π indicates maturity of oligodendrocyte lineage cells with immature cells showing nuclear expression and cytoplasmic expression seen in mature cells. (Tamura et al., 2007) (Kitada et al., 2016; Takeda et al., 2016). We have reported an apoptotic death of mature oligodendrocytes and an increased population of immature oligodendrocytes in lesion WM at 4 weeks after UCAO/JPD (Jalal et al., 2012). By 9 weeks, we observed significantly increased levels of GST-π in vehicle-treated LEC, LIC, and RIC compared with sham-operated rats. Although we do not know the intracellular location of the increased GST-π in oligodendrocytes, our data demonstrated that treatment with minocycline significantly enhanced GST-π levels in LCC and LEC compared with vehicle treatment. In addition, significantly increased GST-π levels were detected in both vehicle- and minocycline-treated ICs at 9 weeks after UCAO/JPD, compared with sham-operated ones. Taken together, our observations suggest that minocycline promoted oligodendrocyte survival and differentiation, leading to WM repair from damage induced by UCAO/JPD. Further studies have to clarify the identity of immature or mature oligodendrocytes that have been protected by minocycline.
It is important to point out that all of the angiogenic factors, especially VEGF-A and MMP3, detected in vascular cells had a greater expression level in the lesion WM regions where the MRI showed slighter injuries, such as LEC, LIC, RCC, REC, and RIC. Patterns of the angiogenic factors correspond with the protein levels of ZO-1, occludin, and claudin-5 in these WM regions at 4 weeks and at 9 weeks the lesion size was greatly expanded. Correlation of the degree of angiogenesis with the extent of WM lesion supports the role of chronic hypoxia in the induction of angiogenesis, which may be facilitated by long-term treatment with minocycline to prevent WM injury and to promote repair. An important caveat is that further studies of molecular signaling pathways will be needed to delineate the underlying mechanisms associated with the angiogenesis and WM repair, and with the protective role of long-term anti-inflammation therapeutic strategy, such as use of minocycline.
In conclusion, we have demonstrated that sham SHR/SPs have abnormal TJP expression at 16 weeks of age, and that chronic hypoxia induced by UCAO/JPD significantly increased BBB permeability by disrupting TJPs, resulting in WM damage and triggering angiogenesis. Our results support the role of hypoperfusion, BBB disruption, oxidative stress, and inflammation in SIVD, and establish this animal model in the testing of therapies to reduce pathogenic mechanisms (Iadecola, 2013). Minocycline treatment reduced WM damage by blocking BBB disruption, and facilitated repair through TJ formation and angiogenesis. While much work has been done on white matter injury due to acute hypoxia, there is very little on the effects of chronic hypoxic hypoperfusion. Our results show the impact of loss of tight junction proteins and the changes that occur in blood vessels with recovery. It is important to understand the long-term changes in order to decide when an intervention would be helpful, and when it could interfere with recovery. We propose that this chronic inflammatory model can clarify the molecular and cellular mechanisms by which chronic hypoxia induces WM damage, and can be used to develop potential treatments for SIVD.
Supplementary Material
Highlights.
Chronic hypoperfusion-induced white matter (WM) damage reflects the extent of injury corresponding to the BBB permeability increases in a rat model of vascular cognitive impairment.
Chronic hypoperfusion opens the BBB by disrupting endothelial tight junctions in injured WM.
Chronic hypoperfusion initiates angiogenesis in injured WM, contributing to the repair process.
Long-term treatment with minocycline prevents BBB damage, enhanced expression of tight junction proteins, and angiogenesis, ultimately preserving oligodendrocytes.
These results in this chronic inflammatory model highlight the potential treatments for this common form of dementia.
Acknowledgments
The study was supported by grants from the NIH (RO1NS045847) to GAR.
Footnotes
Author Contributions
YY conceived and designed the experiments and wrote the manuscript. YY, SKO, JFT, VMS, MC performed biochemical and histologic studies and data analysis. YY performed the UCAO surgery. YY, SKO, and JFT performed MRI scans and data analysis. JFT and SKO helped with the manuscript writing. LR performed histological experiments. FJ participated in animal model preparation. GAR obtained the funding, provided scientific advice in the execution of the study, and participated in the writing of the manuscript.
Conflict of interest
The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adair JC, et al. Measurement of gelatinase B (MMP-9) in the cerebrospinal fluid of patients with vascular dementia and Alzheimer disease. Stroke. 2004;35:e159–62. doi: 10.1161/01.STR.0000127420.10990.76. [DOI] [PubMed] [Google Scholar]
- Ahn SM, et al. Olig2-induced neural stem cell differentiation involves downregulation of Wnt signaling and induction of Dickkopf-1 expression. PLoS One. 2008;3:e3917. doi: 10.1371/journal.pone.0003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009;117:481–96. doi: 10.1007/s00401-009-0483-6. [DOI] [PubMed] [Google Scholar]
- Boero JA, et al. Increased brain capillaries in chronic hypoxia. J Appl Physiol 1985. 1999;86:1211–9. doi: 10.1152/jappl.1999.86.4.1211. [DOI] [PubMed] [Google Scholar]
- Bolton SJ, et al. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. 1998;86:1245–57. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- Brittain JF, et al. An MRI-histological study of white matter in stroke-free SHRSP. J Cereb Blood Flow Metab. 2013;33:760–3. doi: 10.1038/jcbfm.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011;37:56–74. doi: 10.1111/j.1365-2990.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair J, Edmonds E, Prestopnik J, Wills J, Rosenberg G. Matrix Metalloproteinases are Associated with Increased Blood-Brain Barrier Opening in Vascular Cognitive Impairment. Stroke. 2011 doi: 10.1161/STROKEAHA.110.600825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, et al. The blood brain barrier in Alzheimer’s disease. Vascul Pharmacol. 2017;89:12–18. doi: 10.1016/j.vph.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, LaManna JC. Physiologic angiodynamics in the brain. Antioxid Redox Signal. 2007;9:1363–71. doi: 10.1089/ars.2007.1713. [DOI] [PubMed] [Google Scholar]
- Ewing JR, et al. Patlak plots of Gd-DTPA MRI data yield blood-brain transfer constants concordant with those of 14C-sucrose in areas of blood-brain opening. Magn Reson Med. 2003;50:283–92. doi: 10.1002/mrm.10524. [DOI] [PubMed] [Google Scholar]
- Fagan SC, et al. Minocycline Development for Acute Ischemic Stroke. Transl Stroke Res. 2011;2:202–208. doi: 10.1007/s12975-011-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810–7. doi: 10.1161/HYPERTENSIONAHA.113.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushi J, et al. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell. 2004;15:3580–90. doi: 10.1091/mbc.E04-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, et al. Vascular Contributions to Cognitive Impairment and Dementia: A Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini U, et al. New insights into brain damage in stroke-prone rats: a nuclear magnetic imaging study. Stroke. 2002;33:825–30. doi: 10.1161/hs0302.104111. [DOI] [PubMed] [Google Scholar]
- Hachinski V, et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.STR.0000237236.88823.47. [DOI] [PubMed] [Google Scholar]
- Hermann DM, Zechariah A. Implications of vascular endothelial growth factor for postischemic neurovascular remodeling. J Cereb Blood Flow Metab. 2009;29:1620–43. doi: 10.1038/jcbfm.2009.100. [DOI] [PubMed] [Google Scholar]
- Huang L, et al. Decreased serum levels of the angiogenic factors VEGF and TGF-beta1 in Alzheimer’s disease and amnestic mild cognitive impairment. Neurosci Lett. 2013;550:60–3. doi: 10.1016/j.neulet.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–66. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal FY, et al. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. 2012;43:1115–22. doi: 10.1161/STROKEAHA.111.643080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal FY, et al. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungner M, et al. Rosuvastatin in experimental brain trauma: improved capillary patency but no effect on edema or cerebral blood flow. Microvasc Res. 2013;88:48–55. doi: 10.1016/j.mvr.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Kitada M, et al. Regulation of DM-20 mRNA expression and intracellular translocation of glutathione-S-transferase pi isoform during oligodendrocyte differentiation in the adult rat spinal cord. Histochem Cell Biol. 2016;146:45–57. doi: 10.1007/s00418-016-1421-z. [DOI] [PubMed] [Google Scholar]
- Krock BL, et al. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–33. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger M, et al. Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 2015;35:292–303. doi: 10.1038/jcbfm.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, et al. Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009;276:4622–35. doi: 10.1111/j.1742-4658.2009.07174.x. [DOI] [PubMed] [Google Scholar]
- Li L, et al. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia. 2010;58:1610–9. doi: 10.1002/glia.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, et al. Leydig cells express the myelin proteolipid protein gene and incorporate a new alternatively spliced exon. Gene. 2009;436:30–6. doi: 10.1016/j.gene.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, et al. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol (Berl) 2000a;100:323–31. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- Liebner S, et al. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000b;79:707–17. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- Lippoldt A, et al. Structural alterations of tight junctions are associated with loss of polarity in stroke-prone spontaneously hypertensive rat blood-brain barrier endothelial cells. Brain Res. 2000;885:251–61. doi: 10.1016/s0006-8993(00)02954-1. [DOI] [PubMed] [Google Scholar]
- McCaffrey G, et al. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem. 2009;110:58–71. doi: 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencl S, et al. Early microvascular dysfunction in cerebral small vessel disease is not detectable on 3.0 Tesla magnetic resonance imaging: a longitudinal study in spontaneously hypertensive stroke-prone rats. Exp Transl Stroke Med. 2013;5:8. doi: 10.1186/2040-7378-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, et al. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol. 1999a;145:579–88. doi: 10.1083/jcb.145.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, et al. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999b;147:185–94. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwelt EA, et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–82. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, et al. Normal and reactive NG2+ glial cells are distinct from resting and activated microglia. J Neurosci Res. 1997;48:299–312. doi: 10.1002/(sici)1097-4547(19970515)48:4<299::aid-jnr2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- O’Brien JT, et al. Vascular cognitive impairment. Lancet Neurol. 2003;2:89–98. doi: 10.1016/s1474-4422(03)00305-3. [DOI] [PubMed] [Google Scholar]
- Ogunshola OO, Al-Ahmad A. HIF-1 at the blood-brain barrier: a mediator of permeability? High Alt Med Biol. 2012;13:153–61. doi: 10.1089/ham.2012.1052. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Stallcup WB. Pathological angiogenesis is reduced by targeting pericytes via the NG2 proteoglycan. Angiogenesis. 2004;7:269–76. doi: 10.1007/s10456-004-4182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- Patlak CS, et al. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, et al. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–8. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- Schreiber S, et al. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? - New insights from a rat model. Exp Transl Stroke Med. 2013;5:4. doi: 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, et al. The pathologic cascade of cerebrovascular lesions in SHRSP: is erythrocyte accumulation an early phase? J Cereb Blood Flow Metab. 2012;32:278–90. doi: 10.1038/jcbfm.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Sironi L, et al. Analysis of pathological events at the onset of brain damage in stroke-prone rats: a proteomics and magnetic resonance imaging approach. J Neurosci Res. 2004;78:115–22. doi: 10.1002/jnr.20219. [DOI] [PubMed] [Google Scholar]
- Sood R, et al. Increased apparent diffusion coefficients on MRI linked with matrix metalloproteinases and edema in white matter after bilateral carotid artery occlusion in rats. J Cereb Blood Flow Metab. 2009;29:308–16. doi: 10.1038/jcbfm.2008.121. [DOI] [PubMed] [Google Scholar]
- Steed E, et al. Dynamics and functions of tight junctions. Trends Cell Biol. 2010;20:142–9. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Taheri S, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011;42:2158–63. doi: 10.1161/STROKEAHA.110.611731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri S, Sood R. Partial volume effect compensation for improved reliability of quantitative blood-brain barrier permeability. Magn Reson Imaging. 2007;25:613–25. doi: 10.1016/j.mri.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Takeda K, et al. The expression of PLP/DM-20 mRNA is restricted to the oligodendrocyte-lineage cells in the adult rat spinal cord. Histochem Cell Biol. 2016;145:147–61. doi: 10.1007/s00418-015-1384-5. [DOI] [PubMed] [Google Scholar]
- Tamura Y, et al. Intracellular translocation of glutathione S-transferase pi during oligodendrocyte differentiation in adult rat cerebral cortex in vivo. Neuroscience. 2007;148:535–40. doi: 10.1016/j.neuroscience.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Tansey FA, Cammer W. A pi form of glutathione-S-transferase is a myelin- and oligodendrocyte-associated enzyme in mouse brain. J Neurochem. 1991;57:95–102. doi: 10.1111/j.1471-4159.1991.tb02104.x. [DOI] [PubMed] [Google Scholar]
- Toth P, et al. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312:H1–H20. doi: 10.1152/ajpheart.00581.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells. 1998;3:569–73. doi: 10.1046/j.1365-2443.1998.00212.x. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–73. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci. 2000;915:129–35. doi: 10.1111/j.1749-6632.2000.tb05235.x. [DOI] [PubMed] [Google Scholar]
- Virgintino D, et al. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- Weaver J, et al. Tissue oxygen is reduced in white matter of spontaneously hypertensive-stroke prone rats: a longitudinal study with electron paramagnetic resonance. J Cereb Blood Flow Metab. 2014;34:890–6. doi: 10.1038/jcbfm.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, et al. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia. 2004;48:1–13. doi: 10.1002/glia.20049. [DOI] [PubMed] [Google Scholar]
- Yan J, et al. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci. 2012;69:115–28. doi: 10.1007/s00018-011-0731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yang Y, et al. Tissue inhibitor of metalloproteinases-3 mediates the death of immature oligodendrocytes via TNF-alpha/TACE in focal cerebral ischemia in mice. J Neuroinflammation. 2011;8:108. doi: 10.1186/1742-2094-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Hypoxia Inducible Factor 1alpha Promotes Endogenous Adaptive Response in Rat Model of Chronic Cerebral Hypoperfusion. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011;762:333–45. doi: 10.1007/978-1-61779-185-7_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12:26. doi: 10.1186/s12974-015-0245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–14. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WL, et al. Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1alpha accumulation and VEGF expression. Mol Pharmacol. 2007;72:440–9. doi: 10.1124/mol.107.036418. [DOI] [PubMed] [Google Scholar]
- Yenari MA, et al. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–93. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.