

Abstract
The chemokine MIP3α (CCL20) binds to CCR6 on immature dendritic cells. Vaccines fusing MIP3α to gp100 have been shown to be effective in therapeutically reducing melanoma tumor burden and prolonging survival in a mouse model. Other studies have provided evidence that IL-10 neutralizing antibodies (αIL-10) enhance immunological melanoma therapies by modulating the tolerogenic tumor microenvironment. In the current study, we have utilized the B16F10 syngeneic mouse melanoma model to demonstrate for the first time that a therapy neutralizing IL-10 enhances the anti-tumor efficacy of a MIP3α-gp100 DNA vaccine, leading to significantly smaller tumors, slower growing tumors, and overall increases in mouse survival. The additive effects of αIL-10 were not shown to be correlated to vaccine-specific tumor infiltrating lymphocytes (TILs), total TILs, or regulatory T-cells. However, we discovered an upregulation of IFNα-4 transcripts in tumors and a correlation of increased plasmacytoid dendritic cell numbers with reduced tumor burden in αIL-10 treated mice. Interferon alpha receptor knockout (IFNαR1−/−) mice received no benefit from αIL-10 treatment, demonstrating that the additional therapeutic value of αIL-10 is primarily mediated by type I interferons. Efficient targeting of antigen to immature dendritic cells with a chemokine fusion vaccine provides an effective anti-cancer therapeutic. Combining this approach with an IL-10 neutralizing antibody therapy enhances the anti-tumor efficacy of the therapy in a manner dependent upon the activity of type I interferons. This combination of a vaccine and immunomodulatory agent provides direction for future optimization of a novel cancer vaccine therapy.
Keywords: Chemokine CCL20, Therapeutic cancer DNA vaccine, Immunotherapy, Interleukin 10, Interferon-alpha
BACKGROUND
Our previous work has characterized the efficacy and immunogenicity of a novel therapeutic melanoma DNA vaccine platform in which amino acids 25–235 of the human version of melanoma-associated antigen gp100 are fused to a dendritic-cell attracting chemokine MIP3α1. The human and mouse sequences of gp100 are well conserved (76% similarity), and prophylactic xenograft DNA immunizations with the human sequence have proven to be more efficacious in the B16F10 mouse model2. In our therapeutic model, MIP3α-gp100 enhanced immunogenicity over antigen-only vaccination and provided significant anti-tumor efficacy, whereas antigen-only vaccination did not1. Despite enhanced efficacy, the vaccine was unable to induce tumor regression. It was hypothesized that the ultimate inability of the vaccine to induce tumor regression and clearance was due to tolerogenic factors in the tumor microenvironment3. The microenvironment is complex with many factors influencing tolerance, such as regulatory T-cells4, immunosuppressive cytokines5, T-cell checkpoint inhibitors4, directed evolution of tumor cells6, and others5.
One generally immunosuppressive cytokine shown to play an essential role in melanoma immune tolerance is interleukin-10 (IL-10) 7. IL-10 suppresses mechanisms of immune surveillance in melanoma8 and promotes melanoma growth by inhibition of macrophage function and induction of tumor proliferation, both directly and through enhanced angiogenesis7. IL-10 down-regulates MHC/HLA type I and antigen presentation machinery in melanomas9,10. Some B16 cell lines produce IL-1011,12, and, in human melanomas, tumoral production of IL-10 is a poor prognostic factor13. Neutralizing IL-10 enhanced IFN-γ production14 and the efficacy of a prophylactic dendritic cell vaccine in B16 melanoma15. Knocking out IL-10 led to mice being more resistant to B16 tumors14 and more sensitive to treatment with a B16-specific Fab-targeted superantigen16. Finally, tellurium-based compounds sensitize B16 tumors to chemotherapies by inhibiting IL-10 autocrine signaling11,17.
IL-10 has thus become a target for anti-cancer therapies targeting many different cancer types. Blocking IL-10 enhanced the delayed-type hypersensitivity response of BCG anti-bladder cancer vaccination18–20. An aptamer specifically neutralizing IL-10 provided significant protection against the CT26 colon carcinoma model 21. Dendritic cells pulsed with tumor lysate antigens that were either IL-10-deficient or wild type but combined with a neutralizing IL-10 antibody treatment in both cases established sufficient immune memory to protect against subsequent tumor challenge in a mouse prostate cancer model, whereas wild type, pulsed dendritic cells did not 22. IL-10 is a “late cytokine,” and it is hypothesized that its primary physiological role is to prevent excessive immune reactivity by contributing to peripheral tolerance in settings of excessive inflammation resulting from antigen persistence23. The cited studies point to this IL-10 function being pivotal in maintenance of tolerance in cancer systems, including melanoma. The current study examines the role of IL-10 in our therapeutic vaccine model system.
METHODS
Animals and Tumor Model
5–6 week old female C57BL/6 (H-2b) mice were purchased from Charles River Laboratories (Wilmington, MA) and maintained in a pathogen-free micro-isolation facility in accordance with the National Institutes of Health guidelines for the humane use of laboratory animals. Interferon-alpha receptor 1 knockout mice (IFNαr1−/−) carry the B6.129S2-IFNαr1tm1Agt/Mmjax genotype maintained by Jackson Laboratories (Bar Harbor, ME). All experimental procedures involving mice were approved by the IACUC of the Johns Hopkins University (Protocol numbers MO13H219 and MO16H147). B16F10 mouse melanoma cells were a generous gift from Dr. Arya Biragyn (NIH, Baltimore, MD). 6–8 week old wild type and 6–12 week old knockout mice were challenged in the left flank subcutaneously with a lethal dose (5×104 cells) of B16F10 melanoma. Tumor size was recorded as square mm, representing tumor length × width (opposing axes) measured by calipers every 1–3 days. Mice were kept in the study until one of the following occurred: mouse death, tumor diameter eclipsing 20mm, or extensive tumor necrosis resulting in excessive bleeding.
Plasmids, Vaccination, and αIL-10 Therapy
Vaccine consisted of purified plasmid DNA in endotoxin-free PBS. The plasmid encoded the MIP3α-hgp100 fusion sequence, where the antigen includes amino acids 25–235 of human gp100, as described1,24. Vaccination plasmid was extracted from E. coli using Qiagen® (Germantown, MD) EndoFree® Plasmid Maxi, Mega, and Giga Kits. Vaccine DNA purity, quality, and quantity were verified by gel electrophoresis, restriction enzyme analysis, Nanodrop® spectrophotometry, and insert sequencing (JHMI Synthesis and Sequencing Facility, Baltimore, MD). Mock vaccinations were comprised of endotoxin-free PBS only. DNA injections were administered into the hind leg tibialis muscle. Immediately following injection, the muscle was pulsed using an ECM 830 Electro Square Porator™ with 2-Needle Array™ Electrode (BTX Harvard Apparatus®; Holliston, MA) under the following parameters: 106V; 20ms pulse length; 200ms pulse interval; 8 total pulses. Vaccinations of 50ug/dose were delivered at days noted in figure legends. As described by others, vaccination for the therapeutic model began on day three1,25,26. Antibody against mouse IL-10 (Clone JES5.2A5; BioXcell, West Lebanon, NH) was administered intratumorally at 150μg per dose. A typical time course of therapy begins on day 5 post tumor induction and is given every three days for a maximum of 6 doses.
Extraction of Splenocytes and TILs
Spleen and tumor cell suspensions were prepared by grinding sterile excised tissue between the frosted ends of microscope slides and then passing the tissue through a sterile 60 μM mesh. Splenocytes were processed by lysing red blood cells and washing with sterile PBS. For plasmacytoid dendritic cell analysis, tumor lysate was washed with sterile PBS and processed by lysing red blood cells prior to staining. For tumor lymphocyte analysis, tumor lysate was washed with sterile PBS, and the mononuclear cell fraction (including TILs) was enriched by Lympholyte®-M Cell Separation Media (Cedarlane®, Burlington, NC) according to the manufacturer’s protocol. Prior to use all cells were counted by a Z1™ Coulter Counter® (Beckman Coulter, Inc.; Brea, CA) and/or a hemocytometer with Gibco™ Trypan Blue solution 0.4% (Life Technologies, Carlsbad, CA) with cell viability routinely >90%.
Intracellular Cytokine Staining and Flow Cytometry
The following protocol was utilized for intracellular cytokine staining. Enriched splenocytes or TILs were seeded onto Falcon® Multiwell 24-well tissue culture treated plates (Corning, Inc.; Corning, NY) at 1×106 cells per well (or all cells if total is less) and stimulated for 3–4 hours at 37°C with the known immunodominant gp10025–33 (KVPRNQDWL) peptide or control HA (YPYDVPDYA) peptide (JHU School of Medicine Synthesis & Sequencing Facility; Baltimore, MD) combined with Protein Transport Inhibitor Cocktail and costimulatory anti-CD28 and anti-CD49d agonizing antibodies (eBioscience, Inc. San Diego, Ca). Cells were collected, washed, fixed, permeabilized, and stained using standard laboratory protocols for intracellular staining. Fixation and permeabilization buffers from Mouse Regulatory T Cell Staining Kit #2 (eBioscience, Inc. San Diego, CA) were used. For plasmacytoid DC analysis, single cell suspensions were stained and fixed using standard laboratory protocols for flow cytometry. Stains utilized were the following anti-mouse mAbs: PercPCy5.5 conjugated anti-CD3, PE-Cy7-mPDCA1, PE-B220, APC-IFNγ, FITC-CD8, PE-CD4, FITC-PD-1, APC-Foxp3, PE-Granzyme, AF700-Foxp3 (eBioscience, Inc. San Diego, CA), Pacific Blue-CD8, PercPCy5.5-CD25, PE-Cy7-IFN-γ, Pacific Blue-CD44, PercPCy5.5-CD8, APC-CD62L (BioLegend, San Diego, CA), Pacific Orange-CD4, AmCyan-Live/Dead Aqua (Life Technologies, Carlsbad, CA), APC-CD11c (BD Biosciences, San Jose, CA), and Live/Dead Near-IR (Invitrogen by Thermo Fisher Scientific, Carlsbad, CA). FACSCalibur™ (BD Biosciences, San Jose, CA), LSRII™ (BD Biosciences, San Jose, CA), and the Attune™ NxT (Thermo Fisher Scientific, Waltham, MA) flow cytometers were utilized. Flow data were analyzed by FlowJo Software (FlowJo, LLC Ashland, OR).
RNA Extraction and RT-PCR
Samples were taken one day after the dose of αIL-10 where more than 80% of mice have harvestable tumors (size > 30mm2). Mice were sacrificed and portions of tumor weighing less than 150mg were harvested. Tumor minced as finely as possible and added to Trizol® (Ambion® by Life Technologies, Carlsbad, Ca) at 1ml Trizol® per 50mg tumor. RNA was extracted utilizing the manufacturer’s protocol and including a 75% ethanol wash step. The pellet was air dried and resuspended in nuclease-free water. The cDNA Reverse Transcription reaction was performed with 1μg extracted RNA and the High Capacity cDNA Reverse Transcription Kit with random primers (Applied Biosystems™ by Thermo Fisher, Halethorpe, MD) utilizing the manufacturer’s protocol. Real-Time quantitative Reverse Transcription-PCR (qRT-PCR) performed utilizing TaqMan® Gene Expression Master Mix and TaqMan® Gene Expression Assays (Applied Biosystems™ by Thermo Fisher, Halethorpe, MD) with probes specific for GAPDH (expression control), IFNα4, IFNα2/11, H2-ab1, and H2-k1. qRT-PCR ran and analyzed with StepOnePlus™ machine and software (Applied Biosystems™ by Thermo Fisher, Halethorpe, MD).
Statistics and Data
Tumor size, immunologic, RT-PCR, and flow cytometric analyses were statistically tested by one-way anova with Tukey’s multiple comparisons test if multiple groups or by Student’s t-test if two groups. Mouse survival studies were statistically tested by the log-rank test. Tumor time course regressions were analyzed within the window of 8 or 10–17 days post challenge, where treatment groups had tumors growing linearly, and were tested by mixed effect linear regression models. Correlation of plasmacytoid dendritic cells and tumor size was tested by linear regression. STATA v11.2 (StataCorp, College Station, TX) and Prism 7 (GraphPad Software, Inc. San Diego, CA) were utilized for statistical analyses and figure creation. A significance level of α≤0.05 was set for all experiments.
RESULTS
Effect of αIL-10 on Therapeutic Vaccination Model
Intratumoral administration of neutralizing αIL-10 antibodies was given alone and in conjunction with a chemokine-fusion DNA vaccine (MIP3α-gp100) with known anti-tumor efficacy in the mouse B16F10 melanoma model1. Figure 1a shows that αIL-10 as a monotherapy provides significant anti-tumor efficacy with a 43% reduction in average tumor size at day 14 post challenge (p<0.001). However, the tumor burden benefit of the monotherapy disappears at day 17 (figure 1b), and this late, accelerated growth results in loss of a significant effect on the tumor growth rate as measured between days 10 and 17 post challenge (figure 1c). The monotherapy did provide enough efficacy to have significantly enhanced survival (figure 1d; p<0.01), with an increase in median survival time of 11%. These data support the hypothesis that neutralization of IL-10 provides a benefit in the mouse melanoma system.
Figure 1.
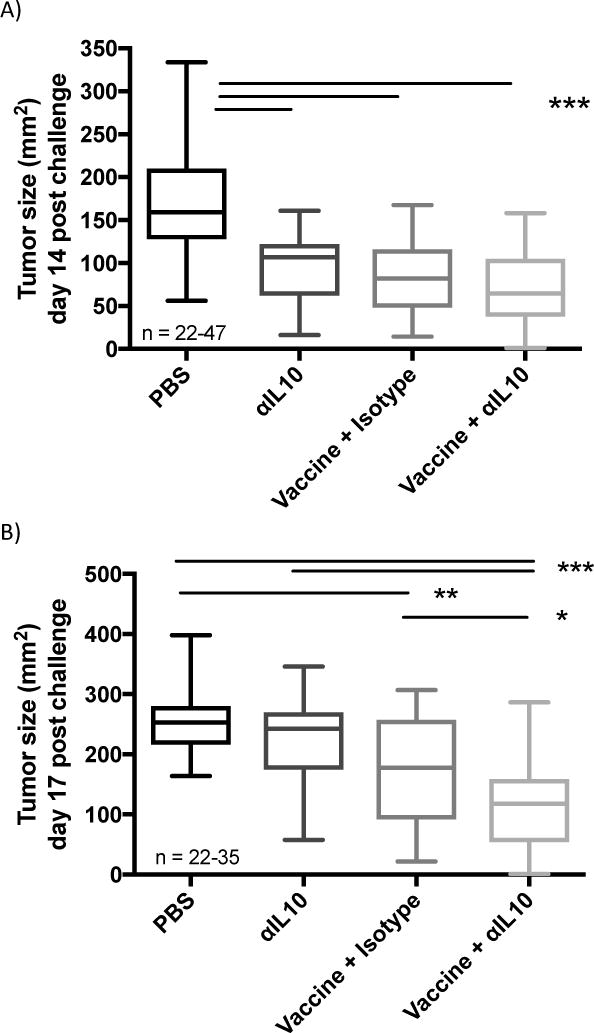
Effect of αIL-10 on tumors in therapeutic vaccination model. Vaccinations occurred on days 3, 10, and 17 post challenge. Doses of αIL-10 began on day 5 post challenge and continued every three days for a total of six doses. A) Tumor burden at day 14 post challenge B) Tumor burden at day 17 post challenge. The data of panels a-b are representative of 3–8 experiments of 5–9 mice per group per experiment, with range of sample sizes noted in the graph. Significance determined by Anova with Tukey’s multiple comparison test. Boxplot whiskers delineate the range, and the box represents the middle 50% with median line. C) Tumor growth rate was assessed between days 10 and 17, at which point tumor growth of the negative control group approached when mice began to be censored due to attainment of tumor size endpoints. Graph shows data points with standard errors and linear regression lines. Numerical slopes are indicated adjacent to their respective regression lines. The data are representative of 4–9 experiments with 5–9 mice per experiment for sample sizes ranging from 28–53 per group. The group combining vaccination with αIL-10 therapy shows significantly slower tumor growth than all other treatments as evaluated by a statistical mixed effects regression model. D) Effect of αIL-10 on mouse survival. Mice were removed from the study at the following endpoints: death, tumor diameter surpassing 2cm, or excessive tumor bleeding and ulceration. Data are representative of three to four experiments comprising a total of 22–29 mice per group and were evaluated by log-rank test. *p<0.05; **p<0.01; ***p<0.001
Figures 1a–d confirm results from our previous study that vaccination with MIP3α-gp100 provides consistent clinical benefit in the B16F10 mouse model, as measured by tumor size, tumor growth rate, and mouse survival1. When combining the vaccine regimen with αIL-10 therapy, clinical benefit at early time-points was equivalent to both monotherapies, as seen in figure 1a. Over time, the combination therapy continued to provide anti-tumor efficacy whereas the two monotherapies began to lose efficacy. As seen in figure 1b, by day 17, tumors of the combination therapy group were on average 46% smaller than αIL-10 monotherapy (p<0.001) and 30% smaller than vaccination (p<0.05).
Further, figure 1c shows that the combination therapy group contains tumors that grow significantly more slowly, with a 31% reduction in growth rate compared to vaccine alone (p<0.01) and a 51% reduction compared to αIL-10 monotherapy (p<0.001). Finally, combination therapy provides significant enhancement in mouse survival compared to vaccine (p<0.05) and antibody (p<0.001) monotherapies (figure 1d), with median survival enhancements of 10% and 21%, respectively.
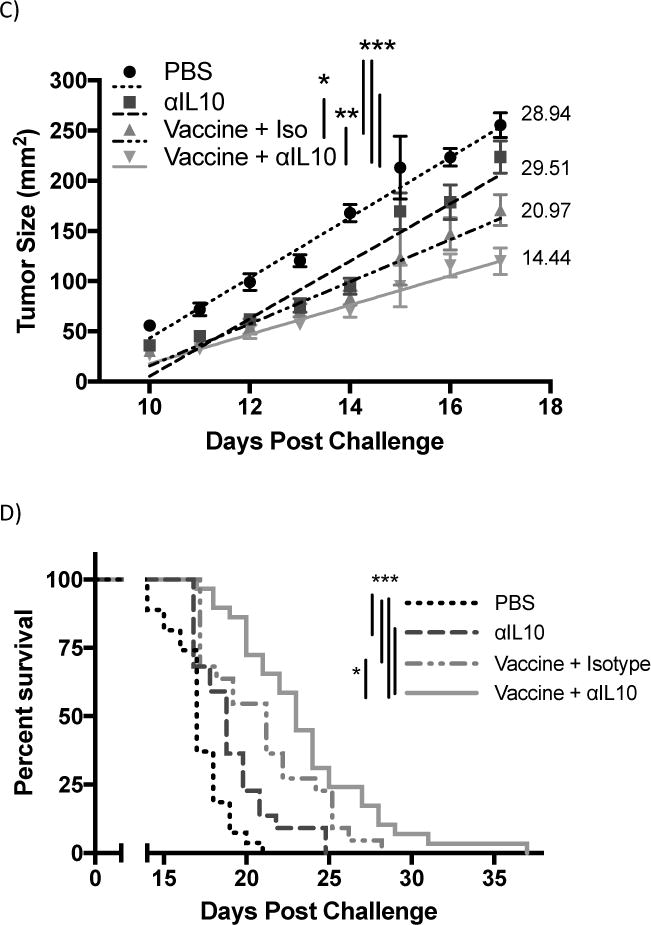
Analysis of Infiltrating Lymphocytes
The potential mechanism for the enhanced efficacy of αIL-10 therapy seen in figure 1 was initially hypothesized to be that IL-10 was inhibiting influx of effector T-cells from blood vessels into the tumor microenvironment, as described by others 27,28. In this model, anti-IL10 therapy should increase the infiltration of lymphocytes. However, while both vaccination alone and vaccination combined with αIL-10 dramatically increased the number of infiltrating lymphocytes compared to the negative control (p<0.001), the two regimens were indistinguishable by this parameter (figure 2a). Similarly, the number (figure 2a) and percentage (figure 2b) of CD8+ infiltrating cells that produced IFN-γ upon ex vivo stimulation with gp10025–33 peptide did not differ significantly between these two treatment groups.
Figure 2.
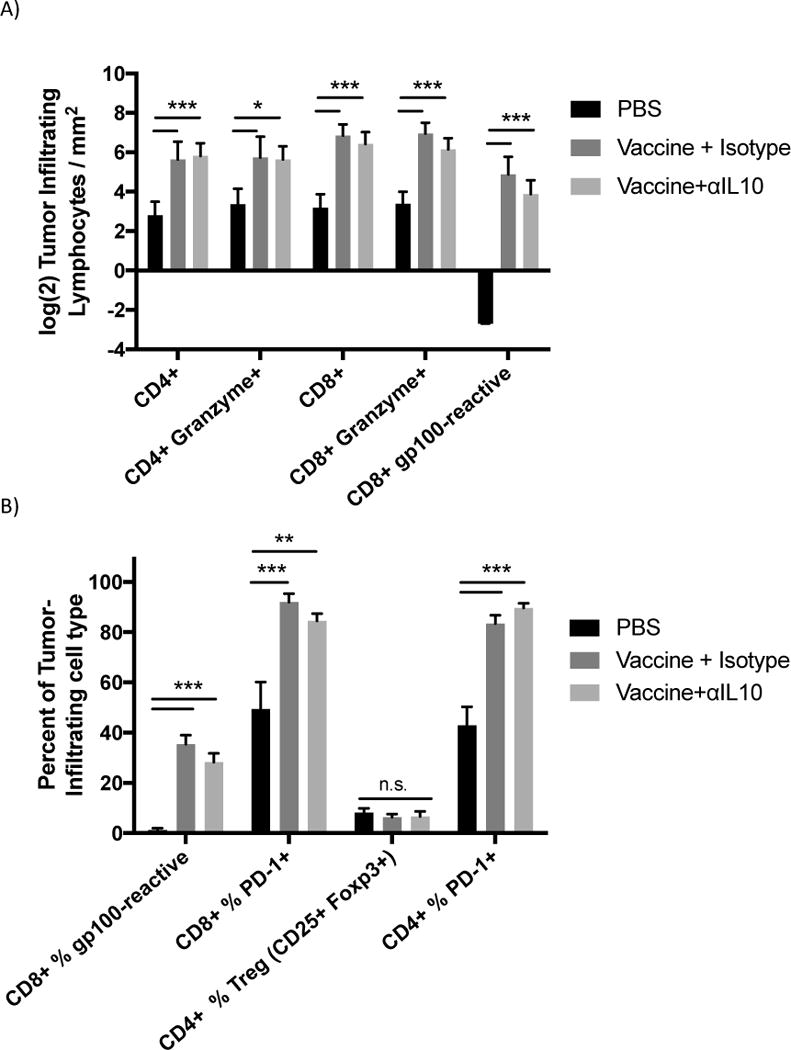
Flow cytometric analysis of tumor infiltrating lymphocytes (TILs). Vaccinations occurred on days 3 and 10 post challenge. Doses of αIL-10 began on day 5 post challenge and continued every three days for a total of four doses. Mice were euthanized on day 17, and tumor tissue was extracted for analysis by flow cytometry as outlined in the methods. Cells were gated on live/dead status and forward vs. side scatter of standard immune cell parameters. A) Analysis of TILs by numbers of cells normalized to tumor size. B) Analysis of TILs by percentage of CD4+ or CD8+ T-cells exhibiting the analyzed characteristic. C) Panel shows the ratio of CD8+ TILs to regulatory CD4+ TILs. All data are from 2–3 independent experiments of 3–8 mice each. Groups were evaluated by Anova with Tukey’s multiple comparisons test. Error bars represent the standard error. *p<0.05; **p<0.01; ***p<0.001.
The second hypothesis for the enhanced efficacy observed with addition of αIL-10 was that IL-10 was negatively affecting T-cell effector function by inhibiting granzyme expression or by enhancing regulatory T-cell numbers in the environment, as seen in a similar model system15. Administration of vaccine alone or combined with αIL-10 did not significantly alter the number of CD4+Granzyme+ or CD8+Granzyme+ cells infiltrating the tumor (figure 2a). Similarly, the difference in percentage of CD4+ and CD8+ infiltrating T-cells with surface expression of the activation/exhaustion marker Programmed Death ligand-1 (PD-1) was not significant between the vaccine alone and vaccine plus αIL-10 groups (figure 2b). Also, in contrast to Kalli, et al. 15, no difference was seen in the percentage of infiltrating CD4+ T-cells that had a regulatory phenotype (figure 2b). The ratio between infiltrating CD8+ T-cells and regulatory CD4+ cells, a parameter generally correlating with T-cell based anti-tumor efficacy29,30, was also not significantly different between groups receiving vaccine alone and groups receiving both vaccine and αIL-10 (figure 2c).
Expression and Cellular Source of IL-10 Responsive Genes
Since αIL-10 did not appear to be affecting T-cells directly, intratumoral expression levels were assessed for some of the genes that are both known to be regulated by IL-10 and capable of indirectly enhancing infiltrating T-cell responses. Genes such as H2-K1 (MHC-I) 10,31,32, H2-ab1 (MHC-II) 33, and Ifnα (Interferon-alpha) 34,35 are known to be negatively regulated by IL-10 under certain conditions. The transcriptional levels of these candidate genes in tumor lysates were analyzed by real-time quantitative reverse-transcription PCR (qRT-PCR) one day after administration of αIL-10. Relative fold-expression values were calculated as noted in the methods and figure legend. Expression levels of H2-k1, H2-ab1, and Ifnα2/11 remained relatively unchanged in vaccine alone versus vaccine plus αIL-10 (figure 3a). Expression levels of Ifnα4 were upregulated 2.8-fold (p<0.05) when adding αIL-10 to the vaccine (figure 3a). In addition, Ifnα4 in the vaccine only group was upregulated 6.7-fold compared to negative control, so the combination group was upregulated 18.7-fold compared to the negative control.
Figure 3.
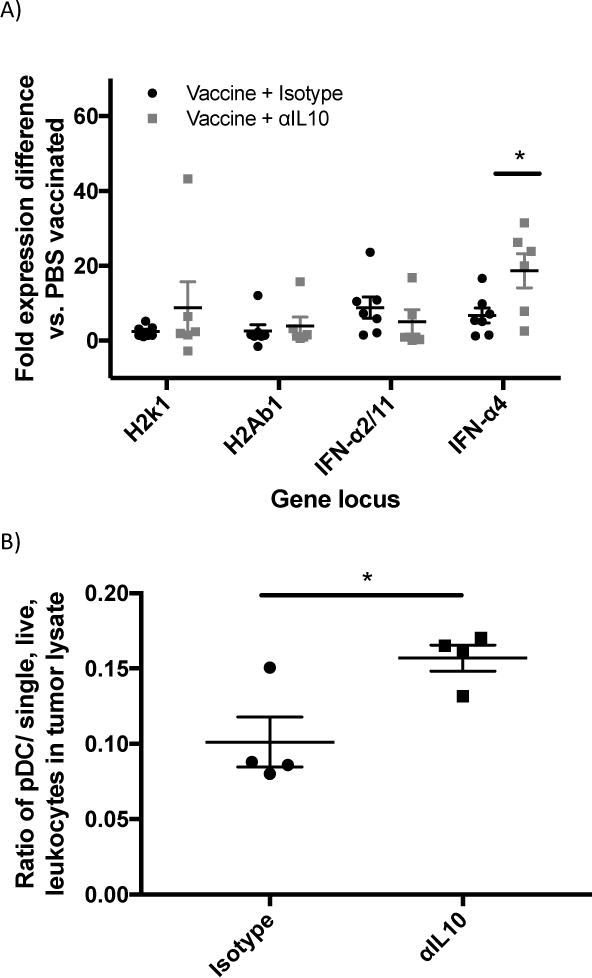
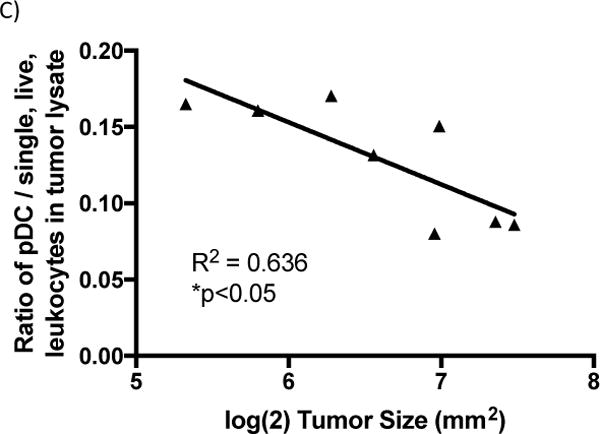
Analysis of genes affected by αIL-10 treatment and the cell types known to produce the resulting proteins. A) Real-time quantitative reverse-transcription PCR (qRT-PCR) analysis of loci potentially affected by αIL-10 treatment. Vaccinations occurred on days 3 and 10 post challenge. Doses of αIL-10 (150μg intratumorally) began on day 5 post challenge and continued every three days until harvest. Mice were euthanized and tumors harvested one day after the αIL-10 dose where >80% of tumors were above minimum cutoff of 30 mm2. qRT-PCR data was analyzed by the ΔΔCt method. Samples were normalized to GAPDH housekeeping gene control to form ΔCt values and then subtracted from the ΔCt of a PBS vaccinated mouse to create the ΔΔCt values. Data shown as fold expression difference as compared to PBS vaccinated mouse by the formula 2(−ΔΔCt). Outliers more than two standard deviations greater than group mean were removed from the dataset. Data are representative of two independent experiments with 3–4 mice per group and were evaluated by Anova with Tukey’s multiple comparisons test. B) Flow cytometry analysis of infiltrating pDCs. Mice were challenged with 5×104 B16F10 cells, and doses of αIL-10 (150μg intratumorally) were given on days 5, 8, and 11 post challenge. Mice were sacrificed on day 13 post challenge, and tumor tissue was harvested, processed, and stained as described in the methods. To define the population, dead cells and cellular aggregates were excluded, and pDCs were gated on FSC/SSC potential leukocytes, B220+, CD11c+, and mPDCA1+. Data are presented as a ratio of pDCs per single, live, leukocytes in the tumor lysate. Data are representative of one experiment with four mice per group and were analyzed by Student’s t test. C) pDC correlation to tumor size. Utilizing the flow cytometry dataset, log transformed tumor size was compared to the pDC ratios presented in figure 3c. Data are representative of one experiment with eight mice and were analyzed by linear regression. Error bars represent the standard error. *p<0.05
It is known in the literature that plasmacytoid dendritic cells (pDCs) are primary producers of type I interferons36. Therefore, the presence of pDCs infiltrating the tumors were analyzed two days after the third αIL-10 treatment. For these data, the system was simplified to treatments of αIL-10 only, without vaccinations. Intratumoral pDCs were increased by 55.0% (p<0.05) at this time point when mice were given αIL-10 (figure 3b). Further, the concentration of pDCs had a significant correlation with reduced tumor size (figure 3c; r2=0.636; p<0.05).
IFNαr1−/− Knockout Mice
The previous experiments indicate that IFNα4 correlates with the anti-tumor efficacy of αIL10 treatment and that the cell type known for producing type I interferons has elevated levels in tumors of αIL-10 treated mice; however, the results do not differentiate between IFNα4 and pDCs being associated with and directly causing the enhanced anti-tumor efficacy. To address this issue, the standard therapeutic protocol was assessed utilizing mice with IFNα-receptor 1 knocked out (IFNαr1−/−). Mice deficient in the receptor are unable to react to the IFNα family of cytokines 37. In figure 4a, tumor sizes at day 17 post challenge of wild type vaccine+αIL-10 treated mice were on average 41.1% smaller compared to wild type vaccine only (p<0.05), consistent with previous results. Wild type mice receiving vaccine only bore tumors that were not significantly different in size compared to IFNαr1−/− mice that were vaccinated alone or were additionally given αIL-10. Importantly, the tumor sizes of the combination treatment in wild type mice were 51.1% smaller than those of IFNαr1−/− mice given the combination treatment (figure 4a; p<0.001).
Figure 4.
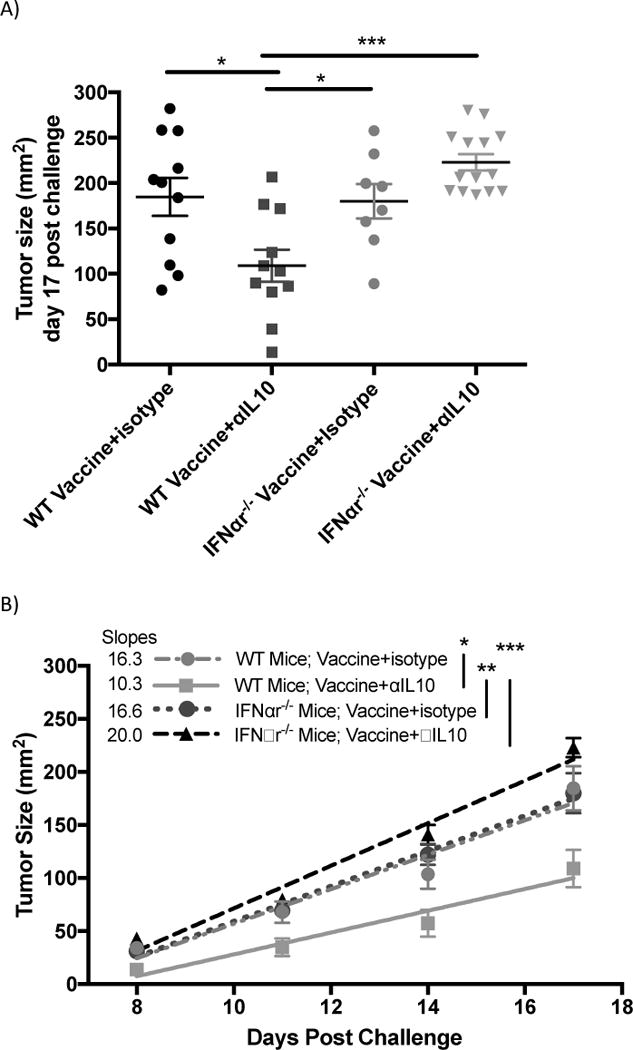
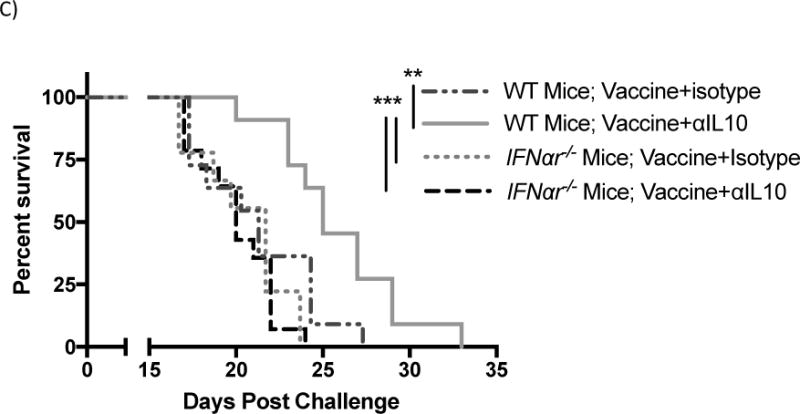
Effects of Interferon alpha receptor knockout (IFNαr1−/−) mice on tumors of mice given vaccinations and αIL-10 treatment. Vaccinations occurred on days 3, 10, and 17 post challenge. Doses of αIL-10 began on day 5 post challenge and continued every three days for a total of five doses. A) Tumor size data shown at day 14 post challenge – the final time-point including all mice. Groups were evaluated by Anova with Tukey’s multiple comparisons test. Error bars represent the standard error. B) Tumor growth shown between day 8, when tumors began growing steadily, and 17, after which >10% of mice had been removed from the study. Groups were evaluated by mixed effects linear regression. Error bars represent the standard error, linear regression lines are included, and numerical slopes are indicated. C) Survival effects of IFNαr1−/− mice on vaccination and αIL-10 treatment. Mice were removed from the study at the following endpoints: death, tumor size surpassing 2cm in any dimension, or excessive tumor bleeding and ulceration. Groups were evaluated by log-rank test. Data represent two to three experiments of 4–6 mice per group per experiment. *p<0.05; **p<0.01; ***p<0.001.
Tumor growth rates followed a similar course (figure 4b). Wild type combination treatment mice had significantly lower tumor growth rates than wild type vaccine-only (36.8% less, p<0.05), IFNαr1−/− vaccine-only (38.0% less, p<0.01), and IFNαr1−/− combination treatment groups (51.5% less, p<0.001). Similarly, survival analysis (figure 4c) demonstrated that wild type mice receiving both treatments survived significantly longer than wild type vaccine-only mice (p<0.01), IFNar1−/− vaccine-only mice (p<0.001), and IFNar1−/− mice receiving both treatments (p<0.001). The group of wild type mice with both treatments showed an increase in median survival of 19.0% compared to the wild type vaccine-only group, 13.6% compared to the IFNar1−/− vaccine-only group, and 25.0% compared to the IFNar1−/− group with both treatments. The wild type vaccine only group showed no significant difference in growth or survival compared to the IFNar1−/− group receiving either vaccine alone or vaccine plus αIL-10.
DISCUSSION
In this study, neutralizing IL-10 at the tumor site enhances the anti-tumor efficacy of a DNA MIP3α-gp100 therapeutic vaccine. The αIL10 therapy does not appear to directly alter T-cell tumor infiltration, T-cell differentiation, or vaccine immunogenicity, as measured by vaccine peptide-reactive tumor infiltrating CD8+ T-cells. Blockade of IL-10 has been shown to enhance T-cell responses to an otherwise ineffective DNA vaccine in a lymphocytic choriomeningitis virus model 38. IL-10 neutralization also enhanced a dendritic cell-based anti-melanoma prophylactic vaccine and the αIL-10 monotherapy correlated with enhanced CD4+ granzyme+ tumor infiltrating lymphocytes (TILs) and reduction of regulatory CD4+ Foxp3+ TILs 15. However, in the present model, levels of CD8+ TILs that produced IFN-γ upon vaccine peptide stimulation did not significantly change when adding αIL-10 treatments to vaccinated mice, nor did levels of CD4+ granzyme+ TILs or regulatory CD4+ Foxp3+ TILs. It is possible that this difference is due to analysis of the monotherapy 15 versus the combination with vaccine as analyzed here. Overall levels of TILs did not change significantly either, suggesting that αIL-10 was not altering T-cell migration or endothelial cell permeability.
Instead, the mechanism by which αIL10 therapy enhances anti-tumor efficacy is associated with intratumoral expression of IFNα4. IFNα4 and 11 are highly active in mice39, and so levels of both were analyzed. While Ifnα2/11 saw no increase in expression, Ifnα4 expression was increased almost 3-fold in combination treatment compared to vaccine-only and approximately 18-fold compared to the negative control. Additionally, the cell type that primarily produces type I interferons, pDCs36, was shown to be enhanced in the tumor environment. Levels of pDCs significantly correlated with reduced tumor growth across treatment and control groups, indicating that this parameter may be important for B16F10 tumor control. This correlation was then shown to be the primary driver responsible for the efficacy of αIL-10 treatment. Knocking out a chain of the common receptor for interferon alpha cytokines (Ifnαr1−/−) caused loss of the enhanced vaccine efficacy phenotype associated with αIL-10 therapy. Ifnαr1−/− mice receiving vaccination and αIL-10 treatment did not show significant differences compared to wild type or Ifnαr1−/− mice that were given vaccine only, but did have larger tumors, a faster tumor growth rate, and reduced survival compared to wild type mice given both vaccine and αIL-10. Clearly, αIL-10 efficacy in combination with a MIP3α-gp100 vaccine is primarily mediated by type I interferons and the responses they induce. Unexpectedly, the course of B16F10 progression remained the same between wild type and Ifnαr1−/− mice that received vaccine only, indicating that baseline vaccine efficacy is independent of type I interferons.
Identification of the role of type I IFNs in the observed anti-IL10 effects facilitates development of a model that explains the patterns of tumor growth observed in the different treatment groups. An increase in type I IFNs could provide a more general benefit by enhancing expression of anti-angiogenic factors40, apoptosis or cell cycle arrest41, macrophage cytotoxicity42, lymphocyte cytotoxicity43, and/or dendritic cell activity44,45. It is possible that the slight reduction in tumor size due to the direct anti-tumor effects of type I IFNs at early time-points in the combined treatments group allowed the vaccine response to achieve better efficacy over time due to the reduced tumor burden. However, this hypothesis is unlikely in the vaccine and αIL-10 combination group, considering that it is not until day 17 post challenge that the combination group significantly separates from the monotherapies. If the enhanced efficacy of the combination group was due to direct anti-tumor effects, one would expect to see earlier separation.
The tumor growth patterns in the IL-10 treated groups of mice suggest two distinct effects, both of which have been previously associated with the anti-tumor efficacy of type I IFNs. The early suppression of tumor growth observed in the mice treated with anti-IL-10 alone could be readily attributable to the direct anti-tumor effects of type I IFNs, as indicated. However, this effect is attenuated over time and by Day 15 the slope of the tumor growth rate in the anti-IL-10 treated mice sharply increases. A recent report suggests that loss of type I IFN activity against melanoma cells can be reversed with treatment with 5-aza-2′–deoxycitidine, suggesting that the loss of direct cell killing activity is attributable to DNA methylation events affecting interferon-stimulated genes 46.
However, the well-described effects of IFN’s on enhanced DC function and T cell priming 44,45 would likely have occurred prior to the attenuation of type I IFN function but would not be manifest until the vaccine-induced immune response becomes functional at the later stages of tumor growth. This delayed expression of the early type I IFN effects on the inductive stage of the immune response likely account for the enhanced extended efficacy observed in the combined treatment group compared to treatment with either vaccine or anti-IL10 alone.
While it is acknowledged that the current enhancements in efficacy are modest, new therapy alternatives are essential for the growth of the field. This study shows that this flexible and novel dendritic-cell targeting vaccine platform can be enhanced by immunomodulating agents. Future studies will focus on other immunodulators currently in the clinic, alone or combined with αIL-10, such as αPD-1, αPDL-1, αCTLA-4, αLAG-3, and/or others47. Since the effect of αIL-10 is mediated by type I IFNs, we also plan on directly administering IFNα to determine whether vaccine efficacy can be enhanced further by this pathway. Notably, this mechanism has clinical relevance. IFN-α2b treatment has shown benefit in patients with early stage II/III melanomas48, and clinical studies have elucidated that adjuvant IFNα therapy significantly increased disease-free survival and sometimes overall survival49. IFNα adjuvant therapy became the first immunotherapeutic agent to show significant benefits in survival in a phase III randomized controlled trial and is a part of the current standard of care for patients with advanced stage II or stage III melanoma49,50. This model identifies paths by which further enhancement of tumor growth inhibition might be achieved and provides direction for future studies.
IL-10 is a complex cytokine with pleiotropic effects. However, the results of this study conclusively show that intratumoral neutralization of IL-10 provides therapeutic efficacy against B16 melanoma alone, and that the efficacy is further enhanced when used in combination with a DNA vaccine. This study demonstrates that Ifnα4 expression is upregulated in the tumor and that plasmacytoid dendritic cells are present at higher levels in the tumor after αIL-10 doses. Finally, action of type I IFNs is necessary for αIL-10 treatment efficacy. Further study could guide investigators into understanding the specific contexts that provide the best anti-tumor efficacy from IL-10 neutralization.
Acknowledgments
We would like to acknowledge Dr. TC Wu (Johns Hopkins School of Medicine, Baltimore, MD) for allowing us to utilize his electroporator. We would like to acknowledge Dr. Ada Tam and the Flow Cytometry and Immune Monitoring Core Facility (Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD) for flow cytometry assistance. We would like to acknowledge support for the statistical analysis from the National Center for Research Resources and the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health through Grant Number 1UL1TR001079, and more specifically Carol Thompson of the Johns Hopkins Biostatistics Center.
Funding
We have no funding sources to disclose.
List of Abbreviations
- MIP3α
Macrophage Inflammatory Protein-3α
- gp100
glycoprotein 100 common melanoma differentiation antigen and vaccine target
- APC
Antigen Presenting Cell
- DC
Dendritic Cell
- MHC
Major Histocompatibility Complex
- B16F10 B16
Mouse melanoma model
- IL-10
Interleukin 10
- αIL-10
IL-10 neutralizing antibody
- pDC
plasmacytoid dendritic cell
Footnotes
Ethics Approval
As reported in the Methods, mice were purchased from Charles River Laboratories (Wilmington, MA) and maintained in a pathogen-free micro-isolation facility in accordance with the National Institutes of Health guidelines for the humane use of laboratory animals. All experimental procedures involving mice were approved by the IACUC of the Johns Hopkins University (Protocol numbers MO13H219 and MO16H85).
Competing Interests
J. Gordy and R. Markham are inventors on pending patents using the vaccine platform described in this manuscript. R. Markham has equity interest in a company that has rights to this vaccine platform. No non-financial conflicts of interest exist for any of the authors.
Authors’ Contributions
JG performed, designed, and analyzed all the experiments and was the primary author of the manuscript. KL provided assisted with design and implementation of the mouse studies. CD provided scientific direction, expertise, and help in data analysis. BF contributed to the conception and design of these experiments. RM contributed to the conception, design, analysis of data, and the writing of the manuscript. All authors read and approved the manuscript.
References
- 1.Gordy JT, Luo K, Zhang H, et al. Fusion of the dendritic cell-targeting chemokine MIP3α to melanoma antigen Gp100 in a therapeutic DNA vaccine significantly enhances immunogenicity and survival in a mouse melanoma model. J Immunother Cancer. 2016;4:96. doi: 10.1186/s40425-016-0189-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawkins WG, Gold JS, Dyall R, et al. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery. 2000;128(2):273–80. doi: 10.1067/msy.2000.107421. [DOI] [PubMed] [Google Scholar]
- 3.Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Current oncology reports. 2008;10:439–446. doi: 10.1007/s11912-008-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spranger S, Spaapen R, Zha Y, et al. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Science Translational Medicine. 5(200):1–10. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Croci D, Fluck M, Rico M, et al. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunology, Immunotherapy. 2007;56(11):1687–1700. doi: 10.1007/s00262-007-0343-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jäger E, Ringhoffer M, Altmannsberger M, et al. Immunoselection in vivo: Independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. International Journal of Cancer. 1997;71(2):142–147. doi: 10.1002/(SICI)1097-0215(19970410)71:2<142::AID-IJC3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 7.García‐Hernández M, Hernández‐Pando R. Interleukin‐10 promotes B16‐melanoma growth by inhibition of macrophage functions and induction of tumour and vascular cell proliferation. Immunology. 2002;105:231–243. doi: 10.1046/j.1365-2567.2002.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biggs M, Eiselein J. Suppression of immune surveillance in melanoma. Medical Hypotheses. 2001;56(6):648–652. doi: 10.1054/mehy.2000.1211. [DOI] [PubMed] [Google Scholar]
- 9.Yue F, Dummer R, Geertsen R, et al. Interleukin‐10 is a growth factor for human melanoma cells and down‐regulates HLA class‐I, HLA class‐II and ICAM‐1 molecules. International Journal of Cancer. 1997;71(4):630–637. doi: 10.1002/(SICI)1097-0215(19970516)71:4<630::AID-IJC20>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 10.Kurte M, López M, Aguirre A, et al. A synthetic peptide homologous to functional domain of human IL-10 down-regulates expression of MHC class I and Transporter associated with Antigen Processing 1/2 in human melanoma cells. Journal of immunology. 2004;173:1731–1737. doi: 10.4049/jimmunol.173.3.1731. [DOI] [PubMed] [Google Scholar]
- 11.Sredni B, Weil M, Khomenok G, et al. Ammonium trichloro(dioxoethylene-o,o’)tellurate (AS101) sensitizes tumors to chemotherapy by inhibiting the tumor interleukin 10 autocrine loop. Cancer Res. 2004;64(5):1843–52. doi: 10.1158/0008-5472.can-03-3179. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Daniel V, Maher D, et al. Production of IL‐10 by melanoma cells: Examination of its role in immunosuppression mediated by melanoma. International Journal of Cancer. 1994;56(5):755–760. doi: 10.1002/ijc.2910560524. [DOI] [PubMed] [Google Scholar]
- 13.Mahipal A, Terai M, Berd D, et al. Tumor-derived interleukin-10 as a prognostic factor in stage III patients undergoing adjuvant treatment with an autologous melanoma cell vaccine. Cancer Immunology, Immunotherapy. 2011;60:1039–1045. doi: 10.1007/s00262-011-1019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchi LH, Paschoalin T, Travassos LR, et al. Gene therapy with interleukin-10 receptor and interleukin-12 induces a protective interferon-γ-dependent response against B16F10-Nex2 melanoma. Cancer Gene Ther. 2011;18(2):110–22. doi: 10.1038/cgt.2010.58. [DOI] [PubMed] [Google Scholar]
- 15.Kalli F, Machiorlatti R, Battaglia F, et al. Comparative analysis of cancer vaccine settings for the selection of an effective protocol in mice. Journal of Translational Medicine. 2013;11(1):120. doi: 10.1186/1479-5876-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litton M, Dohlsten M, Rosendahl A, et al. The distinct role of CD4+ and CD8+ T-cells during the anti-tumour effects of targeted superantigens. British journal of cancer. 1999;81(2):359–66. doi: 10.1038/sj.bjc.6690701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sredni B. Immunomodulating tellurium compounds as anti-cancer agents. Seminars in cancer biology. 2011;22(1):60–9. doi: 10.1016/j.semcancer.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Nadler R, Luo Y, Zhao W, et al. Interleukin 10 induced augmentation of delayed‐type hypersensitivity (DTH) enhances Mycobacterium bovis bacillus Calmette‐Guérin (BCG) mediated antitumour activity. Clinical & Experimental Immunology. 2003;131(2):206–216. doi: 10.1046/j.1365-2249.2003.02071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bockholt NA, Knudson MJ, Henning JR, et al. Anti-Interleukin-10R1 Monoclonal Antibody Enhances Bacillus Calmette-Guérin Induced T-Helper Type 1 Immune Responses and Antitumor Immunity in a Mouse Orthotopic Model of Bladder Cancer. The Journal of Urology. 2012;187(6):2228–2235. doi: 10.1016/j.juro.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Luo Y. Blocking IL-10 enhances bacillus Calmette-Guérin induced T helper Type 1 immune responses and anti-bladder cancer immunity. OncoImmunology. 2012;1(7):1183–1185. doi: 10.4161/onci.20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berezhnoy A, Stewart A, James M, et al. Isolation and Optimization of Murine IL-10 Receptor Blocking Oligonucleotide Aptamers Using High-throughput Sequencing. Molecular Therapy. 2012;20(6):1242–1250. doi: 10.1038/mt.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murugaiyan G, Agrawal R, Mishra GC, et al. Functional dichotomy in CD40 reciprocally regulates effector T cell functions. Journal of immunology. 2006;177(10):6642–9. doi: 10.4049/jimmunol.177.10.6642. [DOI] [PubMed] [Google Scholar]
- 23.Sabat R, Grütz G, Warszawska K, et al. Biology of interleukin-10. Cytokine & Growth Factor Reviews. 2010;21(5):331–344. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Schiavo R, Baatar D, Olkhanud P, et al. Chemokine receptor targeting efficiently directs antigens to MHC class I pathways and elicits antigen-specific CD8+ T-cell responses. Blood. 2006;107(12):4597–605. doi: 10.1182/blood-2005-08-3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miguel A, Herrero M, Sendra L, et al. Comparative Antitumor Effect of Preventive versus Therapeutic Vaccines Employing B16 Melanoma Cells Genetically Modified to Express GM-CSF and B7.2 in a Murine Model. Toxins. 2012;4(11):1058–1081. doi: 10.3390/toxins4111058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan J, Pankhong P, Shin TH, et al. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunology Research. 2013;1:179–189. doi: 10.1158/2326-6066.CIR-13-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshima T, Laroux FS, Coe LL, et al. Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res. 2001;61(1):130–43. doi: 10.1006/mvre.2000.2288. [DOI] [PubMed] [Google Scholar]
- 28.Motz GT, Santoro SP, Wang L-PP, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–15. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali OA, Lewin SA, Dranoff G, et al. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol Res. 2016;4(2):95–100. doi: 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curran M, Montalvo W, Yagita H, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersson M, Charo J, Salazar-Onfray F, et al. Constitutive IL-10 production accounts for the high NK sensitivity, low MHC class I expression, and poor transporter associated with antigen processing (TAP)-1/2 function in the prototype NK target YAC-1. Journal of immunology. 1998;161:2099–2105. [PubMed] [Google Scholar]
- 32.Kundu N, Fulton AM. Interleukin-10 Inhibits Tumor Metastasis, Downregulates MHC Class I, and Enhances NK Lysis. Cellular Immunology. 1997;180:55–61. doi: 10.1006/cimm.1997.1176. [DOI] [PubMed] [Google Scholar]
- 33.De Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174(4):915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito S, Ansari P, Sakatsume M, et al. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood. 1999;93(5):1456–63. [PubMed] [Google Scholar]
- 35.Payvandi F, Amrute S, Fitzgerald-Bocarsly P. Exogenous and endogenous IL-10 regulate IFN-alpha production by peripheral blood mononuclear cells in response to viral stimulation. J Immunol. 1998;160(12):5861–8. [PubMed] [Google Scholar]
- 36.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 37.Uze G, Schreiber G, Piehler J, et al. The receptor of the type I interferon family. Curr Top Microbiol Immunol. 2007;316:71–95. doi: 10.1007/978-3-540-71329-6_5. [DOI] [PubMed] [Google Scholar]
- 38.Brooks D, Lee A, Elsaesser H, et al. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. The Journal of Experimental Medicine. 2008;205(3):533–541. doi: 10.1084/jem.20071948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Pesch V, Lanaya H, Renauld J-CC, et al. Characterization of the murine alpha interferon gene family. J Virol. 2004;78(15):8219–28. doi: 10.1128/JVI.78.15.8219-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albini A, Marchisone C, Grosso F, et al. Inhibition of Angiogenesis and Vascular Tumor Growth by Interferon-Producing Cells A Gene Therapy Approach. Am J Pathology. 2000;156(4):1381–1393. doi: 10.1016/S0002-9440(10)65007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sangfelt O, Erickson S, Castro J, et al. Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines. Cell Growth Differ. 1997;8(3):343–52. [PubMed] [Google Scholar]
- 42.Hervas-Stubbs S, Perez-Gracia J, Rouzaut A, et al. Direct Effects of Type I Interferons on Cells of the Immune System. Clinical Cancer Research. 2011;17(9):2619–27. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 43.Sikora AG, Jaffarzad N, Hailemichael Y, et al. IFN-alpha enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. Journal of immunology. 2009;182(12):7398–407. doi: 10.4049/jimmunol.0802982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–16. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucarini V, Buccione C, Ziccheddu G, et al. Combining Type I Interferons and 5-Aza-2′-Deoxycitidine to Improve Anti-Tumor Response against Melanoma. Journal of Investigative Dermatology. 2017;137:159–169. doi: 10.1016/j.jid.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 47.Dempke WCMC, Fenchel K, Uciechowski P, et al. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer. 2017;74:55–72. doi: 10.1016/j.ejca.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Kirkwood J, Strawderman M, Ernstoff M, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. Journal of clinical oncology. 1996;14(1):7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 49.Raaijmakers M, Rozati S, Goldinger S, et al. Melanoma immunotherapy: historical precedents, recent successes and future prospects. Immunotherapy. 2013;5(2):169–182. doi: 10.2217/imt.12.162. [DOI] [PubMed] [Google Scholar]
- 50.Melanoma Skin Cancer [American Cancer Society website] 2016 May 20; Available at: https://www.cancer.org/cancer/melanoma-skin-cancer.html. Accessed October 17, 2017.