

Abstract
Aldo-keto reductase family 1 member D1 (AKR1D1) is a Δ4-3-oxosteroid 5β-reductase required for bile acid synthesis and steroid hormone metabolism. Both bile acids and steroid hormones, especially glucocorticoids, play important roles in regulating body metabolism and energy expenditure. Currently, our understanding on AKR1D1 regulation and its roles in metabolic diseases is limited. We found that AKR1D1 expression was markedly repressed in diabetic patients. Consistent with repressed AKR1D1 expression, hepatic bile acids were significantly reduced in diabetic patients. Mechanistic studies showed that activation of peroxisome proliferator-activated receptor-α (PPARα) transcriptionally down-regulated AKR1D1 expression in vitro in HepG2 cells and in vivo in mice. Consistently, PPARα signaling was enhanced in diabetic patients. In summary, dysregulation of AKR1D1 disrupted bile acid and steroid hormone homeostasis, which may contribute to the pathogenesis of diabetes. Restoring bile acid and steroid hormone homeostasis by modulating AKR1D1 expression may represent a new approach to develop therapies for diabetes.
Keywords: AKR1D1, diabetes, bile acid synthesis, steroid hormone metabolism, PPARα signaling
1. Introduction
Aldo-keto reductase family 1, member D1 (AKR1D1) is a Δ4-3-oxosteroid 5β-reductase, which catalyzes the reduction of steroids with a 3-oxo-4-ene structure in the presence of nicotinamide adenine dinucleotide phosphate (NADPH) as a hydride donor (Kondo et al., 1994). AKR1D1 is required for cholesterol metabolism into bile acids. In the bile acid synthesis pathway, AKR1D1 reduces the double bond in the A ring of the bile acid intermediates to eventually produce primary bile acids chenodeoxycholic acid (CDCA) and cholic acid (CA) (Russell, 2003; Jin et al., 2014). AKR1D1 is also involved in metabolism and clearance of steroid hormones with 3-oxo-4-ene structure including glucocorticoids (Jin et al., 2014, Chen and Penning, 2014). Since 5β-reduction is a common transformation and major inactivation pathway for steroid hormones, and AKR1D1 is the only enzyme in humans capable of catalyzing a 5β-reduction in those steroids, AKR1D1 plays a critical role in regulating and maintaining the homeostasis of steroid hormones, including glucocorticoids. Therefore, AKR1D1 plays critical roles in bile acid synthesis and steroid hormone metabolism and inactivation.
Bile acids are actively participated in regulating metabolism and energy expenditure. A large body of evidence from human clinical (Bennion and Grundy, 1977; de Leon et al., 1978; Steiner et al., 2011; Zhang et al., 2015) and animal model (Nervi et al., 1978; Uchida et al., 1979) investigations demonstrate that bile acid homeostasis is disrupted in patients with metabolic disease and diabetes while moderating bile acid enterohepatic circulation has beneficial effects on diabetes (Prawitt et al., 2014; Flynn et al., 2015; Ferrannini et al., 2015). On the other hand, steroid hormones, especially glucocorticoids, play important roles in regulating body metabolism and energy expenditure (Vegiopoulos and Herzig, 2007; de Guia et al., 2014). Studies with human and various rodent models have linked dysregulation of glucocorticoids to the development of metabolic disease and diabetes (Pivonello et al., 2010; Chan et al., 2003; Perez et al., 2014).
As a key player in bile acid synthesis and steroid hormone metabolism, our current understanding on AKR1D1’s roles in metabolic disease and diabetes is limited (Valanejad et al., 2017). In this study, we found that AKR1D1 expression was markedly repressed in diabetic patients. In line with repressed AKR1D1 expression, hepatic bile acid levels were significantly reduced in diabetic patients compared with non-diabetic control subjects. Mechanistic studies revealed that activation of peroxisome proliferator-activated receptor-α (PPARα) markedly repressed AKR1D1 expression in vitro and in vivo. Furthermore, it was revealed that PPARα signaling was enhanced in diabetic patients, which provides an explanation for the repressed expression of AKR1D1 in diabetic patients.
2. Materials and Methods
2.1. Chemicals and Supplies
CDCA, CA, deoxycholic acid (DCA), lithocholic acid (LCA), ursodeoxycholic acid (UDCA), glycine or taurine conjugated bile acids, dimethyl sulfoxide (DMSO), DNA oligonucleotides, fetal bovine serum (FBS), charcoal-stripped FBS, pioglitazone (PGZN) and insulin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Luciferase assay kits were from Promega (Madison, WI, USA). Restriction enzymes were from New England BioLabs (Ipswich, MA, USA). Activated charcoal was from Fisher Scientific (Pittsburgh, PA, USA). GW4064, GW7647, GW3965 and GW0742 were purchased from Tocris Bioscience (Minneapolis, MN, USA). Recombinant human AKR1D1 protein was purchased from Novus Biologics (Littleton, CO, USA).
2.2. Liver samples
Liver samples: Twenty-two liver tissues of type 2 diabetes mellitus patients were obtained from Sckisui XenoTech (Kansas City, KS, USA). The quality of those liver tissues were ensured with initial intent for liver transplantation. Twenty non-diabetic normal control liver samples were obtained through the Cooperative Human Tissue Network (CHTN). The quality of the liver tissues provided by CHTN were ensured through the prospective collection model as the quality management system (Grizzle et al., 2015). The characteristics of individual liver tissues from diabetic and non-diabetic control subjects were provided in Table 1 and 2. The protocol for using human tissues was approved by the Institutional Review Board (IRB) at the University of Rhode Island (URI).
Table 1.
Characteristics of diabetic subjects
| ID # | Gender | Age | Race | Cause of Death | Drug Use | Alcoh. Use | NASa Score | Fibrosis Stages | BMIb | Cholesterol (mmol/l) | Type 2 DM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H0403 | Fc | 51 | Ce | Anoxia | Ni | N | 2 | 2 | 70 | 5.6 | Y, took insulin |
| H0412 | Md | 66 | C | CVAg | N | N | 2 | 1 | 27 | 8.1 | Y |
| H0423 | M | 55 | C | Anoxia | N | Yj | 3 | 1 | 28 | 24.3 | Y, for 5 years |
| H0424 | F | 39 | C | CVA | N | Y | 7 | 0 | 36 | 12.5 | Y |
| H0434 | M | 74 | C | CVA | N | N | 7 | 0 | 26 | 6.2 | Y, for 10 years |
| H0436 | F | 72 | C | CVA | N | N | 2 | 3 | 35 | 12.7 | Y |
| H0439 | F | 78 | C | Anoxia | N | N | 1 | 1 | 34 | 7.8 | Y, for 8 years |
| H0462 | M | 55 | C | CVA | N | Y | 0 | 1 | 24 | 9.5 | Y, for 5 years |
| H0465 | M | 50 | Hf | Anoxia | N | N | 1 | 1 | 21 | 11.9 | Y |
| H0469 | F | 56 | H | CVA | N | Y | 3 | 3 | 22 | 7.3 | Y |
| H0476 | F | 46 | C | CVA | N | N | 5 | 1 | 46 | 5.4 | Y, for 6 months |
| H0479 | M | 65 | C | Anoxia | N | N | 2 | 2 | 22 | 16.7 | Y, for 1 month |
| H0502 | M | 49 | C | HTh | N | N | 2 | 1 | 35 | 17.6 | Y, for 8 years |
| H0531 | F | 44 | C | CVA | N | N | 2 | 0 | 43 | 12.1 | Y, for 5 years |
| H0548 | F | 61 | C | Anoxia | N | N | 1 | 0 | 39 | 18.5 | Y, for 4 years |
| H0728 | M | 39 | C | CVA | N | Y | 2 | 1 | 19 | 7.9 | Y, for 20 years |
| H0741 | F | 74 | C | HT | N | N | 2 | 0 | 25 | 19.4 | Y |
| H0750 | F | 53 | C | Anoxia | N | N | 4 | 1 | 44 | 6.6 | Y |
| H0766 | F | 38 | C | Anoxia | N | Y | 4 | 0 | 27 | 10.3 | Y, for 20 years |
| H0772 | M | 46 | C | Anoxia | N | Y | 4 | 1 | 32 | 19.4 | Y |
| H0802 | M | 33 | C | CVA | N | Y | 0 | 1 | 24 | 6.5 | Y |
| H0804 | M | 57 | C | CVA | N | N | 5 | 1 | 31 | 17.8 | Y, for 13 years |
NAS: Non-alcoholic fatty liver disease (NAFLD) activity score;
BMI: Body mass index;
F: Female;
M: Male;
C: Caucasian;
H: Hispanic;
CVA: cerebrovascular accident;
HT: head trauma;
N: No;
Y: Yes.
Table 2.
Characteristics of liver tissues from non-diabetic control subjects
| ID # | Gender | Age | Race | Procurement | BMIa | Liver Condition | Cause of Death |
|---|---|---|---|---|---|---|---|
| 35685 | Female | 74 | Caucasian | Autopsy | 33 | Normal | Respiratory failure |
| 35935 | Male | 65 | Caucasian | Autopsy | 27 | Normal | Infarction |
| 37696 | Female | 61 | Caucasian | Autopsy | NAb | Normal | Respiratory failure |
| 39376 | Female | 77 | Caucasian | Autopsy | 24 | Normal | Pulmonary edema |
| 40093 | Male | 59 | Africa American | Autopsy | NA | Normal | Heart attack |
| 42433 | Female | 86 | Africa American | Autopsy | NA | Normal | Pulmonary disease |
| 42534 | Female | 71 | Caucasian | Surgery | NA | Normal | Not applicable |
| 43946 | Male | 80 | Caucasian | Autopsy | 28 | Normal | Bradycardic/hypotensive |
| 44230 | Male | 76 | Africa American | Autopsy | NA | Normal | Car accident |
| 44588 | Male | 73 | Caucasian | Autopsy | NA | Normal | Respiratory failure |
| 44679 | Female | 73 | Caucasian | Surgery | NA | Normal | Not applicable |
| 44683 | Male | 59 | Caucasian | Autopsy | 46 | Normal | Respiratory failure |
| 45014 | Male | 64 | Caucasian | Autopsy | NA | Normal | Mesothelioma |
| 46001 | Female | 55 | Caucasian | Surgery | NA | Normal | Not applicable |
| 46782 | Male | 72 | Caucasian | Surgery | NA | Normal | Not applicable |
| 46925 | Female | 22 | Africa American | Autopsy | NA | Normal | Prader-willi Syndrome |
| 47574 | Female | 52 | Caucasian | Surgery | NA | Normal | Not applicable |
| 51139 | Female | 60 | Caucasian | Surgery | NA | Normal | Not applicable |
| 51215 | Male | 78 | Caucasian | Surgery | NA | Normal | Not applicable |
| Z2898 | Male | 71 | Caucasian | Autopsy | 18 | Normal | Adrenal insufficiency |
BMI: Body mass index;
NA: Not available.
2.3. Plasmid constructs
Human AKR1D1 promoter reporter phAKR1D1(−5.0kb) was constructed by cloning the 5kb promoter region into the pGL4.10 luciferase vector (Promega). The 5kb promoter was PCR-amplified by forward (5′-TAAGATGCTAATCGTAATCCCCAGGGTAAC-3′) and reverse primer (5′-GGAGTGATTCTGAACACCACAGGGAGTCT-3′) using human genomic DNA as templates. Expression plasmids for human farnesoid x receptor-α2 (FXRα2) was provided by Dr. Matthew Stoner. Expression plasmids for human FXRα1, PPARα, PPARβ, PPARγ and liver x receptor-α (LXRα) were kindly provided by Dr. Bingfang Yan at URI.
2.4. Mice and treatments
Eighteen CD-1 mice were randomly divided into three groups. One group of mice were treated with PPARα agonist GW7647 (10mg/kg). The second group were treated with FXR agonist GW4064 (10mg/kg). The third group were treated with vehicle propanediol as controls. Mice were injected i.p. twice a day for 3 days. Twelve hours after the last injection, animal were euthanized and liver tissues were harvested and processed for gene expression analysis. The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at URI.
2.5. HepG2 cells and treatments
HepG2 cells seeded in 12-well plates were treated with GW4064 (1μM), GW7647 (10μM), PGZN (10μM) and insulin (100ng/ml) in phenol-red free medium containing 1% charcoal striped FBS for 30h, followed by analysis of gene expression.
2.6. TaqMan quantitative real-time PCR
Total RNA isolation from liver tissues (human and mice) or HepG2 cells and subsequent TaqMan real-time PCR were carried out as described (Song et al., 2014). Transcript levels of AKR1D1, FXR, PPARα, cholesterol 7α-hydroxylase or cytochrome P450 family 7 subfamily A member 1 (CYP7A1), sterol 12α-hydroxylase (CYP8B1), bile salt export pump (BSEP), sodium-taurocholate cotransporting polypeptide (NTCP), organic anion-transporting polypeptide member 1B1 (OATP1B1), multidrug resistance-associated protein 3 (MRP3), MRP4, cytochrome P450 family 27 subfamily A member 1 (CYP27A1), cytochrome P450 family 7 subfamily B member 1 (CYP7B1) and carnitine palmitoyltransferase-1A (CPT1A) were normalized against β-actin or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels according to the treatments or conditions, which had minimal effects on β-actin or GAPDH (Kroetz et al., 1998; Ma et al., 2013; de la Monte et al., 2011; Zhou et al., 2008; Biswas et al., 2013). Validated TaqMan PCR probes and master mixtures were obtained from Applied Biosystems (Foster City, CA, USA).
2.7. Analysis of bile acids in human liver by liquid chromatography tandem-mass spectrometry (LC-MS/MS)
Bile acid extraction from human liver tissues and subsequent quantification by LC-MS/MS were carried out as described with some modifications (Alnouti et al., 2008). An Acquity UPLC from Waters connected to a Xevo TQ MS mass spectrometry was used for quantification. The system was controlled with MassLynx™ software (V 4.1) and data was processed using TargetLynx™ tool.
For preparation of liver tissue extracts for quantification of bile acids, liver samples were homogenized on ice in 1:4 w/v 50% methanol (Fisher Scientific, AC61513). After addition of 10 μl of mifeprestone internal standard and 2 ml of ice-cold alkaline acetonitrile (5% ammonium hydroxide in acetonitrile) (Fisher Scientific), samples were vortexed and shaken continuously for 1 hour at 4°C. Samples were then centrifuged at 4600×g for 20 minutes at 4°C, supernatants were collected. The pelleted samples were extracted again with 1 ml of ice-cold alkalinizede acetonitrile and the supernatants were collected. The two supernatants were pooled and then evaporated using a Thermo Scientific SpeedVac system, followed by reconstitution of the samples in 200 μl of 50% methanol.
Conjugated and unconjugated bile acids were chromatographically separated on an Acquity UPLC BEH C18 (2.1 mm × 50 mm) column with 1.7 μm particle size. Mobile phase that was consisted of water containing 7.5 mM ammonium acetate (mobile phase A) and 95:5 v:v Methanol : acetonitrile (mobile phase B) was pumped at flow rate of 0.5 ml/min. A 20 μl sample loop was used to deliver aliquots of 10 μl samples in a partial loop injection mode. The total run time was 7 mins. Gradient elution was employed and maintained at 2% mobile phase B for 0.5 min and increased gradually to 80% over 5 min and maintained at this level for one minute. To re-equilibrate the column for the next run, the proportion of mobile phase B was decreased within 0.1 min to the initial condition and kept constant for 0.9 min. The calibration and quality control concentrations of bile acids under the investigation were presented in Supplemental Table 1. The lower limit of quantification (LLOQ) and lower limit of detection (LLOD) values were also presented in Supplemental Table 1.
Unconjugated and taurine conjugated bile acids were monitored in negative ionization mode while glycine conjugated bile acids were monitored in positive ionization mode. Mycophenolic acid (Cerilliant Corp. Round Rock, TX; molecular weight of 320.34 g/mol) was simultaneously monitored in both positive and negative ionization modes and was used as an internal standard for quantification of all bile acids (Supplemental Fig. 1).
2.8. Reporter luciferase assays
Transient transfection and dual-luciferase reporter assays in HepG2 cells were performed as described (Chen et al., 2013). Standard amounts of plasmid DNA used per well in 24-well plates were 100ng for promoter reporter phAKR1D1(−5.0kb), 100ng for nuclear receptor expression plasmid (FXRα1, FXRα2, LXRα, PPARα, PPARβ, or PPARγ), and 10ng for the null-Renilla luciferase plasmid. Sixteen hours post-transfection, cells were treated with DMSO (0.1%), GW4064 (1μM), GW3965 (1 μM), GW0742 (1μM) and GW7647 (10μM) in a phenol red-free medium containing 1% charcoal-stripped FBS for 30 hours. Luciferase activities were assayed as previously described (Song et al., 2013).
2.9. Western blotting
Cell lysates were made from HepG2 cells or liver tissues as described (Chen et al., 2013). Thirty micrograms of total protein were loaded into each well. Membranes were blotted with antibodies against AKR1D1, CYP7A1, BSEP, or PPARα. The same membranes were stripped and re-blotted with antibodies against GAPDH. In addition, a control liver sample was included in all the blots as an external standard to facilitate the comparison of expression levels on different blots. The images were captured using Gel Logic 2200 PRO system and protein bands were quantified using ImageQuant TL software. The expression levels of GAPDH were used to normalize the expression of target genes as the treatments or conditions had minimal effects on the expression of GAPDH proteins (Song et al., 2014; Ma et al., 2013). Primary antibodies against AKR1D1 (sc-365932), CYP7A1 (sc-25536), BSEP (sc-25571), PPARα (sc-398394) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-GAPDH antibodies (G9545) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The specificity of those antibodies against the target proteins was demonstrated and reported previously (Valanejad et al., 2017; Song et al., 2014; He et al., 2011; Bellet et al., 2016). The specificity of AKR1D1 antibodies was evaluated with recombinant human AKR1D1 protein and the results were presented in Supplemental Fig. 2.
2.10. Statistical analysis
Student’s t-test was applied to pair-wise comparison for normally distributed data. One-way ANOVA was applied to analyze data with multiple groups, followed by Tukey post-hoc test for multiple comparisons. Non-parametric Mann-Whitney test was used for pair-wise comparison for non-normally distributed data. A p value of 0.05 or less was considered statistically significant.
3. Results
3.1. Characteristics of diabetic and control subjects
Twenty-two liver tissues from type 2 diabetic patients were obtained from XenoTech. The characteristics of the diabetic subjects were provided in Table 1. Among the 22 patients, eleven subjects (50%) were female. The average age of the patients is 54.6 ± 12.7 years with 91% of Caucasian and 9% of Hispanic. Majority of patients (68% with a body mass index: BMI >25) were either overweight, obese or severe obsess with hypercholesterolemia being detected in 86.4% of the patients. Consistently, 77.7% patients had a non-alcoholic fatty liver disease (NAFLD) activity score (NAS) of 2 or above while only 18.2 % patients had a fibrosis stage 2 or above. Matched with diabetic subjects, among the 20 non-diabetic control subjects, which were obtained through CHTN, ten subjects (50%) were female (Table 2). Similar to the diabetic subjects, the majority of control subjects were Caucasian (80%). The average age of the control subjects were 67.8 ± 13.1, which is 13.2 years older than the diabetic subjects. Thirteen liver tissues were procured from autopsy of subjects died of non-diabetic condition while 7 samples were from surgery. Those liver tissues exhibited normal macro- and microscopic histology. Limited data showed that the subjects had a large range of BMI from 18 to 46, within which 95% of diabetic subjects were.
3.2. AKR1D1 expression was markedly repressed in diabetic patients
AKR1D1 catalyzes reduction of molecules with a 3-oxo-4-ene structure, including glucocorticoids and bile acid intermediates (2–4), thus playing critical roles in bile acid synthesis and glucocorticoid metabolism and clearance (Fig. 1A). To investigate whether AKR1D1 is dysregulated in diabetic patients, thus may contribute to the pathogenesis of diabetes, the expression levels of AKR1D1 were determined. As shown in Fig. 1B, the mRNA levels of AKR1D1 were markedly decreased by 84% in diabetic patients (p=0.0074) when compared with non-diabetic control subjects. Consistently, AKR1D1 protein levels were reduced by 75% in Western blot (p=0.002) (Fig. 1C and 1D). Taken together, the data on AKR1D1 expression at mRNA and protein levels firmly established that AKR1D1 was dysregulated and its expression was markedly repressed in diabetic patients.
Fig. 1. AKR1D1 expression was markedly repressed in diabetic patients.
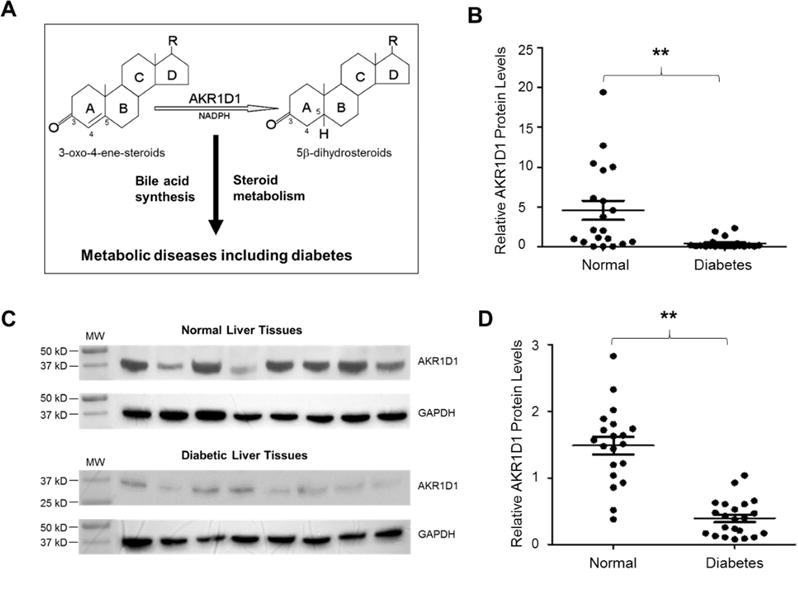
(A) AKR1D1 catalyzes the reduction of molecules with a 3-oxo-4-ene structure in the presence of nicotinamide adenine dinucleotide phosphate (NADPH) as a hydride donor and involved in bile acid synthesis and steroid hormone metabolism, both of which have been implicated in metabolic diseases and diabetes. (B) Hepatic expression levels of AKR1D1 mRNA were quantified by TaqMan real-time PCR in diabetic patients (n=22) and normal control subjects (n=20). The expression levels of GAPDH mRNA were used to normalize AKR1D1 expression. (C) representative Western blots showing the protein expression of AKR1D1 and GAPDH in diabetic and control subjects. (D) quantification of AKR1D1 protein levels on Western blots normalized by the protein levels of GAPDH. The means and standard errors of the group values were indicated by the long and short lines, respectively. ** p<0.01 with Student’s t-test.
3.3. Total hepatic bile acids were markedly reduced in diabetic patients
AKR1D1 is required for bile acid synthesis and glucocorticoid metabolism. With markedly reduced expression of AKR1D1, we hypothesized that bile acid synthesis and glucocorticoid metabolism were compromised in diabetic patients. To provide evidence to support the hypothesis, we systemically characterized total as well as individual hepatic bile acids in diabetic patients. As shown in Fig. 2A, in comparison with control subjects, total hepatic bile acids were markedly reduced in diabetic patients (p=0.0017). The average concentration of total bile acids were decreased by 53% from 83.8 nmoles/g to 39.8 nmoles/g. The decrease is largely due to the reduction in conjugated bile acids (Fig. 2B), which were decreased from 82.9 ± 49.9 nmoles/g to 38.3 ± 26.2 nmoles/g (p=0.0014). On the other hand, unconjugated bile acids were slightly increased from 0.9 ± 0.9 nmoles/g to 1.2 ± 2.5 nmoles/g but did not reach a statistical significance (p=0.89) (Fig. 3C). Among the conjugated bile acids, both glycine and taurine conjugated bile acids were significantly reduced in diabetic patients (Fig. 2D and Fig. 2E). However, the reductions were more severe in glycine conjugated than taurine conjugated bile acids. The average glycine conjugated bile acid level was decreased by 57% from 63.9 ± 43.3 nmoles/g to 27.3 ± 18.7 nmoles/g (p=0.0017). Taurine conjugated bile acids were reduced by 42% from 19.0 ± 9.2 nmoles/g to 11.0 ± 10.7 nmoles/g (p=0.02). Taken together, total hepatic bile acids were markedly reduced in diabetic patients, mainly due to reduction of conjugated bile acids, especially glycine conjugated bile acids.
Fig. 2. Total hepatic bile acids were markedly reduced in diabetic patients.
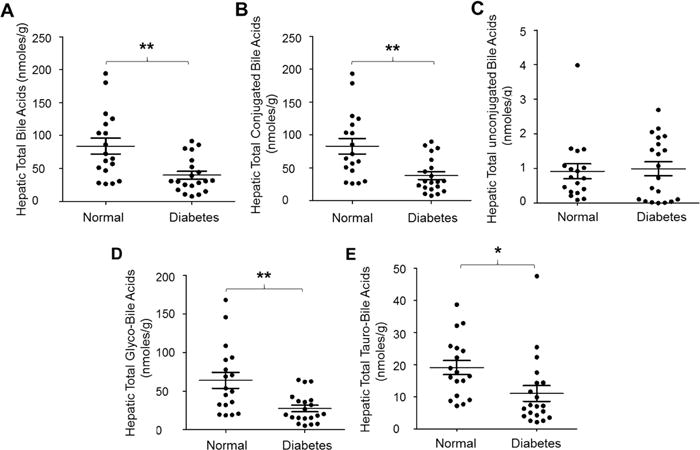
The concentrations of (A) total hepatic bile acids, (B) conjugated bile acids, (C) unconjugated bile acids, (D) glycine conjugated bile acids (glyco-bile acids) and (E) taurine conjugated bile acids (tauro-bile acids) were compared between diabetic patients and control subjects. Total bile acids were extracted from human liver tissues from diabetic patients (n=22) and control subjects (n=20), and quantified by LC-MS/MS. Means with standard error of mean (SEM) were indicated. * p<0.05 and ** p<0.01.
Fig. 3. Hepatic CDCA concentrations were significantly reduced in diabetic patients.
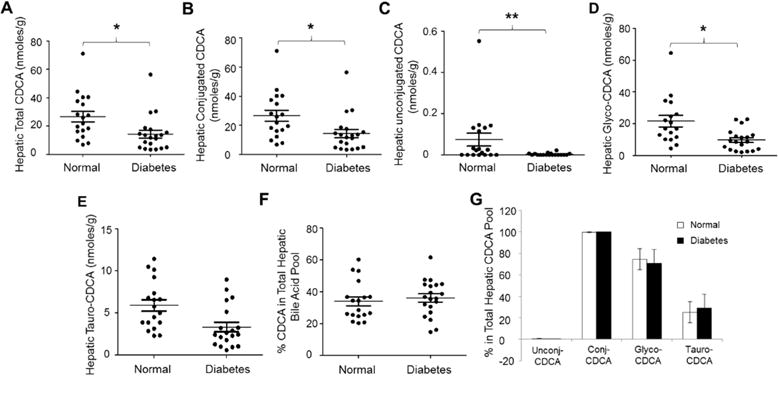
The concentrations of (A) total hepatic CDCA, (B) conjugated CDCA, (C) unconjugated CDCA, (D) glycine conjugated CDCA (glyco-CDCA), and (E) taurine conjugated CDCA (tauro-CDCA) were compared between diabetic patients and control subjects. Total bile acids were extracted from human liver tissues. The concentrations of CDCA, glyco-CDCA and tauro-CDCA were quantified by LC-MS/MS. Means with standard error of mean (SEM) were indicated. (F) The percentages of CDCA in the total hepatic bile acid pools and (G) the compositions of the CDCA pools were compared between diabetic patients and control subjects. * p<0.05 and ** p<0.01.
3.4. Hepatic CDCA concentrations were significantly reduced in diabetic patients
As shown in Fig. 3A, total hepatic CDCA levels were significantly reduced in diabetic patients (p=0.012). The average total CDCA level was decreased by 46% from 26.7 ± 15.9 nmoles/g to 14.4 ± 12.4 nmoles/g. Both conjugated and unconjugated CDCA were significantly reduced in diabetic patients (Fig. 3B and 3C). Conjugated CDCA levels were decreased from 26.6 ± 15.9 nmoles/g to 14.4 ± 12.4 nmoles/g (p=0.013) while unconjugated CDCA concentrations were reduced from 0.075 ± 0.13 nmoles/g to 0.003 ± 0.005 nmoles/g (p=0.009). Among the conjugated CDCA, glycine conjugated CDCA (glyco-CDCA) was decreased by 54 % from 20.8 ± 14.6 nmoles/g to 9.6 ± 6.5 nmoles/g (p=0.0042) (Fig. 3D). Decreases in taurine conjugated CDCA (tauro-CDCA) were also detected but did not reach a statistical significance (p=0.56) (Fig. 3E). The percentages of CDCA in the total hepatic bile acid pool were not significantly altered (p=0.59), 34% and 36% in control and diabetic subjects, respectively (Fig. 3F). Similarly, the percentages of unconjugated, conjugated, glyco- and tauro-CDCA in the total hepatic CDCA pool were not significantly altered (Fig. 3G). Taken together, total hepatic CDCA concentrations were significantly reduced, mainly due to marked decreases in glycine conjugated CDCA in diabetic patients. The percentages of CDCA in the total hepatic bile acid pool and the relative compositions of the hepatic CDCA pool were minimally changed in diabetic patients.
3.5. Hepatic CA homeostasis was disrupted in diabetic patients
Consistent with reduced total bile acid levels, diabetic patient exhibited markedly decreased total hepatic CA levels (p=0.0037) (Fig. 4A). The average concentration of CA was decreased by 61% from 33.6 ± 26.7 nmoles/g to 13.2 ± 10.9 nmoles/g. Both conjugated and unconjugated CA were markedly reduced in diabetic patients (Fig. 4B and 4C). Total conjugated CA concentrations were decreased from 33.2 ± 26.2 nmoles/g to 13.1 ± 10.9 nmoles/g (p=0.0037) while unconjugated CA levels were reduced from 0.39 ± 0.79 nmoles/g to 0.06 ± 0.06 nmoles/g (p=0.04). Among the conjugated CA, both glyco-CA and tauro-CA were severely reduced (Fig. 4D and 4E). Glyco-CA levels were decreased by 61% from 23.6 ± 20.1 nmoles/g to 9.1 ± 7.9 nmoles/g (p=0.0059). Tauro-CA levels were reduced by 59% from 9.7 ± 6.9 nmoles/g to 4.0 ± 3.6 nmoles/g (p=0.0032). However, the percentages of CA in the total hepatic bile acid pool were not significantly altered (p=0.39) (Fig. 4F). The hepatic bile acid pools consisted of 37% CA in control subjects and 33% CA in diabetic patients. Likewise, no significant alterations in the compositions of the hepatic CA pool were detected (Fig. 4G). Taken together, total hepatic CA concentrations were markedly reduced while the percentages of CA in the total hepatic bile acid pool and the compositions of the hepatic CA pool were not altered in diabetic patients.
Fig. 4. Hepatic CA homeostasis was disrupted in diabetic patients.
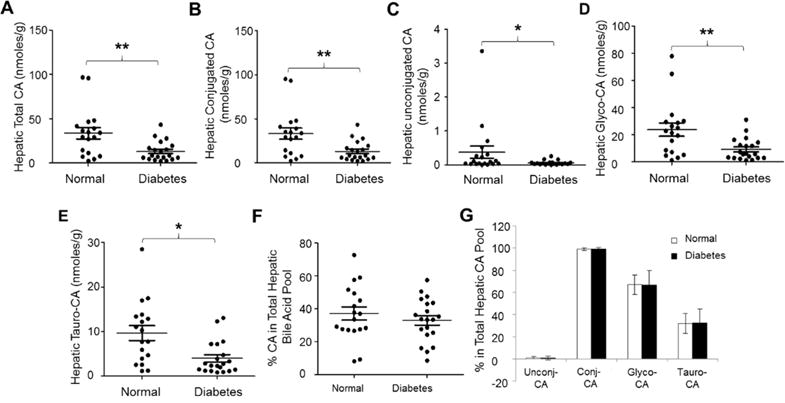
The concentrations of (A) total hepatic CA, (B) conjugated CA, (C) unconjugated CA, (D) glycine conjugated CA (glyco-CA), and (E) taurine conjugated CA (tauro-CA) were compared between diabetic patients and control subjects. Total bile acids were extracted from human liver tissues, followed by quantifying the concentrations of CA, glyco-CA and tauro-CA by LC-MS/MS. Means with standard error of mean (SEM) were indicated. (F) The percentages of CA in the total hepatic bile acid pools and (G) the compositions of the CA pools were compared between healthy subjects and diabetic patients. * p<0.05 and ** p<0.01.
3.6. Alterations of hepatic DCA homeostasis in diabetic patients
In contrast to CDCA and CA, total hepatic DCA levels showed a trend of reduction but without reaching a statistical significance in diabetic patients (p=0.061) (Fig. 5A). However, the concentrations of conjugated DCA were significantly decreased by 53% from 23.0 ± 22.1 nmoles/g to 10.8 ± 11.7 nmoles/g (p=0.04) (Fig. 5B). The unconjugated DCA levels were slightly increased but did not reach a statistical difference (p=0.12) (Fig. 5C). Among the conjugated DCA, glyco-DCA but not tauro-DCA were markedly reduced (Fig. 5D and 5E). The average glyco-DCA levels were decreased by 56% from 19.5 ± 19.1 nmoles/g to 8.6 ± 10.2 nmoles/g (p=0.034). Similar to CA and CDCA, the percentages of DCA in the total hepatic bile acid pools remained comparable with 28.9% in control and 31.0% in diabetic subjects (p=0.72) (Fig. 5F). In contrast to CDCA and CA, the compositions of hepatic DCA pools were significantly altered (Fig. 5G). The percentages of unconjugated DCA in the DCA pools were significantly increased from 3.6% in control subjects to 12.9% in diabetic patients (p=0.02). The percentages of conjugated DCA were significantly decreased from 96.4% in healthy subjects to 87.1% in diabetic patients (p=0.02), due to a significant reduction of glyco-DCA from 80.7% to 67% (p=0.021). On the other hand, the percentages of tauro-DCA were slightly increased without reaching a statistical significance (p=0.38). Taken together, conjugated DCA, mainly glyco-DCA, was significantly decreased and the compositions of hepatic DCA pools were altered in diabetic patients.
Fig. 5. Alterations of hepatic DCA homeostasis in diabetic patients.
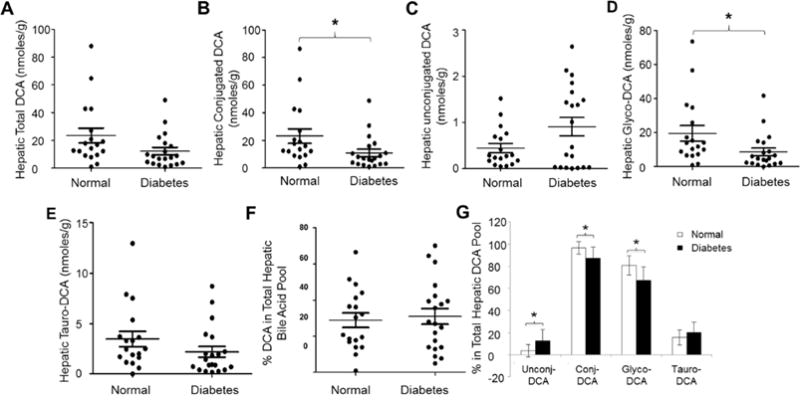
The concentrations of (A) total hepatic DCA, (B) conjugated DCA, (C) unconjugated DCA, (D) glycine conjugated DCA (glyco-DCA), and (E) taurine conjugated DCA (tauro-DCA) were compared between diabetic patients and control subjects. Total bile acids were extracted from human liver tissues. The concentrations of DCA, glyco-DCA and tauro-DCA were quantified by LC-MS/MS. Means with standard error of mean (SEM) were indicated. (F) The percentages of DCA in the total hepatic bile acid pools and (G) the compositions of the DCA pools were compared between healthy subjects and diabetic patients. * p<0.05
It should also be mentioned that the levels of hepatic LCA and UDCA were very low and only detected in a few liver samples, excluding them being reliably analyzed in this study with LLOQ of 1 μg/L and LLOD of 0.2 μg/L. We also attempted to detect hepatic glucocorticoids (cortisol and cortisone) and their 5β-reduced metabolites. However, their levels were too low to be detected in the liver.
3.7. No significant alterations in the expression of CYP7A1 and BSEP in diabetic patients
CYP7A1 is the rate limiting enzyme in hepatic bile acid synthesis while biliary excretion of bile acids through BSEP is the rate-limiting step in the enterohepatic circulation of bile acids (Chiang, 2013; Trauner and Boyer, 2003). To determine whether CYP7A1 and BSEP are dysregulated in diabetic patients, thus contributing to reduced hepatic bile acid levels in diabetic patients, the expression levels of CYP7A1 and BSEP in diabetic patients and control subjects were quantified. As shown in Fig. 6A, CYP7A1 mRNA levels were not significantly altered in diabetic patients compared with control subjects. Consistently, the protein levels of CYP7A1 were minimally changed (Fig. 6B and 6C). The results suggested that CYP7A1 did not contribute to the reduced hepatic bile acids in diabetic patients. BSEP mRNA levels were actually decreased in diabetic patients (Fig. 6D), which presumably decreases biliary secretion of bile acids, resulting in hepatic accumulation of bile acids. To determine whether such decrease in mRNA levels is consistent with the protein levels, Western blots were carried out to determine the levels of membrane-associated BSEP protein. In contrast to the mRNA levels, BSEP protein abundances were actually slightly increased in the diabetic patients without reaching a statistical significance (Fig. 6E and 6F), indicating a post-transcriptional regulation of BSEP in diabetic patients. The data suggested that biliary secretion of bile acids through BSEP were not significantly altered in diabetic patients, excluding altered BSEP expression as a mechanism for the dysregulation of hepatic bile acids in diabetic patients.
Fig. 6. No significant alterations in the expression of CYP7A1 and BSEP in diabetic patients.
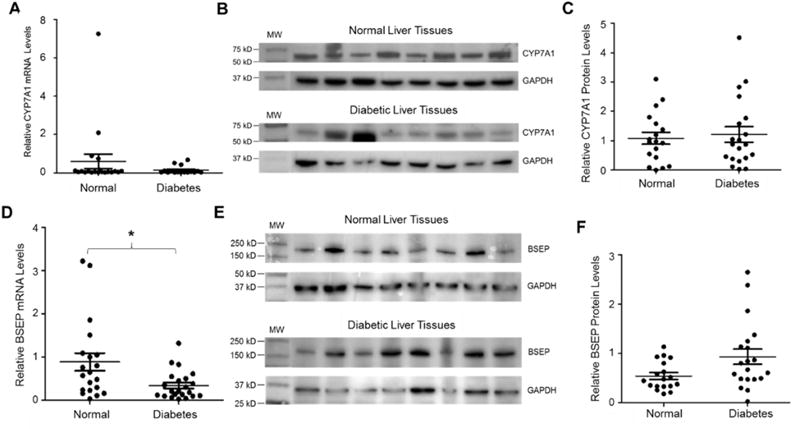
(A) the expression levels of CYP7A1 mRNA in the liver of normal control subjects (n=20) and diabetic patients (n=22) were quantified by TaqMan real-time PCR. (B) the representative Western blots exhibiting the protein expression of CYP7A1 and GAPDH in diabetic and control subjects. (C) quantification of CYP7A1 protein levels on Western blots. (D) the expression levels of BSEP mRNA in the liver of control subjects and diabetic patients were quantified by TaqMan real-time PCR. (E) the representative Western blots exhibiting the protein expression of BSEP and GAPDH in diabetic and control subjects. (F) quantification of BSEP protein levels on Western blots. For both real-time PCR and Western blotting, the expression levels of GAPDH were used to normalize the expression levels of CYP7A1 and BSEP mRNA and protein. The means and standard errors of the group values were indicated by the long and short lines, respectively. * p<0.05 with Student’s t-test.
3.8. Other key enzymes in the bile acid synthesis pathways were minimally altered in diabetic patients
In the bile acid synthesis pathways, in addition to CYP7A1, there are three other key enzymes upstream the reaction catalyzed by AKR1D1, including CYP8B1, CYP27A1 and CYP7B1 (Fig. 7A). CYP8B1 is responsible for the production of primary bile acid CA. As presented previously, hepatic CA levels were significantly reduced in diabetic patients (Fig. 4A). To determine whether CYP8B1 is dysregulated in diabetic patients, the expression levels of CYP8B1 were determined. As shown in Fig. 7B, the CYP8B1 expression levels were comparable in diabetic patients to that in control subjects.
Fig. 7. No significant alterations of other key enzymes in the bile acid synthesis pathways in diabetic patients.
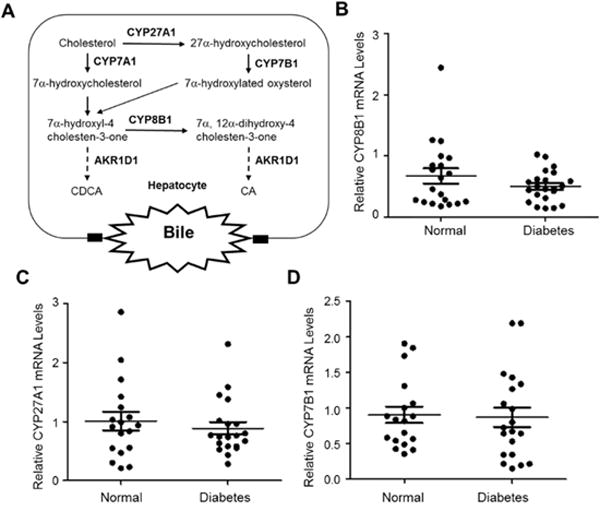
(A) in addition to CYP7A1, other key enzymes are involved in the classical and alternative bile acid synthesis pathways, including CYP8B1, CYP27A1 and CYP7B1. (B) the expression levels of CYP8B1, (B) CYP27A1 and (C) CYP7B1 mRNA in the liver of diabetic (n=20) and control subjects (n=18) were determined by TaqMan real-time PCR. The expression levels of GAPDH mRNA were used to normalize the expression levels of CYP8B1, CYP27A1 and CYP7B1. The means and standard deviations of the group values were indicated by the long and short lines, respectively. No statistical significances were detected by the Student’s t-test between normal and diabetic subjects.
In human, approximately 5% to 10% bile acids are synthesized through the alternative bile acid synthesis pathway (Russell, 2003). To determine whether reduced bile acid synthesis through the alternative pathway contributes to the reduced hepatic bile acids, the expression levels of the two key enzymes in the alternative synthesis pathway, CYP27A1 and CYP7B1, were investigated. As shown in Fig. 7C, the expression levels of CYP27A1 mRNA were minimally changed in diabetic patients. Similarly, no significant alterations in the expression levels of CYP7B1 mRNA were detected (Fig. 7D). The data indicated that bile acid synthesis through the alternative pathway was not compromised in diabetic patients.
3.9. Other hepatic bile acid uptake and efflux transporters were largely unaffected in diabetic patients
Hepatic bile acid transporters play critical roles in maintaining hepatic bile acid homeostasis. In addition to BSEP, there are other hepatic bile acid transporters involved in bile acid transport including NTCP and organic anion-transporting polypeptides (OATPs) and multidrug resistance-associated proteins (MRPs) (Fig. 8A). In the enterohepatic circulation of bile acids, hepatic uptake of bile acids are mediated mainly through NTCP and to a much less extent by OATP1B1 (Trauner and Boyer, 2003; Halilbasic et al., 2013). Under pathological conditions, such as cholestasis, hepatic bile acids can also be transported back into portal blood through basolateral efflux transporters including MRP3 and MRP4 (Halilbasic et al., 2013) (Fig. 8A). To determine whether other hepatic bile acid uptake and efflux transporters are altered in diabetic patients, the expression levels of NTCP, OATP1B1, MRP3 and MRP4 were investigated.
Fig. 8. Other hepatic bile acid uptake and efflux transporters were largely unaffected in diabetic patients.
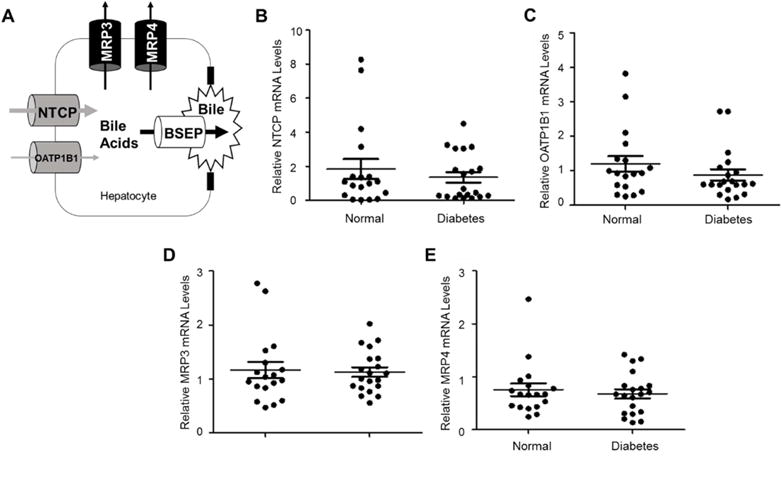
(A) in addition to BSEP, other bile acid transporters including NTCP and OATP1B1 for bile acid uptake and MRP3 and MRP4 for bile acid efflux. (B) the expression levels of NTCP, (C) OATP1B1, (D) MRP3 and (E) MRP4 mRNA in the liver of diabetic (n=20) and control subjects (n=18) were determined by TaqMan real-time PCR. The expression levels of GAPDH mRNA were used to normalize the expression levels of NTCP, OATP1B1, MRP3 and MRP4. The means and standard errors of the group values were indicated by the long and short lines, respectively. No statistical significances were detected by the Student’s t-test between normal and diabetic subjects.
For the bile acid uptake transporters, both NTCP and OATP1B1 mRNA levels were not significantly altered in diabetic patients (Fig. 8B and 8C), indicating that hepatic uptake of bile acids was not compromised in diabetic patients. Similarly, the expression levels of the basolateral efflux transporters MRP3 and MRP4 were comparable in diabetic patients to control subjects (Fig. 8D and 8E). The data thus demonstrated that hepatic bile acid uptake and efflux transporters were largely unaffected in diabetic patients.
Taken together, the investigation revealed minimal alterations in the expression of enzymes involved in bile acid synthesis including CYP7A1, CP8B1, CYP27A1 and CYP7B1, and hepatic bile acid transporters including BSEP, NTCP, OATP1B1, MRP3 and MRP4 in diabetic patients. It is thus most likely that repressed expression of AKR1D1 (Fig. 1), resulting in decreased bile acid synthesis, is responsible for the reduced hepatic bile acids (Fig. 2) in diabetic patients.
3.10. Activation of PPARα signaling repressed AKR1D1 expression in vitro in HepG2 cells
Our current understanding on transcriptional regulation of AKR1D1 is limited. We have previously reported that AKR1D1 is differentially regulated by bile acids through the mitogen-activated protein kinases/c-Jun N-terminal kinases (MAPK/JNK) signaling pathway (Valanejad et al., 2017). CDCA markedly down-regulates while CA upregulates AKR1D1. Considering that both hepatic CDCA and CA levels are reduced in diabetic patients, it is unlikely that repression of AKR1D1 expression in diabetic patients is mediated by bile acids. To uncover the potential underlying mechanisms by which AKR1D1 expression is repressed in diabetic patients, the effects of several nuclear receptor-mediated signaling pathways including FXR, LXR, PPARs and insulin on AKR1D1 expression were investigated. FXR, LXR and PPAR are three master regulators in cholesterol, bile acids, lipids, triglycerides, glucose and energy metabolisms, and have been directly linked to metabolic disease and diabetes (Ding et al., 2014; Jay and Ren, 2007). Among the three PPAR isoforms, PPARα is predominantly expressed in tissues with a high capacity for fatty acid oxidation, including the liver, whereas low levels of PPARγ expression is also detected in the liver. In addition to fatty acid oxidation, PPARα signaling also regulates transcription of multiple genes involved in glucose metabolism (Jay and Ren, 2007).
As shown in Fig. 9A, activation of FXR by synthetic agonist GW4064 had minimal effects on AKR1D1 expression, consistent with our previous report that FXR signaling was not involved in bile acids-mediated regulation of AKR1D1 (Valanejad et al., 2017). Activation of LXR signaling by synthetic agonist GW3965 resulted in a slight increase in AKR1D1 expression without reaching a statistic significance (Fig. 9B). However, activation of PPARα by PPARα-specific agonist GW7647 significantly repressed AKR1D1 mRNA expression by 56% while PPARγ activation by PPARγ-specific agonist PGZN had minimal effects (Fig. 9C). Treatment with insulin at a concentration of 100ng/ml resulted in no changes (Fig. 9D). Consistent with mRNA, AKR1D1 protein levels were significantly decreased by GW7647 treatment (Fig. 9E and 9F). It was thus concluded that AKR1D1 was regulated by PPARα signaling pathway and activation of PPARα markedly decreased AKR1D1 expression in vitro in HepG2 cells.
Fig. 9. Activation of PPARα signaling repressed AKR1D1 expression in vitro in HepG2 cells.
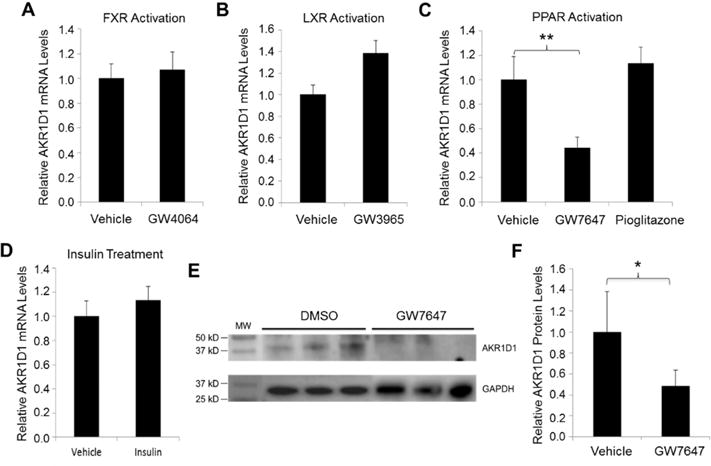
(A) HepG2 cells were treated with synthetic FXR agonist GW4064 (1μM), (B) LXR agonist GW3965 (1μM), (C) PPARα agonist GW7647 (10μM) or PPARγ agonist pioglitazone (10μM) or (D) insulin (100ng/ml) for 30h, followed by detection of AKR1D1 mRNA expression by TaqMan real-time PCR. The expression levels of GAPDH were used to normalize the expression levels of AKR1D1 for the treatments with GW4064, GW3965, GW7647 and pioglitazone. The expression levels of β-actin were used to normalize the expression levels of AKR1D1 for the treatment with insulin. (E) HepG2 cells were treated with PPARa agonist GW7647 for 48h, followed by detection of AKR1D1 protein. A representative Western blot showing protein expression of AKR1D1 and GAPDH. (F) quantification of AKR1D1 protein levels on Western blots normalized by the levels of GAPDH protein. For both real-time PCR assays and Western blot, the experiments were independently repeated at least once with each experiments having triplicates for each treatment. ** p<0.01 with one-way ANOVA followed by Tukey post-hoc test. * p<0.05 with Student’s t-test.
3.11. Activation of α repressed Akr1d1 expression in vivo in mice
To confirm the findings from human HepG2 cells, a mouse study was conducted. Mice were treated with either vehicle, PPARα agonist GW7647 or FXR agonist GW4064 as control, followed by detection of Akr1d1 mRNA and protein. GW7647 treatment markedly repressed Akr1d1 mRNA expression by 46% while FXR activation resulted in a slight increase in Akr1d1 expression without reaching a statistical significance (Fig. 10A). Consistently, significant decreases in Akr1d1 protein levels were detected in mice treated with GW7647 (Fig. 10B and 10C). Taken together, the data from in vitro in HepG2 cells and in vivo in mice established that AKR1D1 was markedly repressed by PPARα activation.
Fig. 10. Activation of PPARα signaling repressed Akr1d1 expression in vivo in mice.
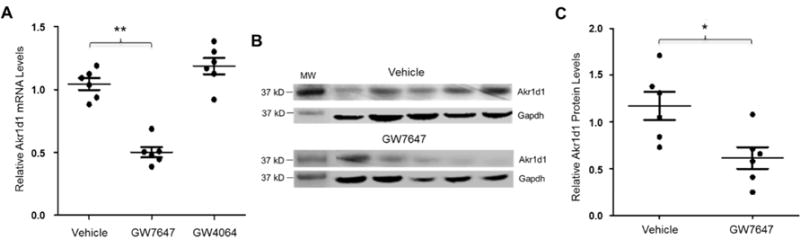
(A) three groups of mice (n=6/group) were treated with either PPARα agonist GW7647 (10mg/kg), FXR agonist GW4064 (10mg/kg) or vehicle propanediol through i.p. twice a day for 3 days. Akr1d1 mRNA levels were quantified by real-time PCR and normalized with the expression levels of Gapdh. (B) Akr1d1 protein were detected by Western blot. (C) quantification of Akr1d1 protein levels on Western blots normalized by the levels of GAPDH protein. The means and standard errors of the group values were indicated by the long and short lines, respectively. ** p<0.01 with one-way ANOVA followed by Tukey post-hoc test. * p<0.05 with Student’s t-test.
3.12. AKR1D1 promoter was significantly transrepressed by PPARα activation
To determine whether reduced expression of AKR1D1 by PPARα activation is achieved at the transcriptional level and further understand the mechanistic insights into PPARα-mediated repression, the AKR1D1 promoter was cloned and characterized in HepG2 cells with the dual luciferase assays. As expected, activation of FXRα1, FXRα2 and LXRα with FXR agonist GW4064 and LXR agonist GW3965 had minimal effects on AKR1D1 promoter activities (Fig. 11A, 11B and 11C). However, activation of PPARα with PPARα-specific agonist GW7647 significantly reduced AKR1D1 promoter transactivation while activation of PPARβ and PPARγ with PPARβ-specific agonist GW0742 or PPARγ-specific agonist PGZN had minimal effects (Fig. 11D, 11E and 11F). The data thus demonstrated that PPARα-mediated repression of AKR1D1 is at the transcriptional level (transrepression), consistent with reduced expression of AKR1D1 in vitro and in vivo by PPARα activation.
Fig. 11. AKR1D1 promoter was transrepressed by PPARα activation.
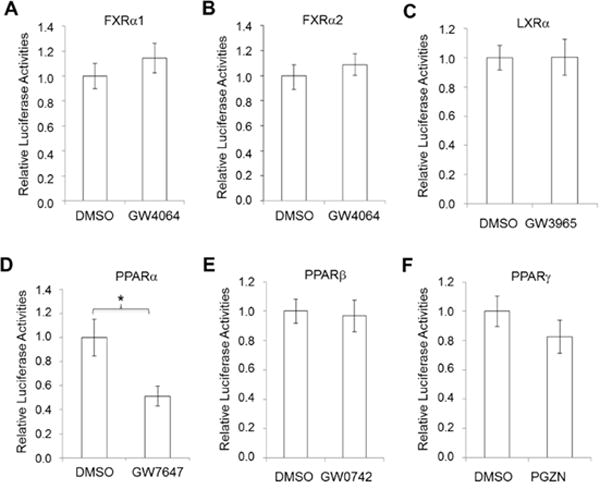
(A) AKR1D1 promoter reporter, phAKR1D1(-5kb), was co-transfected into HepG2 cells with nuclear receptor FXRα1, (B) FXRα2, (C) LXRα, (D) PPARα, (E) PPARβ or (F) PPARγ, followed by treatment with corresponding nuclear receptor agonists GW4064 (1μM) for FXRα1 and FXRα2, GW3965 (1μM) for LXRα, GW7647 (10μM) for PPARα, GW0742 for PPARβ and pioglitazone (PGZN) for PPARγ for 30h. AKR1D1 promoter transactivation levels were quantified by the dual luciferase assays. The experiments were independently repeated at least once with each experiment having triplicates for each treatment. * p<0.05 with Student’s t-test.
3.13. PPARα signaling was enhanced in diabetic patients
With the identification of PPARα signaling as a negative regulator of AKR1D1 and AKR1D1 being markedly repressed in diabetic patients, we investigated the status of PPARα signaling in diabetic patients by evaluating the expression levels of PPARα and its positively regulated target CPT1A (Napal, et al., 2005). As shown in Fig. 12A and 12B, the mRNA expression levels of PPARα were significantly increased whereas minimal alterations in FXR expression were detected in diabetic patients compared with normal control subjects. In line with the mRNA levels, PPARα proteins were augmented in diabetic patients (Fig. 12C and 12D). Furthermore, the expression levels of CPT1A, a PPARα positively regulated target, were significantly elevated in diabetic patients (Fig. 12E). The data thus revealed that PPARα signaling was enhanced in diabetic patients, which provides a mechanistic explanation for the marked repression of AKR1D1 expression in diabetic patients.
Fig. 12. PPARα signaling was enhanced in diabetes.
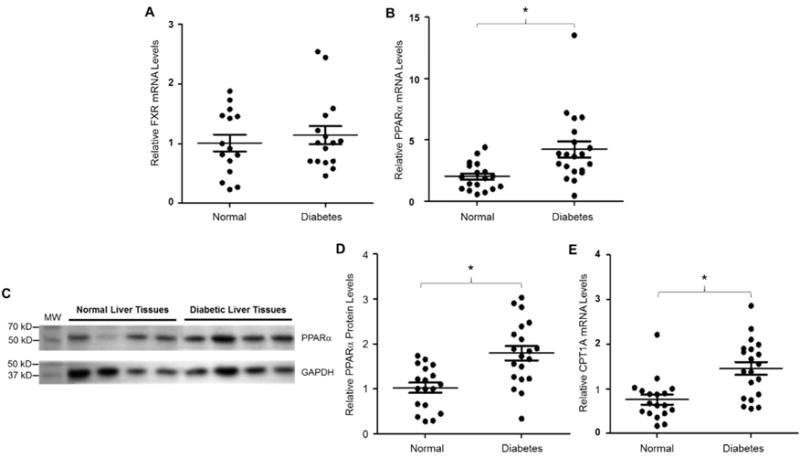
(A) hepatic expression levels of FXR and (B) PPARα mRNA were quantified by TaqMan real-time PCR in normal control (n=18) and diabetic subjects (n=20). The expression levels of GAPDH mRNA were used to normalize FXR and PPARα expression. (C) a representative Western blot showing the protein expression of PPARα and GAPDH in diabetic and control subjects. (D) quantification of PPARα protein levels on Western blots normalized by the protein levels of GAPDH. (E) hepatic CPT1A mRNA levels were detected by real-time PCR in control and diabetic subjects. The expression levels of GAPDH mRNA were used to normalize CPT1A expression. The means and standard errors of the group values were indicated by the long and short lines, respectively. * p<0.05 with Student’s t-test.
4. Discussion
As a key enzyme for both bile acid synthesis and steroid metabolism, AKR1D1 plays critical roles in maintaining bile acids and steroid hormones homeostasis, both of which are involved in regulating body metabolism and energy expenditure. In this study, we revealed that AKR1D1 expression was markedly repressed at mRNA, protein and cellular levels in diabetic patients (Figs. 1 and 2), indicating that AKR1D1 is dysregulated under diabetic condition. Repression of AKR1D1 expression has significant impact on both bile acid and steroid hormone homeostasis. Decrease in bile acid synthesis due to repression of AKR1D1 expression leads to reduction in bile acid levels. On the other hand, slowing down of steroid hormone metabolism and inactivation as a result of AKR1D1 repression leads to an increase in steroid hormone concentrations.
Indeed, consistent with repressed AKR1D1 expression, hepatic bile acid levels were markedly reduced by 53% in diabetic patients (Fig. 3). Bile acids are actively participated in regulating metabolisms and energy expenditure. Clinical studies have revealed that bile acid homeostasis is disrupted in patients with metabolic diseases or diabetes. Serum bile acids are altered in obese and/or diabetic patients (Bennion and Grundy, 1977; de Leon et al., 1978; Steiner et al., 2011; Zhang et al., 2015), and serum bile acids directly correlate with body mass index (BMI) in obese subjects (Vincent et al., 2013; Ma and Patti, 2014). Consistently, changes in bile acids are associated with diabetes in rodent models (Nervi et al., 1978; Uchida et al., 1979). Most previous studies measured serum bile acids. Due to large diurnal changes and individual variations in serum bile acids (Steiner et al., 2011), inconsistent results have been reported for total bile acids or bile acid pool sizes in diabetic patients (Bennion and Grundy, 1977; de Leon et al., 1978; Steiner et al., 2011; Zhang et al., 2015, Vincent et al., 2013). In this study, we directly measured hepatic bile acids. Compared with normal control subjects, hepatic bile acid concentrations exhibited less variations in diabetic patients (Fig. 3). The results represented the first description of hepatic bile acid alterations in diabetic patients. In line with reduced hepatic bile acids (Fig. 3), decreases in total serum bile acids, especially CDCA (33% reduction), were reported in diabetic patients when compared with non-diabetic control subjects (Brufau et al., 2010).
Under the physiological conditions, hepatic bile acid concentrations are maintained mainly through FXR-mediated feedback regulation of CYP7A1, which catalyzes the rate-limiting reaction for bile acid synthesis, and feed-forward regulation of BSEP, which mediates the rate-limiting step for enterohepatic circulation of bile acids (Chiang, 2013; Trauner and Boyer, 2003). In studies with rodent models, Cyp7a1 expression was upregulated under diabetic conditions (Li et al., 2012). Currently, no studies have directly investigated the CYP7A1 expression in diabetic patients. In this study, we found that CYP7A1 expression at both mRNA and protein levels was not significantly altered in diabetic patients (Fig. 4A, 4B and 4C). Species difference may explain the discrepancy. Previous studies with diabetic rodent models showed that BSEP expression was minimally changed with diabetes (van Waarde et al., 2002). In this study, BSEP expression was reduced at the mRNA levels but comparable at protein levels in diabetic patients (Fig. 4D, 4E and 4F), indicating a post-transcriptional regulation of BSEP under the diabetic conditions. The results demonstrated that CYP7A1 and BSEP expression is largely maintained in diabetic patients. Thus, reduced hepatic bile acids were not resulted from alterations in the expression of CYP7A1 and BSEP. In addition to CP7A1 and BSEP, we also investigated the expression of other enzymes involved in bile acid synthesis and hepatic bile acid transport in diabetic patients, including CYP8B1, CYP27A1, CYP7B1, NTCP, OATPB1, MRP-3 and MRP-4. None of those genes exhibited significant alterations in expression in diabetic patients (Fig. 5 and 6). Taken together, the results suggested that repressed expression of AKR1D1, which leads to decrease in bile acid synthesis, was most likely responsible for reduced hepatic bile acids in diabetic patients. The finding also indicated that in addition to CYP7A1, AKR1D1 also plays an important role in regulating bile acid synthesis.
Whether dysregulation of bile acids contributes to the development of diabetes or is an innocent bystander of the diseases is still debatable. However, accumulating evidence supports a view that bile acids are the active players in the development of the disease and increasing bile acid synthesis improves diabetic conditions. First, bile acids serve as hormone-like molecules to regulate lipids, glucose and energy metabolism through FXR and Takeda G-protein receptor 5 (TGR5), and activation of FXR and/or TGR5 improves diabetic conditions (Prawitt et al., 2011; Thomas et al., 2009; Mudaliar et al., 2013). Second, bile acid sequestrants improve insulin resistance and lower glucose levels in diabetic patients by blocking reabsorption of bile acids in the small intestine and increasing bile acid synthesis in the liver (Prawitt et al., 2014; Out et al., 2012). Third, diversion of enterohepatic bile acids circulation by surgery improves diabetic conditions with increased bile acid synthesis and circulation (Flynn et al., 2015; Ferrannini et al., 2015; Kaválková et al., 2016; Kaska et al., 2016). Therefore, dysregulation of AKR1D1, which leads to reduced bile acid synthesis, may contribute to the pathogenesis of diabetes.
AKR1D1 is the only reductase in humans capable of catalyzing 5β-reduction of steroid hormones and metabolizes glucocorticoids (cortisol and cortisone) into their inactive metabolites. Therefore, AKR1D1 plays an important role in controlling the glucocorticoid levels. In addition to their anti-inflammatory activities, glucocorticoids are key regulators of carbohydrate, lipid metabolism and energy balance (Vegiopoulos and Herzig, 2007; de Guia et al., 2014). Indeed, high percentages of patients with diabetes have elevated glucocorticoid levels with characteristic features of glucocorticoid excess (Lee et al., 1999; Rose and Herzig, 2013). Exogenous hypercortisolism resulting from short or long-term exposure of glucocorticoids is associated with increased odd ratio for development of hyperglycemia and new-onset diabetes (Panthakalam et al., 2004; Petersons et al., 2013). Therefore, elevation of glucocorticoids endogenously or exogenously is a risk factor for developing metabolic disease/diabetes. Repression of AKR1D1 expression, which leads to reduced metabolism and inactivation of glucocorticoids, may contribute to the pathogenesis of diabetes.
Currently, our understanding on transcriptional regulation of AKR1D1 under physiological and diabetic conditions is limited. In our previous study, we reported that AKR1D1 was differentially regulated by bile acids (Valanejad et al., 2017). In this study, we found that activation of FXR, LXR and PPARγ had minimal effects on AKR1D1 expression. Similarly, insulin treatment resulted in no significant changes in AKR1D1 expression. However, activation of PPARα markedly decreased AKR1D1 expression in vitro in HepG2 cells and in vivo in mice. The data thus demonstrated that AKR1D1 was transcriptionally regulated by PPARα and activation of PPARα signaling repressed AKR1D1 expression. To explore the possible mechanisms by which AKR1D1 was repressed in diabetic patients, we investigate the status of PPARα signaling in diabetic patients. It was found that PPARα signaling was enhanced in diabetic patients with significantly elevated expression of PPARα and its positively regulated target CPT1A. Thus, it was speculated that enhanced PPARα signaling activation contributed to the repression of AKR1D1 expression in diabetic patients. In line with our findings and speculation, PPARα signaling was upregulated in diabetic rodent models (Asayama et al., 1999; Memon et al., 2000). Furthermore, endogenous ligands that activate PPARα with the high potency, including saturated and unsaturated fatty acids, were often elevated in patients with metabolic disease and diabetes (Liu et al., 2010; Bergman and Ader, 2000).
5. Conclusion
AKR1D1 was down-regulated in diabetic patients, most likely through enhanced PPARα signaling. Dysregulation of AKR1D1 resulted in disturbance of bile acid and steroid hormone homeostasis, which may contribute to the pathogenesis of diabetes. Restoring bile acid and steroid hormone homeostasis by modulating AKR1D1 expression may represent a new approach to develop therapies for diabetes.
Supplementary Material
Highlights.
AKR1D1 expression was markedly repressed in diabetic patients
Hepatic bile acids were significantly reduced in diabetic patients
AKR1D1 was transcriptionally down-regulated by PPARα signaling
PPARα signaling was enhanced in diabetic patients
Acknowledgments
This work was supported by the National Institutes of Health (NIH) Grant R01DK087755. A.F. is supported by NIH grants GM101599 and UH3 TR000963. C.N. is partially supported by the pre-doctoral fellowship from American Foundation for Pharmaceutical Education. We thank Dr. Bingfang Yan (University of Rhode Island) for sharing instruments in his laboratory with us and kindly providing expression plasmids for human FXR, LXR and PPAR. Instrumental supports from the RI-INBRE Core Facility in the College of Pharmacy, University of Rhode Island are greatly appreciated, which is supported by an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the National Institutes of Health under grant P20 GM103430-13. We thank Dr. Matthew Stoner for providing expression plasmid of human FXRα2.
Abbreviations used
- AKR1D1
aldo-keto reductase family 1, member D1
- BMI
body mass index
- BSEP
bile salt export pump
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CPT1A
carnitine palmitoyltransferase-1A
- CYP7A1
cholesterol 7α-hydroxylase or cytochrome P450 family 7 subfamily A member 1
- CYP7B1
cytochrome P450 family 7 subfamily B member 1
- CYP8B1
sterol 12α-hydroxylase or CYP family 8 subfamily B member 1
- CYP27A1
cytochrome P450 family 27 subfamily A member 1
- DCA
deoxycholic acid
- FXR
farnesoid x receptor
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- LCA
lithocholic acid
- LXR
liver x receptor
- LC-MS/MS
liquid chromatography tandem-mass spectrometry
- LLOD
low limit of quantification
- LLOQ
low limit of detection
- MRP3
multidrug resistance-associated protein 3
- MRP4
multidrug resistance-associated protein 4
- NADPH
nicotinamide adenine dinucleotide phosphate
- NTCP
sodium-taurocholate cotransporting polypeptide
- OATP1B1
organic anion-transporting polypeptide member 1B1
- PGZN
pioglitazone
- PPARα
peroxisome proliferator-activated receptor alpha
- SEM
standard error of mean
- TGR5
Takeda G-protein receptor 5
- UDCA
ursodeoxycholic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
Authors have nothing to declare.
References
- Alnouti Y, Csanaky IL, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asayama K, Sandhir R, Sheikh FG, Hayashibe H, Nakane T, Singh I. Increased peroxisomal fatty acid beta-oxidation and enhanced expression of peroxisome proliferator-activated receptor-alpha in diabetic rat liver. Mol Cell Biochem. 1999;194:227–234. doi: 10.1023/a:1006930513476. [DOI] [PubMed] [Google Scholar]
- Bellet MM, Masri S, Astarita G, Sassone-Corsi P, Della Fazia MA, Servillo G. Histone Deacetylase SIRT1 Controls Proliferation, Circadian Rhythm, and Lipid Metabolism during Liver Regeneration in Mice. J Biol Chem. 2016;291:23318–23329. doi: 10.1074/jbc.M116.737114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennion LJ, Grundy SM. Effects of diabetes mellitus on cholesterol metabolism in man. N Engl J Med. 1977;296:1365–1371. doi: 10.1056/NEJM197706162962401. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab. 2000;11:351–356. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- Biswas S, Tapryal N, Mukherjee R, Kumar R, Mukhopadhyay CK. Insulin promotes iron uptake in human hepatic cell by regulating transferrin receptor-1 transcription mediated by hypoxia inducible factor-1. Biochim Biophys Acta. 2013;1832:293–301. doi: 10.1016/j.bbadis.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Brufau G, Bahr MJ, Staels B, Claudel T, Ockenga J, Böker KH, Murphy EJ, Prado K, Stellaard F, Manns MP, Kuipers F, Tietge UJ. Plasma bile acids are not associated with energy metabolism in humans. Nutr Metab (Lond) 2010;7:73. doi: 10.1186/1743-7075-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan O, Inouye K, Riddell MC, Vranic M, Matthews SG. Diabetes and the hypothalamo-pituitary-adrenal (HPA) axis. Minerva Endocrinol. 2003;28:87–102. [PubMed] [Google Scholar]
- Chen M, Penning TM. 5β-Reduced steroids and human Δ(4)-3-ketosteroid 5β-reductase (AKR1D1) Steroids. 2014;83:17–26. doi: 10.1016/j.steroids.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Song X, Valanejad L, Vasilenko A, More V, Qiu X, Chen W, Lai Y, Slitt A, Stoner M, Yan B, Deng R. Bile salt export pump is dysregulated with altered farnesoid X receptor isoform expression in patients with hepatocellular carcinoma. Hepatology. 2013;57:1530–1541. doi: 10.1002/hep.26187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY. Bile acid metabolism and signaling. Compr Physiol. 2013;3:1191–212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guia RM, Rose AJ, Herzig S. Glucocorticoid hormones and energy homeostasis. Horm Mol Biol Clin Investig. 2014;19:117–128. doi: 10.1515/hmbci-2014-0021. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Pang M, Chaudhry R, Duan K, Longato L, Carter J, Ouh J, Wands JR. Peroxisome proliferator-activated receptor agonist treatment of alcohol-induced hepatic insulin resistance. Hepatol Res. 2011;41:386–398. doi: 10.1111/j.1872-034X.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MP, Ferenderes R, Carulli N. Bile lipid composition and bile acid pool size in diabetes. Am J Dig Dis. 1978;23:710–716. doi: 10.1007/BF01072357. [DOI] [PubMed] [Google Scholar]
- Ding L, Pang S, Sun Y, Tian Y, Yu L, Dang N. Coordinated Actions of FXR and LXR in Metabolism: From Pathogenesis to Pharmacological Targets for Type 2 Diabetes. Int J Endocrinol. 2014;2014:751859. doi: 10.1155/2014/751859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, Camastra S, Astiarraga B, Nannipieri M, Castro-Perez J, Xie D, Wang L, Chakravarthy M, Haeusler RA. Increased Bile Acid Synthesis and Deconjugation After Biliopancreatic Diversion. Diabetes. 2015;64:3377–3385. doi: 10.2337/db15-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn CR, Albaugh VL, Cai S, Cheung-Flynn J, Williams PE, Brucker RM, Bordenstein SR, Guo Y, Wasserman DH, Abumrad NN. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun. 2015;6:7715. doi: 10.1038/ncomms8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizzle WE, Gunter EW, Sexton KC, Bell WC. Quality management of biorepositories. Biopreserv Biobank. 2015;13:183–194. doi: 10.1089/bio.2014.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58:155–168. doi: 10.1016/j.jhep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Nishida S, Xu M, Makishima M, Xie W. PXR prevents cholesterol gallstone disease by regulating biosynthesis and transport of bile salts. Gastroenterology. 2011;140:2095–2106. doi: 10.1053/j.gastro.2011.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay MA, Ren J. Peroxisome proliferator-activated receptor (PPAR) in metabolic syndrome and type 2 diabetes mellitus. Curr Diabetes Rev. 2007;3:33–39. doi: 10.2174/157339907779802067. [DOI] [PubMed] [Google Scholar]
- Jin Y, Chen M, Penning TM. Rate of steroid double-bond reduction catalysed by the human steroid 5β-reductase (AKR1D1) is sensitive to steroid structure: implications for steroid metabolism and bile acid synthesis. Biochem J. 2014;462:163–171. doi: 10.1042/BJ20140220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaska L, Sledzinski T, Chomiczewska A, Dettlaff-Pokora A, Swierczynski J. Improved glucose metabolism following bariatric surgery is associated with increased circulating bile acid concentrations and remodeling of the gut microbiome. World J Gastroenterol. 2016;22:8698–8719. doi: 10.3748/wjg.v22.i39.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaválková P, Mráz M, Trachta P, Kloučková J, Cinkajzlová A, Lacinová Z, Haluzíková D, Beneš M, Vlasáková Z, Burda V, Novák D, Petr T, Vítek L, Pelikánová T, Haluzík M. Endocrine effects of duodenal-jejunal exclusion in obese patients with type 2 diabetes mellitus. J Endocrinol. 2016;231:11–22. doi: 10.1530/JOE-16-0206. [DOI] [PubMed] [Google Scholar]
- Kondo KH, Kai MH, Setoguchi Y, Eggertsen G, Sjöblom P, Setoguchi T, Okuda KI, Björkhem I. Cloning and expression of cDNA of human delta 4-3-oxosteroid 5 beta-reductase and substrate specificity of the expressed enzyme. Eur J Biochem. 1994;219:357–363. doi: 10.1111/j.1432-1033.1994.tb19947.x. [DOI] [PubMed] [Google Scholar]
- Kroetz DL, Yook P, Costet P, Bianchi P, Pineau T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J Biol Chem. 1998;273:31581–31589. doi: 10.1074/jbc.273.47.31581. [DOI] [PubMed] [Google Scholar]
- Lee ZS, Chan JC, Yeung VT, Chow CC, Lau MS, Ko GT, Li JK, Cockram CS, Critchley JA. Plasma insulin, growth hormone, cortisol, and central obesity among young Chinese type 2 diabetic patients. Diabetes Care. 1999;22:1450–1457. doi: 10.2337/diacare.22.9.1450. [DOI] [PubMed] [Google Scholar]
- Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, Erickson SK, Chiang JY. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem. 2012;287:1861–1873. doi: 10.1074/jbc.M111.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li Y, Guan C, Li K, Wang C, Feng R, Sun C. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2817–2825. doi: 10.1016/j.jchromb.2010.08.035. [DOI] [PubMed] [Google Scholar]
- Ma H, Patti ME. Bile acids, obesity, and the metabolic syndrome. Best Pract Res Clin Gastroenterology. 2014;28:573–583. doi: 10.1016/j.bpg.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013;30:1447–57. doi: 10.1007/s11095-013-0986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology. 2000;141:4021–4031. doi: 10.1210/endo.141.11.7771. [DOI] [PubMed] [Google Scholar]
- Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–582. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- Napal L, Marrero PF, Haro D. An intronic peroxisome proliferator-activated receptor-binding sequence mediates fatty acid induction of the human carnitine palmitoyltransferase 1A. J Mol Biol. 2005;354:751–759. doi: 10.1016/j.jmb.2005.09.097. [DOI] [PubMed] [Google Scholar]
- Nervi FO, Severín CH, Valdivieso VD. Bile acid pool changes and regulation of cholate synthesis in experimental diabetes. Biochim Biophys Acta. 1978;529:212–223. doi: 10.1016/0005-2760(78)90064-4. [DOI] [PubMed] [Google Scholar]
- Out C, Groen AK, Brufau G. Bile acid sequestrants: more than simple resins. Curr Opin Lipidol. 2012;23:43–55. doi: 10.1097/MOL.0b013e32834f0ef3. [DOI] [PubMed] [Google Scholar]
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J. 2004;49:139–141. doi: 10.1177/003693300404900407. [DOI] [PubMed] [Google Scholar]
- Perez A, Jansen-Chaparro S, Saigi I, Bernal-Lopez MR, Miñambres I, Gomez-Huelgas R. Glucocorticoid-induced hyperglycemia. J Diabetes. 2014;6:9–20. doi: 10.1111/1753-0407.12090. [DOI] [PubMed] [Google Scholar]
- Petersons CJ, Mangelsdorf BL, Jenkins AB, Poljak A, Smith MD, Greenfield JR, Thompson CH, Burt MG. Effects of low-dose prednisolone on hepatic and peripheral insulin sensitivity, insulin secretion, and abdominal adiposity in patients with inflammatory rheumatologic disease. Diabetes Care. 2013;36:2822–2829. doi: 10.2337/dc12-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, Lombardi G, Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl 1):77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- Prawitt J, Caron S, Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diab Rep. 2011;11:160–166. doi: 10.1007/s11892-011-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prawitt J, Caron S, Staels B. Glucose-lowering effects of intestinal bile acid sequestration through enhancement of splanchnic glucose utilization. Trends Endocrinol Metab. 2014;25:235–244. doi: 10.1016/j.tem.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Herzig S. Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol. 2013;380:65–78. doi: 10.1016/j.mce.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- Song X, Chen Y, Valanejad L, Kaimal R, Yan B, Stoner M, Deng R. Mechanistic insights into isoform-dependent and species-specific regulation of bile salt export pump by farnesoid X receptor. J Lipid Res. 2013;54:3030–3044. doi: 10.1194/jlr.M038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vasilenko A, Chen Y, Valanejad L, Verma R, Yan B, Deng R. Transcriptional dynamics of bile salt export pump during pregnancy: mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology. 2014;60:1993–2007. doi: 10.1002/hep.27171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner C, Othman A, Saely CH, Rein P, Drexel H, von Eckardstein A, Rentsch KM. Bile acid metabolites in serum: intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS One. 2011;6:e25006. doi: 10.1371/journal.pone.0025006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- Uchida K, Takase H, Kadowaki M, Nomura Y, Matsubara T, Takeuchi N. Altered bile acid metabolism in alloxan diabetic rats. Jpn J Pharmacol. 1979;29:553–562. doi: 10.1254/jjp.29.553. [DOI] [PubMed] [Google Scholar]
- Valanejad L, Nadolny C, Shiffka S, Chen Y, You S, Deng R. Differential Feedback Regulation of Δ4-3-Oxosteroid 5β-Reductase Expression by Bile Acids. PLoS One. 2017;12:e0170960. doi: 10.1371/journal.pone.0170960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Waarde WM, Verkade HJ, Wolters H, Havinga R, Baller J, Bloks V, Müller M, Sauer PJ, Kuipers F. Differential effects of streptozotocin-induced diabetes on expression of hepatic ABC-transporters in rats. Gastroenterology. 2002;122:1842–1852. doi: 10.1053/gast.2002.33582. [DOI] [PubMed] [Google Scholar]
- Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61. doi: 10.1016/j.mce.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Vincent RP, Omar S, Ghozlan S, Taylor DR, Cross G, Sherwood RA, Fandriks L, Olbers T, Werling M, Alaghband-Zadeh J, Roux CW. Higher circulating bile acid concentrations in obese patients with type 2 diabetes. Ann Clin Biochem. 2013;50:360–364. doi: 10.1177/0004563212473450. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li M, Zhan L, Lu X, Liang L, Su B, Sui H, Gao Z, Li Y, Liu Y, Wu B, Liu Q. Plasma metabolomic profiling of patients with diabetes-associated cognitive decline. PLoS One. 2015;10:e0126952. doi: 10.1371/journal.pone.0126952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.