

Abstract
Proteins that bind to DNA depending on its methylation status play an important role in methylation‐mediated regulation of gene expression. Using a variety of genomics and proteomics approaches, we identify zinc finger and BTB domain‐containing protein 2 (ZBTB2) as a reader of unmethylated DNA in mouse embryonic stem cells. ZBTB2 preferentially binds to CpG island promoters, where it acts as a transcriptional activator. The binding of ZBTB2 to its targets is direct and independent of two other zinc finger proteins, ZBTB25 and ZNF639, which we show to interact with ZBTB2. Our data suggest an anticorrelation between ZBTB2 DNA binding and DNA methylation, indicating that ZBTB2‐binding dynamics in vivo are sensitive to differential DNA methylation. ZBTB2 is intricately interwoven with DNA methylation, as we find not only that its binding to DNA is methylation sensitive, but also that ZBTB2 regulates the turnover of methylated DNA. In ZBTB2 knockout cells, several pluripotency factors are upregulated, inducing a delay in differentiation. We propose that ZBTB2 is a novel DNA methylation‐sensitive transcription factor that regulates cellular differentiation.
Keywords: differentiation, DNA methylation, mouse embryonic stem cells, ZBTB2
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation; Stem Cells
Introduction
Epigenetic modifications are alterations in the interpretation of the genetic information which cause a heritable phenotype without changing the actual DNA sequence. DNA methylation is an important epigenetic modification that takes place at the level of the DNA nucleosides themselves. In vertebrates, it comprises the coupling of a methyl group (CH3) to the 5′ position of the pyrimidine ring of cytosine, resulting in methylcytosine (meC). This reaction is catalysed by DNA methyltransferases (DNMTs): DNMT1 is important for maintenance methylation, whereas DNMT3A and DNMT3B are involved in de novo DNA methylation. DNMT3L lacks inherent DNA methylation activity, but is an important regulatory factor that forms heterodimers with DNMT3A and DNMT3B 1. Impaired activity of the DNMTs leads to loss of meC over several cell divisions, a process called passive DNA demethylation. Alternatively, DNA demethylation can take place through active mechanisms, in which the family of ten‐eleven translocation (TET) methylcytosine dioxygenases plays a key role. These proteins catalyse the oxidation of meC into hydroxymethylcytosine (hmC) 2, which can then be further processed in a variety of ways to yield unmethylated DNA 3. DNMTs and TETs are thus central components of the dynamic DNA methylation network.
Although DNA methylation can occur in other contexts, it usually takes place on cytosines that are followed by a guanine, constituting a self‐complementary CpG dinucleotide. The frequency of CpGs in the genome is much lower than expected by chance, largely due to the mutability of methylated cytosines 4. In contrast, there are short regions with high CpG context, known as CpG islands, which are estimated to occur in around 60% of mouse gene promoters 5. While most CpG sites in the adult vertebrate genome are methylated, the majority of CpG islands remain unmethylated 4. Methylation of CpG island promoters can regulate transcription of associated genes in cis. Traditionally, DNA methylation of promoters has been linked to gene silencing, although recent research has nuanced this view 6.
Proteins that bind to DNA depending on its methylation status, so‐called readers of unmethylated or methylated DNA, are thought to play an important role in DNA methylation‐mediated gene expression. Quantitative mass spectrometry‐based proteomics has emerged as a powerful tool to identify such readers 7, giving rise to the challenge of elucidating the biological mechanisms through which these proteins regulate gene expression. These mechanisms include, but are probably not limited to, recruitment of writers or erasers of histone modifications 8, 9, nucleosome repositioning 10 and the abolishment of sequence‐specific binding as a consequence of methylation 11.
One putative reader of unmethylated DNA is the zinc finger and BTB domain‐containing protein 2 (ZBTB2) 7, 12. ZBTB2 is a protein family member of KAISO and KAISO‐like proteins, which have previously been shown to bind to methylated DNA and play a role in gene regulation 13. Interestingly, the three zinc finger domains of ZBTB2 are clearly distinct from the evolutionarily conserved methyl‐CpG‐binding zinc fingers of KAISO and KAISO‐like proteins 14 (Fig EV1A and B), suggesting that their sensitivity to DNA methylation may be different.
Figure EV1. Genomic localisation of ZBTB2 in mouse embryonic stem cells in vivo (related to Fig 1).
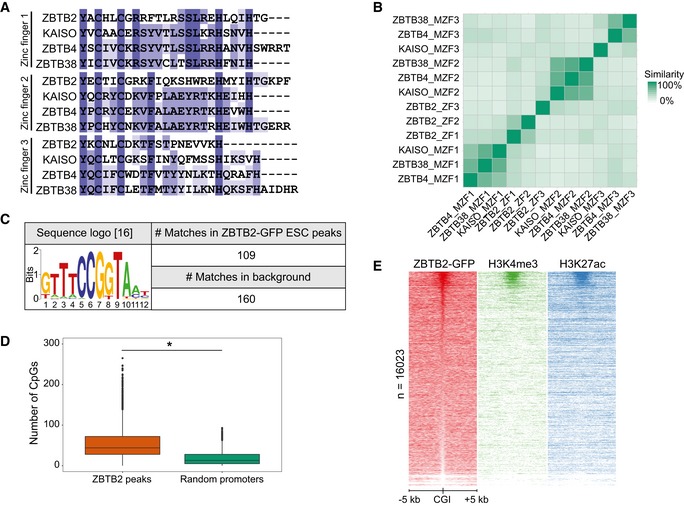
- Comparison of the amino acid sequences of the zinc finger domains of ZBTB2 and the evolutionarily conserved methyl‐CpG‐binding zinc fingers of KAISO and KAISO‐like proteins.
- Matrix of the percentage similarity between each of the zinc finger domains depicted in (A). (M)ZF = (methyl)zinc finger.
- Sequence logo of a recently reported ZBTB2‐binding motif 16 and its occurrence in the called ZBTB2 ChIP‐seq peaks in ESCs and a background consisting of all mouse CGIs.
- The number of CpG dinucleotides counted in the sequences of the ZBTB2 ChIP‐seq peaks in ESCs and in a set of random promoters (n = 4,072) of the same average length (*P < 2.2e‐16, Welch t‐test). Box: median (central line), first and third quartile (box limits); whiskers: 1.5 × interquartile range.
In this study, we used a combination of genomics and proteomics approaches to characterise the genomic localisation and biological function of the zinc finger protein ZBTB2 in mouse embryonic stem cells (ESCs). We found that ZBTB2 preferentially binds to active CpG island promoters in vivo, supporting its role as a reader of unmethylated DNA. DNA methylation seems to at least partially regulate ZBTB2‐binding in vivo, as we found ZBTB2‐binding dynamics to anticorrelate with differential DNA methylation levels. These observations are supported by in vitro assays, which show direct recruitment of ZBTB2 to unmethylated DNA. In turn, ZBTB2 affects DNA methylation through regulation of the TET enzymes, thereby also influencing its own recruitment to DNA. Lastly, our data indicate that ZBTB2 acts mostly as a gene activator and regulates genes that are important for the exit from pluripotency. We hence propose that ZBTB2 is a DNA methylation‐sensitive transcription factor that is involved in cellular differentiation and that influences its own recruitment by tuning the DNA methylation machinery.
Results and Discussion
ZBTB2 binds at active CpG island promoters in mouse embryonic stem cells in vivo
To determine the genome‐wide in vivo DNA binding sites of the putative unmethylated DNA reader ZBTB2, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP‐seq). To this end, we generated a mouse ESC line with a stably integrated bacterial artificial chromosome (BAC) transgene encoding the Zbtb2 gene fused to a C‐terminal green fluorescent protein (GFP) tag under the control of its endogenous promoter 15. Through ChIP‐seq analysis with an antibody against GFP, we identified ± 4,000 ZBTB2‐binding sites in ESCs. Genomic localisation of these peaks revealed that the majority of the ZBTB2‐binding sites (81%) is located within 5 kb of a transcription start site (TSS) (Fig 1A). A more detailed analysis of the genomic distribution of these peaks confirmed the preferential binding of ZBTB2 to TSSs and, in addition, showed that most of these TSSs contain a CpG island (CGI) (Fig 1B). Motif enrichment analysis showed that a recently reported ZBTB2‐binding motif 16 was not found to be enriched in our ZBTB2 peaks in ESCs compared to all CGIs (Fig EV1C), suggesting that in vitro and in vivo binding sites may differ. In addition, de novo motif searches in our ChIP‐seq data set did not identify any clear consensus motif for binding of ZBTB2, but did indicate that one or multiple CpG dinucleotides are usually present. To substantiate this finding, we compared the number of CpG dinucleotides at ZBTB2‐binding sites to a set of random promoters of the same average length and found that ZBTB2 peaks indeed contain significantly more CpGs (Fig EV1D). However, not all TSSs or even CGIs are bound by ZBTB2, suggesting that CpG dinucleotides are important but not sufficient for ZBTB2 recruitment (Figs 1C and EV1E). In order to identify other factors that determine ZBTB2 binding, we examined whether ZBTB2 peaks are enriched for particular histone marks. This analysis revealed that ZBTB2 primarily interacts with TSSs and CGIs that are marked by H3K4me3 and H3K27ac. Since these histone marks are associated with active promoters 17, 18 (Figs 1C and EV1E), our data thus demonstrate that ZBTB2 preferentially binds to active CpG island promoters in ESCs in vivo.
Figure 1. Genomic localisation of ZBTB2 in mouse embryonic stem cells in vivo .

- Histogram depicting the genomic localisation relative to a TSS of the called ZBTB2 ChIP‐seq peaks in ESCs.
- Genomic distribution of the called ZBTB2 ChIP‐seq peaks in ESCs.
ZBTB2‐binding dynamics anticorrelate with differential DNA methylation in vivo
Since most CGIs are hypomethylated 4, our finding that ZBTB2 binding is enriched at CGIs in vivo is in line with ZBTB2 being a reader of unmethylated DNA. To examine whether ZBTB2 occupancy is influenced by DNA methylation in vivo, we altered global DNA methylation levels in two ways. Firstly, we induced global DNA hypomethylation in ZBTB2‐GFP BAC ESCs by adding two small‐molecule kinase inhibitors targeting MEK and GSK3β to the culture medium (2i) 19. Secondly, we differentiated ZBTB2‐GFP BAC ESCs into neural progenitor cells (NPCs), thereby changing the DNA methylation landscape in a cell‐type‐specific manner 20. ChIP‐seq analyses on these cell lines revealed that genome‐wide ZBTB2 binding increases after addition of 2i and decreases upon differentiation into NPCs (Fig 2A). Importantly, ZBTB2 protein levels remain within the same range for all these conditions (Fig EV2A and B), even if mRNA levels seem to be more variable upon addition of 2i (Fig EV2C), indicating that these binding dynamics are not solely caused by changes in abundance. To investigate the extent to which the observed dynamics in ZBTB2 binding are related to changes in DNA methylation, we used publicly available methylation profiles from ESCs grown in serum/LIF or in complete 2i conditions 21 or ESCs and NPCs 20 as a proxy for genome‐wide DNA methylation levels in our cell lines. We took the union of all ZBTB2 peaks that were called in either one of our ChIP‐seq experiments, grouped them according to their genomic annotation, and then compared the difference in DNA methylation as predicted by the methylation profiles to the fold change in ZBTB2 binding as determined by ChIP‐seq (Fig EV2D and E). These analyses suggest that on average, sites where the most dynamics in ZBTB2 binding occur also show the largest change in DNA methylation. To validate these findings, we selected a couple of loci containing a CGI and performed ChIP–qPCR and methylated DNA immunoprecipitation (MeDIP)–qPCR to measure ZBTB2 and DNA methylation levels, respectively (Figs 2B and C, and EV2F and G). Also in our cell lines, we observed an anticorrelation between ZBTB2 binding on the one hand and DNA methylation on the other, suggesting that ZBTB2‐binding dynamics in vivo are sensitive to differential DNA methylation.
Figure 2. Comparison of ZBTB2 occupancy and differential DNA methylation in vivo .
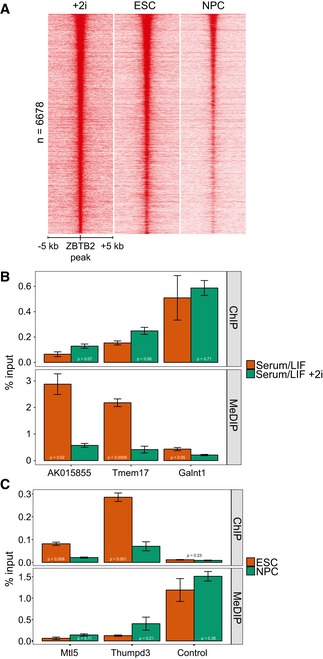
-
AHeat map showing the ChIP‐seq read density (normalised on RPKM) for ZBTB2 in ZBTB2‐GFP BAC ESCs cultured in serum/LIF conditions (middle) or serum/LIF + 2i conditions (left), and in ZBTB2‐GFP BAC NPCs (right), centred on the union of the ZBTB2 peaks in these three conditions.
-
B, CComparisons of ZBTB2 occupancy (assessed by ChIP–qPCR) and DNA methylation levels (measured by MeDIP–qPCR) at three loci (see Fig EV2F and G), between ZBTB2‐GFP BAC ESCs in serum/LIF and serum/LIF + 2i conditions (B) or between ZBTB2‐GFP BAC ESC and NPCs (C). Data are depicted as mean ± SEM of three biological replicates (P = as determined by Welch t‐test).
Figure EV2. Comparison of ZBTB2 occupancy and differential DNA methylation (related to Fig 2).
-
A, BWestern blot analyses of endogenous and GFP‐tagged ZBTB2 levels in ZBTB2‐GFP BAC ESCs in serum/LIF or serum/LIF + 2i conditions (A) or ZBTB2‐GFP BAC ESCs or NPCs (B). Histone H3 was used as a loading control. NE = nuclear extract (nuclear soluble fraction), NP = nuclear pellet (chromatin‐bound fraction).
-
CqRT–PCR analysis of Zbtb2 levels in ZBTB2‐GFP BAC ESCs cultured without or with 2i. Data are depicted as mean ± SEM of three biological replicates (*P = 0.02, Welch t‐test).
-
D, EComparison of ZBTB2‐binding dynamics and differential DNA methylation 20, 21 at the union of ZBTB2 peaks that were called in our three ChIP‐seq experiments, presented as the average values for each genomic category. ZBTB2‐binding dynamics (plotted on the x‐axis) are calculated as the fold change in RPKM values obtained from ZBTB2‐GFP ChIP‐seq in +2i versus serum/LIF conditions (D), or in NPCs versus ESCs (E). Differential DNA methylation (plotted on the y‐axis) is the difference in average methylation status between +2i and serum/LIF (D) or NPCs and ESCs (E).
-
F, GUCSC genome browser views of genomic regions that were used for locus‐specific analysis of ZBTB2 binding and DNA methylation (Fig 2B and C). All loci except the negative control locus contain a CGI. qPCR primers (Appendix Table S5) were designed to amplify a 100‐ to 200‐bp region within the dashed lines. BS‐seq = bisulphite sequencing 21, 45.
Source data are available online for this figure.
ZBTB2 recruits a zinc finger module to unmethylated DNA
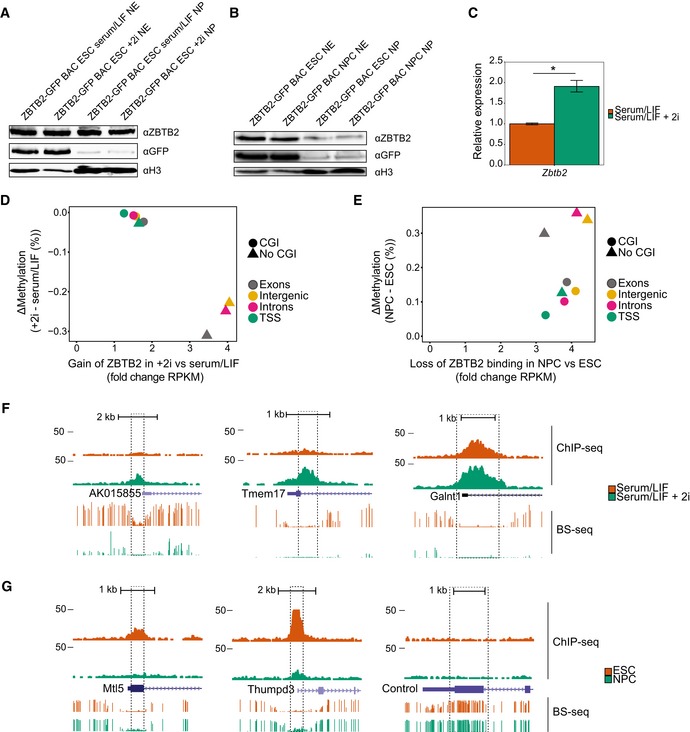
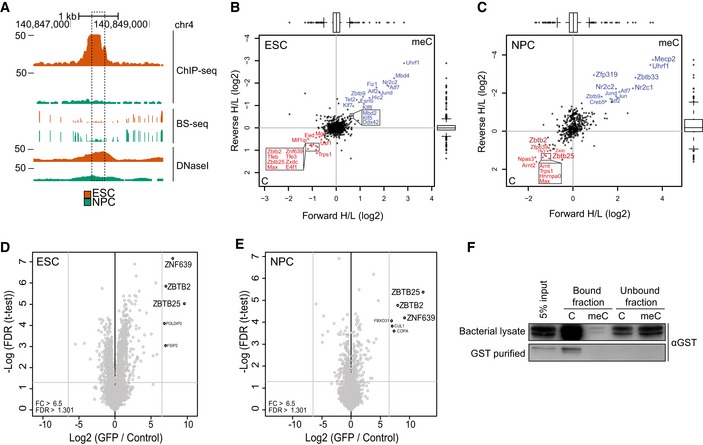
We next performed in vitro binding assays to further elucidate the mechanisms behind ZBTB2 binding to DNA. For this purpose, we selected a locus containing a CGI on the mouse genome where we saw a strong anticorrelation between ZBTB2 occupancy and DNA methylation in ESCs compared to NPCs, while DNase I hypersensitivity data suggest that this genomic region is accessible in both cell types in vivo (Figs 3A and EV3A). To test whether the DNA binding properties of ZBTB2 change upon differentiation into a different cell type, we performed DNA pull‐downs with this genomic sequence containing either unmethylated or methylated CpGs, and using nuclear protein extracts from either ESCs or NPCs. These experiments revealed that in vitro, ZBTB2 is able to bind to the unmethylated sequence independent of the origin of the protein extract, suggesting that the inherent ability of ZBTB2 to bind to unmethylated DNA is unaltered in NPCs (Figs 3B and C and EV3B). We next investigated the possibility that ZBTB2 interacts with different proteins in ESCs and NPCs, which could also affect its recruitment to DNA. Therefore, we prepared nuclear protein extracts from ZBTB2‐GFP BAC ESCs and NPCs and performed GFP pull‐downs to identify ZBTB2 interaction partners. These experiments demonstrated that ZBTB2 interacts with two other zinc finger proteins, ZBTB25 and ZNF639, in both cell types (Fig 3D and E). We validated these protein–protein interactions in ESCs by reciprocal pull‐downs and Western blot analysis (Fig EV3C–E). While the interaction partners of ZBTB2 thus do not change during differentiation, zinc finger proteins are known to have the capacity to bind to DNA 22, giving rise to the possibility that recruitment of ZBTB2 to DNA is mediated by one or both of its interaction partners. We subsequently examined whether ZBTB2 would retain its DNA binding capability in the absence of ZBTB25 and ZNF639. For this purpose, we produced recombinant GST‐fused ZBTB2 protein in Escherichia coli and used this for DNA pull‐downs with generic unmethylated and methylated DNA probes 7. Analysis by Western blot revealed that recombinant ZBTB2 binds to the unmethylated probe with high affinity, while almost no binding can be observed to the methylated probe (Fig 3F). A similar result was obtained when the genomic region indicated in Fig 3A was used (Fig EV3F). This confirms our findings that ZBTB2 preferentially binds to unmethylated DNA and shows that this binding is direct and independent of its interaction with ZBTB25 or ZNF639. In contrast, when we repeated the DNA pull‐down with the genomic region that we described above using nuclear protein extract from Zbtb2 knockout (KO) ESCs, we found that ZBTB25 and ZNF639 are no longer able to bind to this region (Fig EV3G). Although we cannot exclude the possibility that protein levels of ZBTB25 and ZNF639 are affected upon knockout of ZBTB2, RNA‐seq analysis comparing wild‐type and Zbtb2 KO ESCs shows no significant difference in mRNA levels (Fig EV3H). Thus, we conclude that ZBTB2 forms a module with ZBTB25 and ZNF639 and that the capacity of this module to bind to unmethylated DNA is dependent on ZBTB2.
Figure 3. ZBTB2 reads unmethylated DNA directly and independently of its interaction partners.
- A
-
B, CScatterplots of DNA pull‐downs with the genomic region indicated in (A), using nuclear protein extract from wild‐type ESCs (B) or NPCs (C). Proteins binding specifically to the unmethylated or methylated sequence are depicted in the lower left or upper right quadrant, respectively. Significant outliers were determined through box plot statistics. In (C), ZBTB2 and ZNF639 were just below the statistical cut‐offs and have been indicated in dark red. All pull‐downs were performed in duplicate and included a label‐swap. Box: median (central line), first and third quartile (box limits); whiskers: 1.5 × interquartile range.
-
D, EVolcano plots of label‐free GFP pull‐downs using nuclear extracts from ZBTB2‐GFP BAC ESCs (D) or NPCs (E). Label‐free quantification (LFQ) intensity of the experiment relative to the control [fold change (FC)] is plotted on the x‐axis; FDR‐corrected t‐test values are plotted on the y‐axis. Grey lines indicate statistical cut‐offs. All pull‐downs were performed in triplicate.
-
FWestern blot analysis of DNA pull‐downs using generic unmethylated or methylated DNA probes with unpurified (upper panel) or GST‐purified (lower panel) recombinant GST‐ZBTB2. Bound fraction consists of proteins that bound to the DNA probes, and unbound fraction is 5% of the remaining supernatant.
Source data are available online for this figure.
Figure EV3. ZBTB2 reads unmethylated DNA directly and independently of its interaction partners (related to Fig 3).

- Comparison of ZBTB2 occupancy (assessed by ChIP–qPCR) and DNA methylation levels (measured by MeDIP–qPCR) between ZBTB2‐GFP BAC ESC and NPCs, at the genomic locus depicted in Fig 3A. Data are depicted as mean ± SEM of three biological replicates (P = as determined by Welch t‐test).
- Western blot validation of the DNA pull‐downs depicted in Fig 3B and C. Bound fraction consists of proteins that bound to the DNA probes, and unbound fraction is 5% of the remaining supernatant.
- Western blot validation of the GFP pull‐downs depicted in Fig 3D. GFP pull‐down on ZBTB2‐GFP yields the GFP‐tagged (upper band) as well as the endogenous protein (lower band).
- Western blot analysis of endogenous ZBTB25 pull‐downs with wild‐type ESC nuclear protein extract. Pull‐downs with IgG are used as a control.
- Western blot analysis of FLAG pull‐downs with nuclear protein extract from ESCs transiently transfected with a ZNF639‐FLAG construct. FLAG pull‐downs with wild‐type ESC nuclear extract are used as a control.
- Western blot analysis of DNA pull‐downs using the unmethylated or methylated genomic region indicated in Fig 3A with unpurified (upper panel) or GST‐purified (lower panel) recombinant GST‐ZBTB2. Bound fraction consists of proteins that bound to the DNA probes, and unbound fraction is 5% of the remaining supernatant.
- Scatterplot of DNA pull‐downs with the genomic region indicated in Fig 3A, using nuclear protein extract from Zbtb2 KO ESCs. All pull‐downs were performed in duplicate and included a label‐swap. Box: median (central line), first and third quartile (box limits); whiskers: 1.5 × interquartile range.
- RNA‐seq analysis of Zbtb25 and Znf639 levels in Zbtb2 KO ESCs, relative to wild‐type ESCs. Data are depicted as mean of two biological replicates.
Source data are available online for this figure.
ZBTB2 binding and expression and DNA methylation are intertwined
While the previous results suggest that DNA methylation can influence ZBTB2 binding, we wondered whether ZBTB2 levels can conversely also affect DNA methylation levels. To investigate this, we measured global levels of meC and hmC in wild‐type ESCs, Zbtb2 KO ESCs and ZBTB2‐GFP BAC ESCs. We found that global meC levels are lower in Zbtb2 KO ESCs and higher in ZBTB2‐GFP BAC ESCs compared to wild‐type ESCs, while the reverse is true for global hmC levels (Fig 4A). To gain insight into the underlying mechanism of this effect, we looked in our RNA‐seq data comparing wild‐type and Zbtb2 KO ESCs whether the expression of any known regulators of DNA methylation or demethylation is changed in Zbtb2 KO ESCs compared to wild‐type cells. Indeed, we found the DNA methyltransferases Dnmt3A, Dnmt3B and Dnmt3L, as well as the demethylases Tet1 and Tet2, to be differentially expressed in Zbtb2 KO ESCs (Fig EV4A). Together, this suggests that the DNA methylation network is enhanced in Zbtb2 KO ESCs, resulting in a higher turnover of methylated DNA. We validated these results by qRT–PCR in wild‐type and Zbtb2 KO ESCs and also showed that the expression levels can be restored towards wild‐type levels in a transient overexpression of ZBTB2 in the Zbtb2 KO line (Fig 4B). In addition, we found that the changes in expression levels of TET1 are reflected in its occupancy at CpG islands (Fig EV4B), suggesting an active role for ZBTB2 in regulating meC and hmC levels at CGIs. Given our previous findings that ZBTB2 binding is sensitive to DNA methylation, this is particularly interesting, as it suggests that ZBTB2 is involved in a feedback loop to regulate its own recruitment to DNA. We thus speculate that ZBTB2 binding to unmethylated CGI promoters is regulated by a complex interplay between DNA methylation levels and the expression of DNA methylases and demethylases, in which ZBTB2 itself plays a key role.
Figure 4. Effect of ZBTB2 on genome‐wide DNA methylation levels.
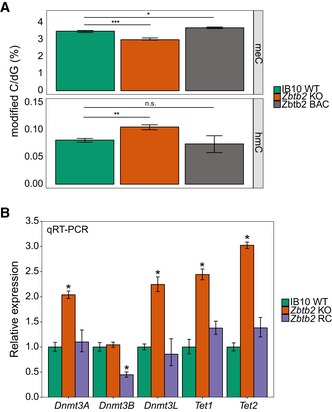
- Global meC and hmC levels in wild‐type, Zbtb2 knockout and ZBTB2‐GFP BAC ESCs. Data are depicted as mean ± SEM of five biological replicates (*P = 0.01857, **P = 0.003181, ***P = 0.0007674, n.s. = not significant, Welch t‐test).
- qRT–PCR analysis of gene expression levels in wild‐type, Zbtb2 knockout (KO) and Zbtb2 KO ESCs transiently transfected with ZBTB2 [Zbtb2 rescue (RC)]. Data are depicted as mean ± SEM of three biological replicates (*P < 0.05 compared to WT, Welch t‐test).
Figure EV4. Effect of ZBTB2 on DNA (de)methylases abundance and TET1 occupancy (related to Fig 4).
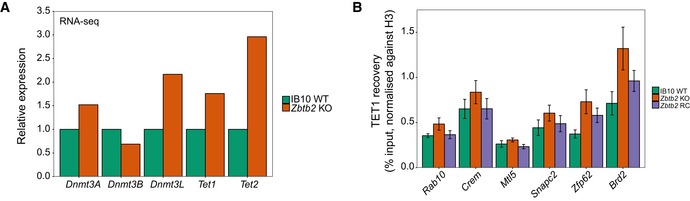
- RNA‐seq analysis of gene expression levels in Zbtb2 KO ESCs, relative to wild‐type ESCs. Data are depicted as mean of two biological replicates.
- ChIP–qPCR analysis of TET1 binding at selected CpG islands in wild‐type, Zbtb2 knockout (KO) and Zbtb2 KO ESCs transiently transfected with ZBTB2 [Zbtb2 rescue (RC)]. Data are depicted as mean ± SEM of three biological replicates (P > 0.05 for all pairwise comparisons, Welch t‐test).
ZBTB2 is a gene activator and regulates the exit from pluripotency
To explore the biological significance of ZBTB2 occupancy at unmethylated CGI promoters in ESCs, we more deeply analysed our RNA‐seq data of wild‐type and Zbtb2 KO ESCs. Since DNA methylation has generally been associated with gene silencing, we expected that ZBTB2 would act as a gene activator rather than a repressor. Supporting this notion, we found that in Zbtb2 KO ESCs, ± 850 genes are differentially regulated compared to wild‐type ESCs, with the majority becoming downregulated. To investigate whether these differentially expressed genes are direct ZBTB2 target genes, we integrated our RNA‐seq and ChIP‐seq data sets (Dataset EV2). This resulted in the identification of ± 300 genes that are bound by ZBTB2 in wild‐type ESCs and whose expression is significantly changed in Zbtb2 KO ESCs, suggesting that ZBTB2 directly regulates the expression of these genes (Fig 5A). The majority of these directly regulated genes are downregulated in Zbtb2 KO compared to wild‐type ESCs. Furthermore, when comparing the ratio of expressed to silence genes in the subset of ZBTB2‐bound genes to the whole transcriptome, as determined by RNA‐seq in wild‐type ESCs, we found that this ratio is significantly higher for ZBTB2‐bound genes (Fig EV5A). Altogether, these results propose a role for ZBTB2 as a gene activator, consistent with our understanding of DNA methylation as a repressive epigenetic mark.
Figure 5. ZBTB2 acts as a gene activator and stimulates cellular differentiation.
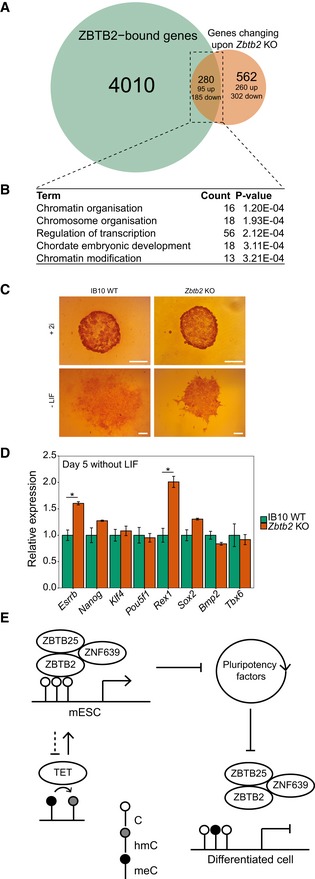
- Venn diagram comparing genes that are bound by ZBTB2 in ESCs (as identified by ChIP‐seq, green) and genes whose expression changes significantly in Zbtb2 KO ESCs (as identified by RNA‐seq, orange).
- Top five hits of GO terms enrichment analysis of genes that were identified in (A) to be directly regulated by ZBTB2.
- Alkaline phosphatase staining of wild‐type and Zbtb2 KO ESCs after culturing for 5 days in the absence of LIF to induce differentiation. Cells that were cultured for 5 days in the presence of 2i served as a reference for completely undifferentiated cells. Pictures are representative of eight biological replicates. Scale bars: 100 μm.
- qRT–PCR analysis of pluripotency and differentiation markers in wild‐type and Zbtb2 KO ESCs after culturing for 5 days in the absence of LIF. Data are depicted as mean ± SEM of three biological replicates (*P < 0.05, Welch t‐test).
- Proposed model of ZBTB2‐binding dynamics. In ESCs, ZBTB2 recruits a zinc finger module to unmethylated CpG island promoters, thereby regulating genes that are important for differentiation. ZBTB2 influences its own binding by regulating TET enzymes and consequently the rate of DNA demethylation. Upon differentiation, binding of the zinc finger module is reduced by increased DNA methylation levels.
Figure EV5. ZBTB2 acts as a gene activator and stimulates cellular differentiation (related to Fig 5).
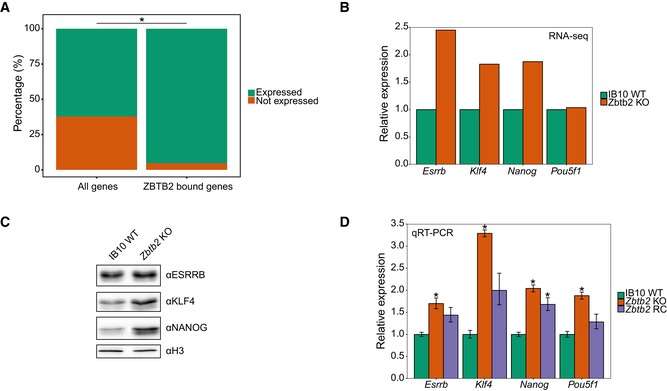
- Percentages of expressed and not expressed genes in the total transcriptome of wild‐type ESCs and the subset of ZBTB2‐bound genes (*P = 8.348e‐09, Fisher's exact test).
- RNA‐seq analysis of gene expression levels in Zbtb2 KO ESCs, relative to wild‐type ESCs. Data are depicted as mean of two biological replicates.
- Western blot analyses of protein levels in wild‐type and Zbtb2 KO ESCs. Histone H3 was used as a loading control.
- qRT–PCR analysis of gene expression levels in wild‐type, Zbtb2 knockout (KO) and Zbtb2 KO ESCs transiently transfected with ZBTB2 [Zbtb2 rescue (RC)]. Data are depicted as mean ± SEM of three biological replicates (*P < 0.05 compared to WT, Welch t‐test).
Source data are available online for this figure.
Next, we performed gene ontology (GO) term enrichment analysis on genes that are directly regulated by ZBTB2 and found that one of the processes that these genes are primarily associated with is embryonic development (Fig 5B). In line with this, our RNA‐seq data revealed that a number of pluripotency factors (such as ESRRB, KLF4 and NANOG) are significantly upregulated in Zbtb2 KO ESCs compared to wild‐type cells (Fig EV5B), suggesting that the pluripotency network in Zbtb2 KO cells is perturbed. We validated this by Western blot and qRT–PCR in wild‐type and Zbtb2 KO ESCs and also showed that a transient overexpression of ZBTB2 in the Zbtb2 KO line causes expression levels to decrease towards wild‐type levels (Fig EV5C and D). To investigate whether a higher expression of these pluripotency factors induces delayed differentiation in Zbtb2 KO ESCs, we induced differentiation in these cells by culturing them in the absence of LIF and assessed their pluripotency by alkaline phosphatase staining and qRT–PCR. We found that after 5 days in medium without LIF, Zbtb2 KO ESCs display a less differentiated morphology compared to wild‐type cells that were treated equally (Fig 5C). Furthermore, qRT–PCR analysis at day five after LIF withdrawal showed that several pluripotency factors remain more highly expressed in Zbtb2 KO ESCs compared to wild‐type cells, while the opposite was observed for differentiation makers (Fig 5D). Taken together, these observations suggest that Zbtb2 KO ESCs differentiate more slowly and hence imply that ZBTB2 is involved in the regulation of genes that are important for ESC differentiation.
Concluding remarks
On the basis of our findings, we propose a model of ZBTB2‐binding dynamics (Fig 5E) in which ZBTB2, together with ZBTB25 and ZNF639, binds to unmethylated CpG island promoters in ESCs, which is mostly positively correlated with expression of the associated genes. ZBTB2 influences its own binding by regulating the TET enzymes, which catalyse the first step of DNA demethylation by converting meC into hmC. ZBTB2 itself does not bind at the promoters of the TETs, indicating that this regulation is probably indirect. In differentiated cells, increased methylation levels lead to a decreased binding of the zinc finger module.
While this model recapitulates the findings we presented in this study, further research is needed to gain a more complete picture of the biological mechanisms that regulate ZBTB2 binding in the genome and its function. Although we have demonstrated that ZBTB2 binding is sensitive to DNA methylation, DNA methylation dynamics at dynamic ZBTB2‐binding sites are not always very pronounced, suggesting that there may be mechanisms other than DNA methylation that can regulate ZBTB2 binding to genomic loci. Furthermore, it remains to be resolved why ESCs show a delay in differentiation upon Zbtbt2 knockout. The genes that are directly regulated by ZBTB2 include some well‐known pluripotency factors, such as Klf4, Tbx3, Tfcp2l1 and Nr5a2 23, 24. However, these genes are all among the subset of genes whose expression goes up rather than down in the Zbtb2 KO, raising the question through which exact mechanisms ZBTB2 regulates gene expression. Previous studies have identified interactions between ZBTB2 and co‐activator or co‐repressor complexes containing histone acetyltransferase or deacetyltransferase activity, respectively, leading to gene activation or gene repression 25, 26, 27. In addition, ZBTB2 has been reported to interact with numerous other proteins, such as OCT4 and PTIP 28, 29. However, we could not confirm any of these interactions in our GFP‐ZBTB2 interaction proteomics data, suggesting that these interactions are either quite transient, substoichiometric or cell‐type specific. Another putative mechanism of ZBTB2‐regulated gene expression is through inhibition of binding of other transcription factors. Indeed, ZBTB2 has been suggested to compete with Sp1 for binding to G/C box motifs and to prevent p53 binding through a direct protein–protein interaction 26. Comprehensive and integrative analysis of various genomics data sets will enable us to investigate to what extent these observations are representative of a general phenomenon.
Materials and Methods
Cell culture and generation of cell lines
IB10 mouse ESCs were cultured on gelatine‐coated plates in ESC medium [DMEM (Gibco) supplemented with 15% HyClone Fetal Bovine Serum (GE Healthcare Life Sciences), GlutaMAX (Gibco), non‐essential amino acids (Lonza), sodium pyruvate (Gibco), penicillin–streptomycin (Gibco), β‐mercaptoethanol and home‐made leukaemia inhibitory factor (LIF)]. For 2i conditions, the medium was supplemented with 3 μM PD0325901 and 1 μM CHIR99021.
ZBTB2‐GFP ESCs were generated by transfection of a GFP‐tagged Zbtb2 BAC construct 15 into IB10 ESCs using Lipofectamine LTX and PLUS reagents (Invitrogen). Cells were subjected to G418 selection for more than 10 days, after which monoclonal cell lines were generated and assessed by Western blot.
ZBTB2‐GFP ESCs were differentiated into NPCs using N2B27 medium (DMEM/F12 (Gibco), supplemented with Neurobasal medium (Gibco), N2 and B27 supplements (Gibco) and β‐mercaptoethanol. ZBTB2‐GFP NPCs and wild‐type NPCs were maintained in NPC medium [NSA (Euromed) supplemented with GlutaMAX (Gibco), N2 supplement (Gibco), 10 ng/ml bFGF (100‐18C, Perprotech) and 10 ng/ml EGF (236‐EG, R&D Systems)].
The CRISPR/Cas9 system was used to generate a Zbtb2 knockout (KO) cell line in ESCs. A guide RNA targeting the second exon of Zbtb2 (CACCGCTGCAGTAGAATAAGTCCA) was cloned into pSpCas9(BB)‐2A‐Puro (PX459; Addgene 48139), essentially as described 30. The resulting plasmid was verified by sequencing and transfected into IB10 ESCs. After 2 days, the cells were subjected to puromycin selection for 48 h, and then, monoclonal cell lines were generated and analysed by sequencing.
Chromatin preparation
Attached cells were cross‐linked with 1% formaldehyde in buffer A (148 mM NaCl, 1.48 mM EDTA, 0.74 mM EGTA, 74 mM Hepes) for 10 min at room temperature, after which cross‐linking was quenched with 0.125 M glycine. Cells were washed with PBS and then harvested by scraping in buffer B (0.25% Triton X‐100, 10 mM EDTA, 0.5 mM EGTA, 20 mM Hepes). Cell suspensions were centrifuged for 5 min at 400 g at 4°C, then resuspended in buffer C (150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM Hepes) and rotated for 10 min at 4°C. After centrifugation for 5 min at 400 g at 4°C, cells were resuspended in incubation buffer (0.15% SDS, 1% Triton X‐100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM Hepes) with EDTA‐free complete protease inhibitors (CPI) (Roche) at a concentration of 15 million cells/ml. Cell suspension was sonicated with a Bioruptor Pico sonicator (Diagenode) at 4°C using cycles of 30 s on and 30 s off, until the average size was approximately 200–300 bp. Sonicated material was centrifuged for 5 min at 21,000 g at 4°C; supernatant was collected, aliquoted, snap‐frozen in liquid nitrogen and stored at −80°C.
Chromatin immunoprecipitation
Chromatin was incubated overnight with antibodies (Appendix Table S1) at 4°C in incubation buffer (as above) with CPI and 0.1% BSA. A 1:1 mixture of Protein A and G Dynabeads (Invitrogen) was washed twice with incubation buffer plus 0.1% BSA, then added to the chromatin mixture and rotated for 90 min at 4°C. Beads were washed twice with wash buffer 1 (0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X‐100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM Hepes), once with wash buffer 2 (wash buffer 1 with 500 mM NaCl), once with wash buffer 3 (250 mM LiCl, 0.5% sodium deoxycholate, 0.5% NP40, 1 mM EDTA, 0.5 mM EGTA, 20 mM Hepes) and twice with wash buffer 4 (1 mM EDTA, 0.5 mM EGTA, 20 mM Hepes). After washing, beads were rotated for 20 min at room temperature in elution buffer (1% SDS, 0.1 M NaHCO3). Eluted chromatin was decross‐linked with 25 μg proteinase K and 200 mM NaCl for 4 h at 65°C in a thermoshaker at 1,000 rpm. Decross‐linked DNA was purified with a MinElute PCR Purification Kit (Qiagen) and analysed by either deep sequencing or quantitative PCR.
Deep sequencing and data analysis
Libraries were prepared for Illumina sequencing with a Kapa Hyper Prep Kit (Kapa Biosystems), essentially according to the manufacturer's instructions. Briefly, 5 ng of DNA was subjected to end repair and A‐tailing, after which 28 nM NEXTflex DNA barcodes (Bioo Scientific) were ligated. Post‐ligation clean‐up was performed with Agencourt AMPure XP reagent (Beckman Coulter), and libraries were amplified using ten cycles of PCR amplification. A MinElute PCR Purification Kit (Qiagen) was used for post‐amplification clean‐up, and size selection for 300‐bp fragments was done with an E‐Gel Precast Agarose Electrophoresis System (Thermo). Samples were analysed for purity with a high‐sensitivity DNA ChIP run on an Agilent 2100 Bioanalyzer and then deep sequenced on an Illumina HiSeq2000 Sequencing System. Reads were mapped onto the reference mouse genome mm9 (NCBI build 37), using the Burrows‐Wheeler Alignment tool. Mapped reads were filtered on sequence quality, and PCR duplicates were removed. Peak calling was performed using the MACS 2.0 tool against a merge of the 5% input and GFP ChIP in wild‐type cells, which was randomly downscaled to match the number of reads of the GFP ChIP in the BAC line. External files (Appendix Table S2) were downloaded and processed as described above. Heat maps were generated using the Fluff package [preprint: 31]. Genomic localisation was analysed with the GREAT tool 32, and genomic distribution was assessed using an in‐house Python script. Motif enrichment analysis and de novo motif predictions were performed using the GimmeMotifs package 33. A control set of random promoters with the same size as the average ZBTB2 peak was generated by extracting sequences starting from mm9 TSSs (downloaded from the UCSC genome browser). Gene ontology terms analysis was performed using the DAVID tool 34.
Methylated DNA immunoprecipitation
Per reaction, 1 μg of decross‐linked DNA was incubated with 5 mM NaOH in incubation buffer (as above) for 3 min at 95°C and then quickly chilled on ice. 0.1% BSA, 1 μg antibody against meC (Appendix Table S1) and a 1:1 mixture of Protein A and G Dynabeads (Invitrogen) were added to the DNA, after which the mixture was rotated for at least 4 h at 4°C. Beads were washed twice with ChIP wash buffer 1, once with ChIP wash buffer 2, once with ChIP wash buffer 3 and twice with ChIP wash buffer 4 (as above). After washing, beads were resuspended in elution buffer (1% SDS, 0.1 M NaHCO3, 200 mM NaCl) and incubated for 10 min at 1,000 rpm at 65°C. Eluted DNA was purified with a MinElute PCR Purification Kit (Qiagen) and analysed by quantitative PCR.
Nuclear extract preparation
Nuclear extractions were performed essentially as described 35. Briefly, cells were harvested with trypsin, washed twice with PBS and spun down for 5 min at 400 g at 4°C. Cells were resuspended in five volumes of buffer A (10 mM Hepes KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl), incubated for 10 min on ice and then centrifuged for 5 min at 400 g at 4°C. Cells were resuspended in two volumes of buffer A with CPI and 0.15% NP40 and transferred to a Dounce homogeniser. After lysis with 40 strokes of a type B pestle, the suspension was centrifuged for 15 min at 3,200 g at 4°C. The nuclear pellet was washed with PBS, centrifuged for 5 min at 3,200 g at 4°C and then resuspended in two volumes of buffer C (420 mM NaCl, 20 mM Hepes KOH pH 7.9, 20% (v/v) glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, CPI, 0.5 mM DTT). This suspension was rotated for 1 h at 4°C and subsequently centrifuged for 45 min at 20,800 g at 4°C. The supernatant was collected, aliquoted, snap‐frozen in liquid nitrogen and stored at −80°C. The nuclear pellets remaining after nuclear extraction were solubilised by resuspension in four volumes of RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP40, 5 mM MgCl2, 10% glycerol) plus 1,000 U benzonase (Millipore) per 100 μl pellet. Pellets were incubated in a thermoshaker at 37°C at 1,000 rpm until dissolved and then centrifuged for 5 min at 14,000 g at 4°C. The supernatant was collected, aliquoted, snap‐frozen in liquid nitrogen and stored at −80°C.
DNA pull‐downs
For DNA pull‐downs with nuclear extract, biotinylated DNA probes were synthesised by a nested PCR approach using mouse genomic DNA as a template. Primers can be found in Appendix Table S3. Methylated probes were generated by incubation with M. SssI (Zymo Research), according to the manufacturer's protocol. Per pull‐down, 20 μl of Streptavidin Sepharose High Performance 50% bead slurry (GE Healthcare Life Sciences) was used. Beads were washed once with PBS containing 0.1% NP40 and once with DNA binding buffer (DBB: 1 M NaCl, 10 mM Tris pH 8.0, 1 mM EDTA, 0.05% NP40). Probes (1 μg per pull‐down) were diluted to a total volume of 600 μl with DBB and rotated with the beads for 30 min at 4°C. Coupling of DNA to the beads was verified by agarose gel electrophoresis. Beads were washed once with DBB and twice with protein incubation buffer [PIB: 150 mM NaCl, 50 mM Tris pH 8.0, 0.25% NP40, 1 mM DTT and CPI). Nuclear extract (150 μg) and competitors (5 μg poly‐dIdC (Sigma, P4929), 5 μg poly‐dAdT (Sigma, P0883) and 5 μg tRNA (Sigma, R5636)] were diluted to a total volume of 600 μl with PIB and rotated with the beads for 90 min at 4°C. After beads were washed thrice with PIB and twice with PBS, all supernatant was removed using a 30‐G syringe. Beads were then resuspended in 50 μl elution buffer (EB: 2 M urea, 100 mM ammonium bicarbonate, 10 mM DTT) and incubated for 20 min in a thermoshaker at 1,400 rpm at room temperature. After addition of 50 mM iodoacetamide (IAA), beads were incubated for 10 min at 1,400 rpm at room temperature in the dark. Proteins were then on‐bead digested into tryptic peptides by addition of 0.25 μg trypsin and subsequent incubation for 2 h at 1,400 rpm at room temperature. The supernatant was transferred to new tubes and further digested overnight at room temperature with an additional 0.1 μg of trypsin. Tryptic peptides were purified on C18‐StageTips 36, followed by on‐StageTip dimethyl labelling. StageTips were loaded with 300 μl labelling buffer (2% of either CH2O (light) or CD2O (medium) in 10 mM NaH2PO4, 35 mM Na2HPO4, 0.6 M NaBH3CN) and centrifuged. Each pull‐down was performed in duplicate, in which label‐swapping was performed between the replicates to eliminate labelling bias. Finally, StageTips were washed once with buffer A (0.1% formic acid) and stored at 4°C.
GFP pull‐downs
Label‐free GFP pull‐downs were performed in triplicate. Per pull‐down, 15 μl of GFP‐trap or non‐GFP‐trap 50% bead slurry (Chromotek) was used. Beads were washed thrice with buffer C (300 mM NaCl, 20 mM Hepes KOH pH 7.9, 20% (v/v) glycerol, 2 mM MgCl2, 0.2 mM EDTA, 1% NP40, 0.5 mM DTT, CPI). Nuclear extract (450 μg–1 mg) and ethidium bromide (50 μg/ml) were diluted to a total volume of 400 μl with buffer C and rotated with the beads for 90 min at 4°C. After beads were washed twice with buffer C, twice with PBS plus 1% NP40 and twice with PBS, all supernatant was removed using a 30‐G syringe. Beads were then resuspended in 50 μl elution buffer (EB: 2 M urea, 100 mM Tris pH 8.5, 10 mM DTT). On‐bead digestion was performed as described above. Tryptic peptides were acidified with 0.5% TFA and purified on C18‐StageTips.
LC‐MS/MS measurements and data analysis
Tryptic peptides were eluted from StageTips. For dimethyl‐labelled samples, the respective light‐ and medium‐labelled samples were combined into a forward and a reverse reaction. Peptides were then separated on an Easy‐nLC 1000 (Thermo) connected online to an LTQ‐Orbitrap Fusion Tribrid mass spectrometer (Thermo) or an LTQ‐Orbitrap Q‐Exactive mass spectrometer (Thermo), using a gradient of acetonitrile (7–32% or 5–30%, respectively), followed by washes at 50% then 90% acetonitrile, for 140 min or 120 min of total data collection. For samples measured on the Fusion, scans were collected in data‐dependent top‐speed mode of a 3‐s cycle with dynamic exclusion set at 60 s. For samples measured on the Q‐Exactive, the top ten most intense precursor ions were selected for fragmentation. Peptides were searched against the UniProt mouse proteome with MaxQuant v1.5.1.0 37, using default settings, the appropriate dimethyl labels, and re‐quantify and/or match between runs enabled. Data were analysed with Perseus version 1.4.0.0 and in‐house R scripts. Raw data used to generate the plots can be found in Dataset EV1.
Reciprocal pull‐downs
For endogenous pull‐downs, 30 μl of a 1:1 mixture of Protein A and G Dynabeads was used per pull‐down. Beads were washed thrice with PBS containing 0.05% NP40. Antibodies (2 μl per pull‐down) were diluted to a total volume of 150 μl with PBS + 0.05% NP40 and rotated with the beads for 1.5 h at 4°C, after which beads were washed thrice with PBS + 0.05% NP40. Nuclear extract (50 μl) and ethidium bromide (66.7 μg/ml) were diluted to a total volume of 150 μl with PBS with 0.25% NP40 and CPI and rotated with the beads for 2.5 h at 4°C. Beads were washed thrice with PBS + 0.25% NP40 and CPI and then boiled in Laemmli buffer. These samples, together with a 5% input sample, were run on an SDS–PAGE and analysed by Western blot. Antibodies can be found in Appendix Table S1.
For FLAG pull‐downs, 20 μl of anti‐FLAG M2 affinity gel (Sigma, A2220) was used per pull‐down. Beads were washed thrice with buffer C (300 mM NaCl, 20 mM Hepes KOH pH 7.9, 20% (v/v) glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.25% NP40, 0.5 mM DTT, CPI). Nuclear extract (450 μg) and ethidium bromide (100 μg/ml) were diluted to a total volume of 200 μl with buffer C and rotated with the beads for 90 min at 4°C. Beads were washed twice with buffer C (as before, with 0.5% NP40) and then boiled in Laemmli buffer. These samples, together with a 5% input sample, were run on SDS–PAGE and analysed by Western blot. Antibodies can be found in Appendix Table S1.
Recombinant protein expression, DNA pull‐downs and Western blot
Full‐length ZBTB2 protein was cloned from mouse complement DNA into the pGEX‐5X‐1 vector (GE Healthcare Life Sciences). Transformed Bl21 E. coli bacteria were grown at 37°C in a shaker until OD600 reached 0.6–0.8. Protein expression was induced overnight at 16°C by addition of 0.1 mM IPTG (Promega, V3951) and 100 μM ZnCl2. Bacterial cells were lysed by rotation for 30 min at 4°C in bacterial lysis buffer (50 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.05% NP40, 1 mM DTT, 1 mM PMSF, 0.5 mg/ml lysozyme and CPI) and repeated freeze–thawing. After the addition of 0.5% NP40, 5 mM MgCl2 and 25 μg/ml DNase I (Sigma, 04716728001), lysates were rotated for 45 min at 4°C and passed through a needle thrice. The suspension was then centrifuged for 30 min at 21,100 g at 4°C. Supernatant was collected and supplemented with 1 μM ZnCl2. To purify GST‐ZBTB2 from the supernatant, Pierce Glutathione Agarose (Thermo) was washed with equilibration/wash buffer (EWB: 50 mM Tris pH 8.0, 150 mM NaCl) and then rotated with bacterial lysate for 1 h at 4°C. After washing twice with EWB, GST‐tagged proteins were eluted with elution buffer [EWB with 10 mM reduced glutathione (Sigma, G‐4251)]. DNA pull‐downs with the purified bacterial lysate were performed with generic unmethylated and methylated DNA probes (10 μg per pull‐down, sequences as described in 7) or genomic DNA probes (1 μg per pull‐down) essentially as described above, except that PIB was supplemented with 10 μM ZnCl2 and that only poly‐dAdT was used as a competitor. After incubation, the beads were washed twice with PIB and boiled in Laemmli buffer. These samples, together with a 5% input sample and 5% unbound fraction samples, were run on an SDS–PAGE and analysed by Western blot. Antibodies can be found in Appendix Table S1.
RNA sequencing and data analysis
RNA was isolated in duplicate from cells using an RNeasy Mini Kit (Qiagen). Ribosomal RNA was depleted by treatment with a Ribo‐Zero rRNA Removal Kit (Illumina) and fragmented into approximately 200‐bp fragments in fragmentation buffer (200 mM Tris–acetate, 500 mM KCH3COO, 150 mM Mg(CH3COO)2, pH 8.2). Strand‐specific libraries of cDNA were prepared using SuperScript III Reverse Transcriptase (Invitrogen) and a Kapa Hyper Prep Kit, as described above, but with an additional incubation with USER enzyme (NEB) before library amplification to digest the second cDNA strand. Reads were mapped onto the reference mouse genome mm9 (NCBI build 37) using the Genomic Short‐read Alignment Program 38. Differential gene expression was analysed with the Cufflinks v2.2.1 package 39.
Alkaline phosphatase staining
Wild‐type and Zbtb2 KO ESCs were seeded into a 48‐well plate at a density of 100 cells/well. Cells were grown for 5 days in ESC medium, either without LIF or supplemented with 2i drugs (3 μM PD0325901 and 1 μM CHIR99021). At day five, the cells were fixed with 4% paraformaldehyde, and alkaline phosphatase staining was performed using an Alkaline Phosphatase Detection Kit (Millipore), according to the manufacturer's instructions.
Quantitative PCR
RNA was isolated from cells using an RNeasy Mini Kit (Qiagen) and converted into cDNA with an iScript cDNA Synthesis Kit (Bio‐Rad). qRT–PCR or ChIP–qPCR was performed using iQ SYBR Green Supermix (Bio‐Rad) on a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad). Primers can be found in Appendix Tables S4 and S5. For qRT–PCR, 18S was used as a reference gene. For ChIP–qPCR, data were normalised to H3 ChIP or to total DNA after ChIP as a measure of ChIP efficiency.
Analysis of genome‐wide meC and hmC levels
5mC and 5hmC levels were measured as described 40. Briefly, genomic DNA was isolated from five independent samples of approximately 2 million cells per sample and degraded into individual nucleosides using a DNA Degrase Plus Kit (Zymo Research), according to the manufacturer's protocol. Individual nucleosides were separated using high‐performance liquid chromatography (HPLC) on an Acquity UPLC (Waters) and measured by tandem mass spectrometry on a Micromass Quattro Premier XE (Waters). Area‐based linear regression curves were derived from calibration standards containing internal standard solutions and used for quantification.
Cloning and transient transfections
Full‐length ZBTB2 protein was cloned from mouse complement DNA into the pMSCV PIG vector 41. This construct was transiently transfected into Zbtb2 KO ESCs using Lipofectamine LTX and PLUS reagents, or Lipofectamine 3000 (Invitrogen). Cells were harvested after 2 or 3 days.
A FLAG‐tagged ZNF639 plasmid was purchased from OriGene (MR207772) and transiently transfected into wild‐type IB10 ESCs using polyethylenimine (Polysciences). Cells were harvested after 1 day.
Statistical methods
Unless specified otherwise, statistical tests were performed by conducting an unpaired two‐tailed Welch two‐sample t‐test in R, where P < 0.05 was considered to be significant.
For dimethyl‐labelled DNA pull‐downs, outliers were identified in the forward and reverse reactions independently, using box plot statistics with a threshold of 1.5 times the interquartile range. Significant proteins were those that were called outliers in both the forward experiment and the reverse experiment. For GFP pull‐downs, specific interactors were determined through an adapted two‐tailed t‐test in Perseus, using a permutation‐based false discovery rate with 250 randomisations. Volcano plots were generated in which statistical cut‐offs were chosen such that no proteins were present on the control side of the graph.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository 42 with the data set identifier PXD007099. High‐throughput sequencing data have been deposited in the GEO database repository with the data set identifier GSE101802.
Author contributions
IDK and MV conceived the project. IDK performed the experiments and analysed the data. IDK and MV wrote the manuscript. MV supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Acknowledgements
We would like to thank Ina Poser (MPI‐CBG Dresden) for contributing the BAC construct, Ozren Bogdanović (Garvan Institute of Medical Research) for sharing data and generating genome tracks and Guido van Mierlo (RIMLS) for help with the meC and hmC measurements. We are grateful to Arjen Brinkman and Hendrik Marks (RIMLS) for helpful suggestions and discussions, the Sequencing and Bioinformatics teams of the RIMLS Mol(Dev)Bio departments for ChIP‐seq and RNA‐seq support, Pascal Jansen and Marijke Baltissen for technical support and all members of the Vermeulen laboratory for feedback. Work in the Vermeulen laboratory is supported by a European Research Council Starting Grant (no. 309384) and the Netherlands Organization for Scientific Research (NWO Gravitation Program Cancer Genomics Netherlands).
EMBO Reports (2018) 19: e44993 29437775
References
- 1. Chédin F (2011) The DNMT3 family of mammalian de novo DNA methyltransferases. Prog Mol Biol Transl Sci 101: 255–285 [DOI] [PubMed] [Google Scholar]
- 2. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L et al (2009) Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu H, Zhang Y (2014) Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156: 45–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bird A (2011) The dinucleotide CG as a genomic signalling module. J Mol Biol 409: 47–53 [DOI] [PubMed] [Google Scholar]
- 5. Antequera F (2003) Structure, function and evolution of CpG island promoters. Cell Mol Life Sci 60: 1647–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spruijt CG, Vermeulen M (2014) DNA methylation: old dog, new tricks? Nat Struct Mol Biol 21: 949–954 [DOI] [PubMed] [Google Scholar]
- 7. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, Bauer C, Münzel M, Wagner M, Müller M, Khan F et al (2013) Dynamic readers for 5‐(hydroxy)methylcytosine and its oxidized derivatives. Cell 152: 1146–1159 [DOI] [PubMed] [Google Scholar]
- 8. Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr ARW, Deaton A, Andrews R, James KD et al (2010) CpG islands influence chromatin structure via the CpG‐binding protein Cfp1. Nature 464: 1082–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones PL, Veenstra GJC, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19: 187–191 [DOI] [PubMed] [Google Scholar]
- 10. Huff JT, Zilberman D (2014) Dnmt1‐independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 156: 1286–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM (2000) CTCF mediates methylation‐sensitive enhancer‐blocking activity at the H19/Igf2 locus. Nature 405: 486–489 [DOI] [PubMed] [Google Scholar]
- 12. Lafaye C, Barbier E, Miscioscia A, Saint‐Pierre C, Kraut A, Couté Y, Plo I, Gasparutto D, Ravanat J‐L, Breton J (2014) DNA binding of the p21 repressor ZBTB2 is inhibited by cytosine hydroxymethylation. Biochem Biophys Res Commun 446: 341–346 [DOI] [PubMed] [Google Scholar]
- 13. Filion GJP, Zhenilo S, Salozhin S, Yamada D, Prokhortchouk E, Defossez P‐A (2006) A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol 26: 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sasai N, Defossez P‐A (2009) Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int J Dev Biol 53: 323–334 [DOI] [PubMed] [Google Scholar]
- 15. Poser I, Sarov M, Hutchins JRA, Hériché J‐K, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW et al (2008) BAC TransgeneOmics: a high‐throughput method for exploration of protein function in mammals. Nat Methods 5: 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund‐Sayeed S, Das PK, Kivioja T, Dave K, Zhong F et al (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356: eaaj2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T‐Y, Peng W, Zhang MQ et al (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A et al (2013) Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB et al (2008) Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HHD, Matarese F, Lepikhov K, Gut M, Brun‐Heath I, Hubner NC et al (2013) Whole‐genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 13: 360–369 [DOI] [PubMed] [Google Scholar]
- 22. Klug A, Rhodes D (1987) ‘Zinc fingers’: a novel protein motif for nucleic acid recognition. Trends Biochem Sci 12: 464–469 [Google Scholar]
- 23. Dunn S‐J, Martello G, Yordanov B, Emmott S, Smith AG (2014) Defining an essential transcription factor program for naive pluripotency. Science 344: 1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heng J‐CD, Feng B, Han J, Jiang J, Kraus P, Ng J‐H, Orlov YL, Huss M, Yang L, Lufkin T et al (2010) The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 6: 167–174 [DOI] [PubMed] [Google Scholar]
- 25. Jung SY, Malovannaya A, Wei J, O'Malley BW, Qin J (2005) Proteomic analysis of steady‐state nuclear hormone receptor coactivator complexes. Mol Endocrinol 19: 2451–2465 [DOI] [PubMed] [Google Scholar]
- 26. Jeon BN, Choi WI, Yu MY, Yoon AR, Kim MH, Yun CO, Hur MW (2009) ZBTB2, a novel master regulator of the p53 pathway. J Biol Chem 284: 17935–17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim M‐Y, Koh D‐I, Choi W‐I, Jeon B‐N, Jeong D‐Y, Kim K‐S, Kim K, Kim S‐H, Hur M‐W (2015) ZBTB2 increases PDK4 expression by transcriptional repression of RelA/p65. Nucleic Acids Res 43: 1609–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, Choudhary J (2010) An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 6: 382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Nuland R, Smits AH, Pallaki P, Jansen PWTC, Vermeulen M, Timmers HTM (2013) Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol 33: 2067–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ran FA, Hsu PPD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Georgiou G, van Heeringen SJ (2016) Fluff: exploratory analysis and visualization of high‐throughput sequencing data. PEERJ 4: e2209 https://doi.org/10.7717/peerj.2209 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Heeringen SJ, Veenstra GJC (2011) GimmeMotifs: a de novo motif prediction pipeline for ChIP‐sequencing experiments. Bioinformatics 27: 270–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang DW, Lempicki RA, Sherman BT (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- 35. Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption / ionization, nanoelectrospray, and LC / MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- 37. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 38. Barnett DW, Garrison EK, Quinlan AR, Str̈mberg MP, Marth GT (2011) Bamtools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27: 1691–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kroeze LI, Aslanyan MG, Van Rooij A, Koorenhof‐Scheele TN, Massop M, Carell T, Boezeman JB, Marie JP, Halkes CJM, De Witte T et al (2014) Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood 124: 1110–1118 [DOI] [PubMed] [Google Scholar]
- 41. Mayr C, Bartel DP (2009) Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J et al (2013) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 41: 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T‐K, Koche RP et al (2007) Genome‐wide maps of chromatin state in pluripotent and lineage‐committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J (2014) Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14: 838–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D et al (2011) DNA‐binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495 [DOI] [PubMed] [Google Scholar]
- 46. Deng T, Zhu ZI, Zhang S, Leng F, Cherukuri S, Hansen L, Mariño‐Ramírez L, Meshorer E, Landsman D, Bustin M (2013) HMGN1 modulates nucleosome occupancy and DNase I hypersensitivity at the CpG island promoters of embryonic stem cells. Mol Cell Biol 33: 3377–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository 42 with the data set identifier PXD007099. High‐throughput sequencing data have been deposited in the GEO database repository with the data set identifier GSE101802.