

Abstract
Heterochromatin protein‐1 (HP1) is a key component of heterochromatin. Reminiscent of the cohesin complex which mediates sister‐chromatid cohesion, most HP1 proteins in mammalian cells are displaced from chromosome arms during mitotic entry, whereas a pool remains at the heterochromatic centromere region. The function of HP1 at mitotic centromeres remains largely elusive. Here, we show that double knockout (DKO) of HP1α and HP1γ causes defective mitosis progression and weakened centromeric cohesion. While mutating the chromoshadow domain (CSD) prevents HP1α from protecting sister‐chromatid cohesion, centromeric targeting of HP1α CSD alone is sufficient to rescue the cohesion defects in HP1 DKO cells. Interestingly, HP1‐dependent cohesion protection requires Haspin, an antagonist of the cohesin‐releasing factor Wapl. Moreover, HP1α CSD directly binds the N‐terminal region of Haspin and facilitates its centromeric localization. The need for HP1 in cohesion protection can be bypassed by centromeric targeting of Haspin or inhibiting Wapl activity. Taken together, these results reveal a redundant role for HP1α and HP1γ in the protection of centromeric cohesion through promoting Haspin localization at mitotic centromeres in mammalian cells.
Keywords: centromere, cohesin, Haspin, heterochromatin, HP1
Subject Categories: Cell Cycle; Chromatin, Epigenetics, Genomics & Functional Genomics
Introduction
Chromosomal instability (CIN), a hallmark of many types of cancer, is caused by errors in the segregation of chromosomes during mitosis. The fidelity of chromosome segregation is controlled by a sophisticated cellular network, a key point of which is sister‐chromatid cohesion mediated by the ring‐shaped multi‐subunit cohesin complex 1. Sister‐chromatid cohesion is established during DNA replication in interphase. When vertebrate cells enter mitosis, the bulk of cohesin on chromosome arms is removed by its antagonist Wapl 2, 3, whereas cohesin at centromeres is protected from precocious removal to ensure chromosome bi‐orientation until anaphase onset 4, 5. It remains incompletely understood how centromeric cohesion is protected to the full extent from Wapl‐dependent cohesin release in mitosis 6.
Centromeres in most eukaryotes are composed of repetitive arrays of DNA, the majority of which are embedded in constitutive heterochromatin marked by the addition of methyl groups to histone H3 Lys9 (H3K9me) 7. The primary requirement of heterochromatin for precise mitotic chromosome segregation in S. pombe is the enrichment of cohesin at the centromere 8. Previous studies reported that this occurs through the physical association of Swi6, a fission yeast homolog of heterochromatin protein‐1 (HP1), with the cohesin subunits Rad21 9 and Psc3 10. However, such a direct interaction between HP1 and cohesin has not been demonstrated in higher eukaryotes 11, 12. As a key factor of heterochromatin structure, HP1 consists of an N‐terminal chromodomain (CD) and a C‐terminal chromoshadow domain (CSD) linked by a flexible hinge region 13. While the CD binds to di‐ and trimethylated H3K9 (H3K9me2/3) generated by the histone methyltransferase Suv39h 14, 15, 16, 17, the CSD functions as a dimerization module that forms a binding platform for other effecter proteins 18, 19, 20. Reminiscent to the distribution of cohesin in mitosis, the majority of HP1 proteins in mammals are released from chromosome arms when cells enter mitosis 21, 22, whereas a small amount of HP1 is retained at the centromere region 23, 24. The function of HP1 at mitotic centromeres in mammalian cells remains largely elusive 25, particularly with regard to whether the binding of HP1 CD to H3K9me2/3 is required to localize HP1 at mitotic centromeres 12, 26, 27, and whether and how HP1 is involved in the protection of centromeric cohesion 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39.
Here, using CRISPR/Cas9‐mediated knockout (KO), we clearly demonstrate that HP1 is required for the protection of mitotic centromere cohesion in human cells. We reveal the interaction between HP1 and Haspin, a protein kinase recently identified as an antagonist of Wapl 40, 41, 42, 43, 44. We further provide evidence that HP1 promotes centromeric localization of Haspin to antagonize Wapl‐mediated cohesin release at mitotic centromeres.
Results and Discussion
Double knockout (DKO) of HP1α and HP1γ causes defective mitosis progression
There are three closely related HP1 isoforms, HP1α, HP1β, and HP1γ, in mammals. To study the localization and function of HP1 proteins in mitosis, we used a near chromosomally stable HeLa cell line, which behaves like the non‐transformed retinal pigment epithelial (RPE‐1) cells with regard to few segregation errors in anaphase and the proper maintenance of chromosome bi‐orientation upon metaphase arrest induced by the proteasome inhibitor MG132 (Appendix Fig S1). Immunofluorescence microscopy showed concentrated localization of HP1α and HP1γ at inner centromeres in mitotic HeLa cells arrested with the spindle microtubule poison nocodazole (Fig 1A). Using the same fixation and staining procedure, we were not able to clearly detect endogenous HP1β (Fig 1A), or exogenously expressed EGFP‐fused HP1β (Fig EV1A), at mitotic centromeres. Similar results were obtained in nocodazole‐arrested mitotic RPE‐1 cells (Appendix Fig S2). Using wild‐type (WT) or a nickase mutant of CRISPR/Cas9 45, 46, 47, we established several stable clones, namely 2A4, 3A2, and 4A4, in which HP1α and HP1γ were knocked out with various single‐guide RNAs (sgRNAs) (Appendix Fig S3) 48. Genomic DNA sequencing confirmed indels predicted to cause frameshift mutations (Fig EV1B and C, Appendix Fig S4). Loss of HP1α and HP1γ proteins was confirmed by immunoblotting (Fig 1B) and immunofluorescence microscopy (see later in Fig EV3E).
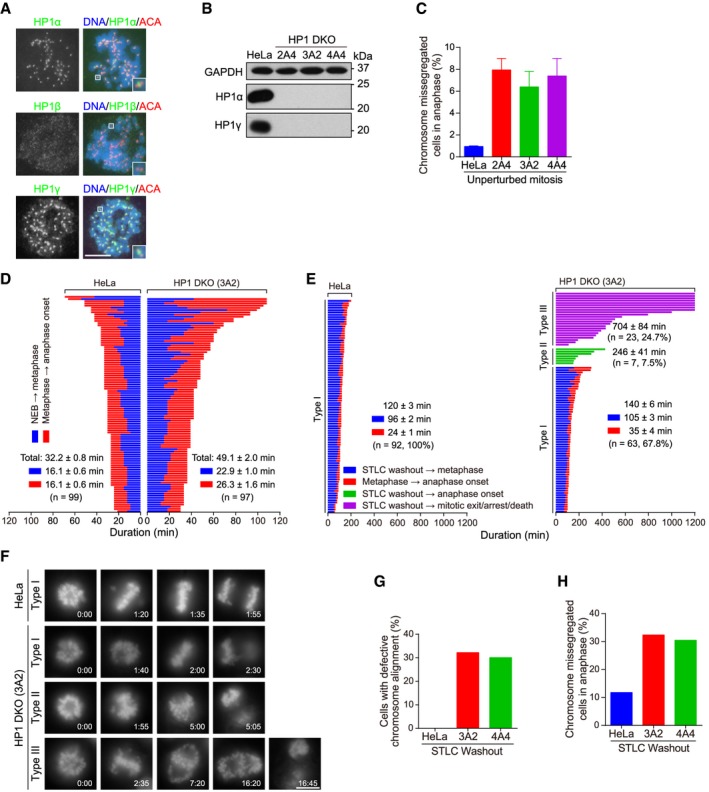
Figure 1. Double knockout (DKO) of HP1α and HP1γ causes defective mitosis progression.
-
AHeLa cells were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained.
-
BAsynchronous HeLa and the indicated HP1 DKO clones were immunoblotted.
-
CHeLa and HP1 DKO clones were fixed and stained with anti‐human centromere autoantibodies (ACA) and DAPI. The percentage of cells with lagging chromosomes was determined in 200 anaphase cells. Means and ranges are shown (n = 2).
-
DThe mitosis progression of HeLa and HP1 DKO clone 3A2 cells stably expressing H2B‐GFP was analyzed by time‐lapse live imaging. The time from nuclear envelope breakdown (NEB) to metaphase chromosome alignment, and from metaphase to anaphase onset, was determined. Fate profiles of cells were determined. See also Movies EV1 and EV2.
-
E, FHeLa and HP1 DKO clone 3A2 cells stably expressing H2B‐GFP were released from 5‐h treatment with STLC, followed by live imaging of mitosis progression. Fate profiles of mitotic cells (E), and the selected frames of the movies (F), are shown. Time stated in hours:minutes. See also Movies EV3,EV4 and EV5.
-
G, HThe percentage of mitotic cells with defective chromosome alignment (G) and chromosome missegregation (H) were quantified in 102 cells (for HeLa and clone 3A2) and 74 cells (for clone 4A4).
Figure EV1. DKO of HP1α and HP1γ causes defective mitosis progression (related to Fig 1).

-
AEGFP‐HP1β was transiently expressed in HeLa cells in which endogenous HP1β was stably knocked out by CRISPR/Cas9. Chromosome spreads prepared from nocodazole‐arrested mitotic cells were immunostained. Scale bar, 10 μm.
-
B, CGenomic DNA sequencing of HeLa‐derived clones in which HP1α (B), or HP1α and HP1γ (C), were knocked out. The genomic DNA PCR fragments were subcloned and sequenced. For clone 1D4 cells, all 12 bacterial colonies showed insertion of 13 bases. For clone 2A4 cells, all eight bacterial colonies showed insertion of one base. For clone 3A2 cells, all 10 bacterial colonies showed insertion of 38 bases. For clone 4A4 cells, 7 out of 12 bacterial colonies showed insertion of 22 bases, whereas the rest five bacterial colonies showed insertion of 57 bases and deletion of two bases.
-
DGenomic DNA sequencing of HeLa‐derived clone 2A5 cells in which HP1β was knocked out. The genomic DNA PCR fragments were subcloned and sequenced. Three out of 12 bacterial colonies showed deletion of 35 bases, whereas the rest nine bacterial colonies showed insertion of 34 bases.
-
E, FAsynchronous HeLa and the indicated HP1β KO clone 2A5 were immunoblotted (E), or were fixed and stained with ACA and DAPI. The percentage of cells with lagging chromosomes was determined in 100 anaphase cells.
-
GThe mitosis progression of HeLa and HP1 DKO clone 3A2 cells stably expressing H2B‐GFP were analyzed by time‐lapse live imaging (related to Fig 1D). The time from NEB to metaphase chromosome alignment, and from metaphase to anaphase onset, was determined (unpaired t‐test). Means and SDs are shown.
Source data are available online for this figure.
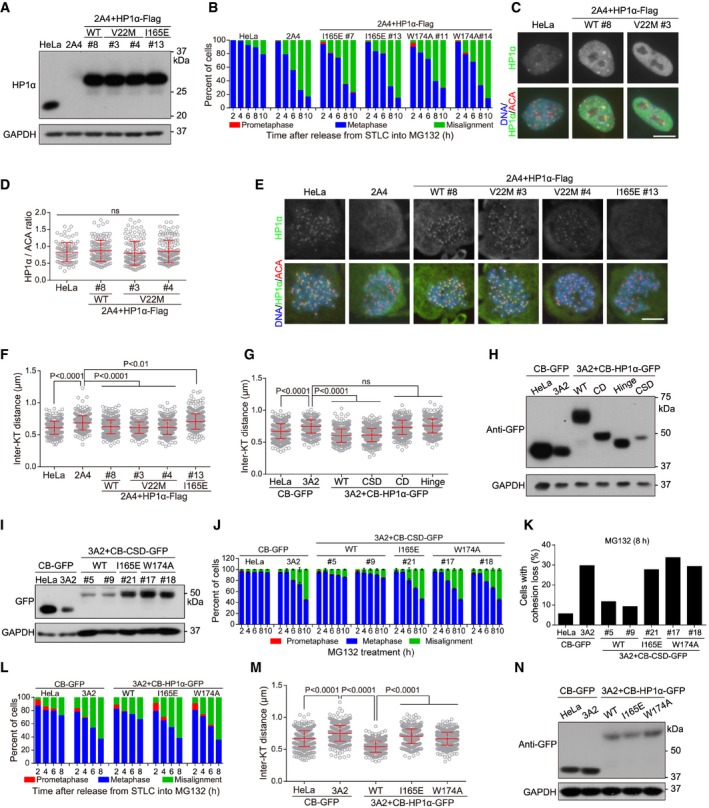
Figure EV3. CSD localization at centromeres is necessary and sufficient for HP1 to protect centromeric cohesion (related to Fig 3).
-
ALysates of asynchronous HeLa and HP1 DKO clone 2A4 cells with or without stable expression of the indicated exogenous HP1α were immunoblotted.
-
BHeLa and the indicated stable cell lines were released from 5‐h treatment with STLC into MG132‐containing medium, fixed at the indicated time points for DNA staining, and then quantified in around 100 cells.
-
CAsynchronous HeLa cells and the indicated stable cell lines were immunostained.
-
D, EHeLa and the indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. The centromeric HP1α/ACA immunofluorescence intensity ratio was determined on over 100 chromosomes in 10 cells (D) (unpaired t‐test). Example images are shown (E).
-
FHeLa and the indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with ACA antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
-
GHeLa and HP1 DKO clone 3A2 cells transiently expressing the indicated fusion proteins were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
-
HLysates of asynchronous HeLa and HP1 DKO clone 3A2 cells transiently expressing the indicated fusion proteins were immunoblotted.
-
ILysates of asynchronous HeLa and HP1 DKO clone 3A2 cells stably expressing the indicated fusion proteins were immunoblotted.
-
JThe indicated stable cell lines were analyzed for metaphase chromosome alignment in around 200 cells. Means and ranges are shown (n = 2).
-
KThe indicated stable cell lines were analyzed for cohesion loss in around 100 cells.
-
LHeLa and HP1 DKO clone 3A2 cells transiently expressing the indicated fusion proteins were released from 5‐h treatment with STLC into MG132‐containing medium, fixed at the indicated time points for DNA staining, and then quantified in around 100 cells.
-
MHeLa and HP1 DKO clone 3A2 cells transiently expressing the indicated fusion proteins were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
-
NLysates of asynchronous HeLa and HP1 DKO clone 3A2 cells transiently expressing the indicated fusion proteins were immunoblotted.
By scoring the percentage of formaldehyde‐fixed cells with lagging chromosomes in anaphase, we observed increased rate of chromosome missegregation in asynchronously growing HP1α and HP1γ DKO (HP1 DKO in short) cells (Fig 1C), but not in HP1β KO cells (Fig EV1D–F). Time‐lapse live imaging of cells stably expressing histone H2B fused to GFP (H2B‐GFP) showed prolonged duration of mitosis in HP1 DKO cells (49.1 ± 2.0 min versus 32.2 ± 0.8 min in control HeLa cells), particularly during the metaphase‐to‐anaphase transition (Figs 1D and EV1G, see Movies EV1 and EV2). Interestingly, there were strong mitosis progression defects in HP1 DKO cells during the recovery from transient mitotic arrest with S‐trityl‐L‐cysteine (STLC), an Eg5 inhibitor that reversibly blocks cells in prometaphase with monopolar spindles (Fig 1E and F, see Movies EV3,EV4 and EV5). As expected, almost all control HeLa cells underwent anaphase onset at 120 ± 3 min on average after STLC washout, following chromosome bi‐orientation. In contrast, HP1 DKO cells showed strong mitotic arrest with complex chromosome behaviors which could be classified into three categories. 67.8% of HP1 DKO cells (type I) behaved like control cells, except for a moderate delay in the establishment of chromosome bi‐orientation. In another 7.5% of cells (type II), anaphase onset initiated, frequently accompanied by chromosome missegregation, without the detectable alignment of all chromosomes at the metaphase plate. The remaining 24.7% of cells (type III) were defective in either establishing bi‐orientation or maintaining it for an appreciable period of time, then underwent mitotic death or slippage, or were persistently arrested in mitosis during the course of the imaging. Following transient mitotic arrest and release, similar defects in chromosome alignment (Fig 1G) and segregation (Fig 1H) were observed in HP1 DKO clones 3A2 and 4A4, as revealed by live cell imaging.
Taken together, these results indicate that HP1 DKO cells undergo prolonged mitosis duration and increased chromosome missegregation.
HP1α and HP1γ are redundantly required to protect mitotic centromere cohesion
We noticed that, when released from STLC into MG132, HP1 DKO cells were strongly impaired in maintaining metaphase chromosome alignment (Fig 2A). Similar defects were observed when HP1 DKO cells were directly subjected to MG132‐induced metaphase arrest (Fig 2B), though to a lesser extent likely due to the lack of STLC‐induced mitotic arrest 43. Time‐lapse live cell imaging further showed that MG132‐arrested metaphase HP1 DKO cells began to exhibit irreversible chromosome scattering from the metaphase plate much earlier than control cells (Fig 2C and D, see Movies EV6 and EV7).
Figure 2. HP1α and HP1γ are redundantly required to protect mitotic centromere cohesion.

-
AHeLa and HP1 DKO clones were released from 5‐h treatment with STLC into MG132‐containing medium and then fixed at the indicated time points for DNA staining. The percentage of mitotic cells in prometaphase, metaphase, and metaphase with some misaligned chromosomes, was determined in around 300 cells (n = 3).
-
BHeLa and HP1 DKO clones were exposed to MG132, then fixed at the indicated time points for DNA staining, and quantified in around 300 cells (n = 3).
-
C, DHeLa and HP1 DKO clones stably expressing H2B‐GFP were exposed to MG132 and subjected to live imaging. The time from the achievement of metaphase chromosome alignment to chromosome scattering was determined (unpaired t‐test) (C). Selected frames of the movies are shown (D). Time stated in hours:minutes. See also Movies EV6 and EV7.
-
E, FHeLa and HP1 DKO clones were exposed to MG132 for 8 h. Using mitotic chromosome spreads, the percentage of cells with cohesion loss was determined in around 100 cells (E). Example images are shown (F).
-
GHeLa and HP1 DKO clone 2A4 cells with or without stable expression of the indicated exogenous HP1 proteins were analyzed for metaphase chromosome alignment in around 300 cells (n = 3) as in (B).
-
HHeLa, HP1α KO, HP1γ KO, and HP1 DKO cells were analyzed for metaphase chromosome alignment in around 300 cells (n = 3) as in (B).
-
IThe indicated cell lines were analyzed for cohesion loss in around 100 cells as in (E).
These results prompted us to investigate whether sister‐chromatid cohesion is compromised in HP1 DKO cells. Inspection of chromosome spreads prepared from HP1 DKO cells arrested in metaphase with MG132 revealed a strong increase in premature chromatid separation (PCS) (Fig 2E and F). For example, after 8‐h treatment with MG132, the percentage of cells with cohesion loss increased from 5.7% in control cells to 21.3–27.4% in HP1 DKO cells (Appendix Fig S5). These results indicate an accelerated “cohesion fatigue” defect 49, 50, which is closely related to CIN 51, 52. We also found that the inter‐kinetochore (inter‐KT) distance between sister kinetochores of chromosome spreads prepared from nocodazole‐arrested mitotic cells was around 15% further apart in HP1 DKO cells than in the control cells (Fig EV2A–C), indicative of compromised cohesion at mitotic centromeres.
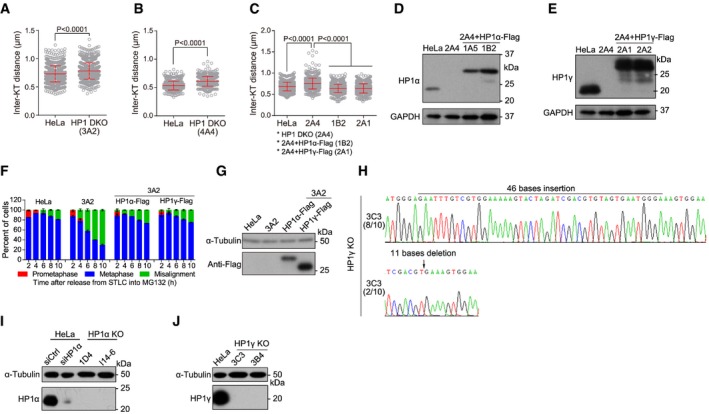
Figure EV2. HP1α and HP1γ are redundantly required to protect mitotic centromere cohesion (related to Fig 2).
-
A–CThe indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C or ACA antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test). Means and SDs are shown.
-
D, ELysates of asynchronous HeLa and HP1 DKO clone 2A4 cells with or without stable expression of the indicated exogenous HP1 were immunoblotted.
-
F, GHeLa and HP1 DKO clone 3A2 cells with or without transient expression of HP1α‐Flag or HP1γ‐Flag were analyzed for metaphase chromosome alignment in around 200 cells. Means and ranges are shown (F; n = 2). Lysates of asynchronous cells were immunoblotted (G).
-
HGenomic DNA sequencing of HP1γ KO clone 3C3 cells. The genomic DNA PCR fragments were subcloned and sequenced. Eight out of 10 bacterial colonies showed insertion of 46 bases, whereas the rest two bacterial colonies showed deletion of 11 bases.
-
I, JLysates of asynchronous HeLa and HP1α or HP1γ KO cells were immunoblotted.
Source data are available online for this figure.
Interestingly, these cohesion defects in HP1 DKO cells were effectively rescued by stable (Figs 2G and EV2C–E) or transient (Fig EV2F and G) expression of exogenous HP1α or HP1γ which was C‐terminally fused to the Flag‐tag and 6xHis tag (HP1‐Flag in short). Furthermore, KO of HP1α or HP1γ alone did not cause detectable defects in maintaining metaphase chromosome bi‐orientation and sister‐chromatid cohesion (Figs 2H and I, and EV2H–J).
Our results indicate that HP1α and HP1γ are redundantly required for the protection of centromeric cohesion in mitosis, which are in line with the recent report that inducible CRISPR/Cas9 KO of HP1α, HP1β, or HP1γ did not cause conspicuous mitotic defects in HeLa cells 53. Indeed, in our hand, small interfering RNA (siRNA)‐medicated knockdown of HP1α did not compromise metaphase chromosome alignment and sister‐chromatid cohesion in HeLa cells and RPE‐1 cells (Appendix Fig S6). Due to unknown reason, this is inconsistent with the previous reports that siRNA‐mediated HP1α knockdown caused cohesion defects 28, 35. In addition, a previous study reported that knockdown of HP1α and HP1γ by siRNA in human cells did not seem to affect the centromeric recruitment of the cohesin subunit Scc1 which was exogenously expressed 29. Compared with our current results, this may be due to the technical difficult to completely and simultaneously deplete HP1α and HP1γ by siRNA.
CSD is necessary for HP1α to protect centromeric cohesion
The restoration of proper centromeric cohesion in HP1 DKO cells by exogenous expression of HP1α alone allowed us to investigate the importance of CD and CSD for HP1α function in cohesion protection. We then stably expressed various mutant forms of exogenous HP1α‐Flag in HP1 DKO cells (Figs 3A and EV3A). Consistent with the importance of CSD for HP1 localization in mitosis 23, 32, we observed the failure in mitotic centromere localization of the CSD mutants of HP1α‐Flag (Fig 3B), which contained the mutations of I165E and W174A that disrupt the formation of CSD dimer and hydrophobic pocket, respectively 18, 20, 54. These two mutants were defective in maintaining metaphase chromosome alignment (Figs 3C and EV3B), sister‐chromatid cohesion (Fig 3D), and proper inter‐KT distance (Fig 3E), indicating the importance of CSD for HP1α to protect centromeric cohesin.
Figure 3. CSD localization at centromeres is necessary and sufficient for HP1 to protect centromeric cohesion.

- Lysates of asynchronous HeLa and HP1 DKO cells with or without stable expression of the indicated exogenous HP1α proteins were immunoblotted.
- HeLa and the indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. Scale bar, 10 μm.
- HeLa and the indicated stable cell lines were exposed to MG132, then fixed at the indicated time points for DNA staining, and quantified in around 300 cells (n = 3).
- HeLa and the indicated stable cell lines were exposed to MG132 for 8 h. Using mitotic chromosome spreads, the percentage of cells with cohesion loss was determined in around 100 cells.
- HeLa and the indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
- HeLa and the indicated stable cell lines were analyzed for metaphase chromosome alignment in around 300 cells (n = 3) as in (C).
- Lysates of asynchronous HeLa and HP1 DKO clone 2A4 cells stably expressing the indicated fusion proteins were immunoblotted.
- The indicated stable cell lines were analyzed for metaphase chromosome alignment in around 300 cells (n = 3) as in (C).
- The indicated stable cell lines were analyzed for cohesion loss in around 100 cells as in (D).
- HeLa and the indicated stable cell lines were analyzed for the inter‐KT distance on over 400 chromosomes in over 20 cells (unpaired t‐test) as in (E).
As expected, the CD mutant of HP1α‐V22M‐Flag, which contained the V22M mutation that selectively disrupts CD binding to H3K9me2/3 55, did not localize at heterochromatin in interphase cells (Fig EV3C). However, this mutant localized normally to mitotic centromeres (Fig EV3D and E) and restored proper centromeric cohesion in HP1 DKO cells (Figs 3F and EV3F). These results indicate that the interaction of CD with H3K9me2/3 is not necessary for HP1α to localize at mitotic centromeres and protect sister‐chromatid cohesion, which is in line with a previous report that Suv39h is dispensable for enrichment and mitotic protection of cohesin at centromeres in mouse embryonic fibroblasts 12.
Centromeric targeting of HP1α CSD alone is sufficient to support proper sister‐chromatid cohesion
We next examined the strength of centromere cohesion in HP1 DKO cells upon centromeric targeting of various domains/regions of HP1α, which were expressed as a fusion protein with the centromeric DNA binding domain of CENP‐B (CB in short where necessary) 56. We found that transient expression of CENP‐B‐fused full‐length HP1α and its CSD, but not CD and Hinge, effectively shortened the inter‐KT distance in HP1 DKO clone 3A2 cells (Fig EV3G and H). Importantly, stable expression of CENP‐B‐fused CSD (CB‐CSD) (Fig 3G), but not the I165E mutant, rescued the defects of HP1 DKO clone 2A4 cells in maintaining metaphase chromosome bi‐orientation (Fig 3H), sister‐chromatid cohesion (Fig 3I), and proper inter‐KT distance (Fig 3J). Similar centromeric cohesion defects were observed in HP1 DKO clone 3A2 cells which either stably expressed the W174A mutant of CB‐CSD (Fig EV3I–K), or transiently expressed the I165E or W174A mutant of CB‐HP1α (Fig EV3L–N). Thus, centromeric targeting of CSD alone effectively rescues sister‐chromatid cohesion defects in HP1 DKO cells.
HP1 CSD interacts with the N‐terminus of Haspin
The incapability of the W174A mutant of CB‐CSD or CB‐HP1α to rescue centromeric cohesion defects in HP1 DKO cells suggests the involvement of CSD‐interacting proteins in HP1‐mediated cohesion protection. In addition, a previous study demonstrated an interaction between Swi6 and Hrk1 (Haspin‐related kinase 1, the Haspin homologue) in fission yeast 57. We then examined whether HP1 associates with Haspin in mammalian cells.
We detected reciprocal co‐immunoprecipitation (co‐IP) of exogenously expressed Haspin and HP1α in nocodazole‐arrested mitotic cell lysates (Fig 4A and B). In contrast, co‐IP of Haspin with the I165E or W174A mutant of HP1α was hardly detectable (Fig 4B and C). Moreover, GST‐fused HP1α and CSD, but not CD and Hinge, selectively pulled down SFB‐Haspin (SFB is a triple tag of S‐tag, Flag‐tag, and streptavidin‐binding peptide) from mitotic cell lysates (Fig EV4A), indicating that CSD is sufficient to bind Haspin. In addition, the N‐terminal non‐catalytic region (residues 1–480) of Haspin tagged with Myc and 6xHis (Haspin‐N480‐Myc in short) was readily pulled down by GST‐HP1α from mitotic cell lysates (Fig EV4B). Importantly, purified recombinant MBP‐HP1α was directly pulled down by GST‐fused residues 1–350 of Haspin (GST‐Haspin‐N350) (Fig 4D). Thus, the CSD of HP1α directly binds the N‐terminus of Haspin in vitro and in human cells.
Figure 4. Haspin binds HP1 CSD and is required for HP1‐dependent cohesion protection.

-
ALysates of nocodazole‐arrested mitotic HeLa cells transiently expressing HP1α‐Flag and Haspin‐GFP were subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies.
-
BLysates of nocodazole‐arrested mitotic HeLa cells transiently expressing SFB‐Haspin and HP1α‐GFP (WT and the indicated mutants) were subjected to immunoprecipitation followed by immunoblotting. S. exp., short exposure.
-
CLysates of nocodazole‐arrested mitotic HeLa cells transiently expressing Haspin‐GFP and HP1α‐Flag (WT and the indicated mutants) were subjected to immunoprecipitation followed by immunoblotting.
-
DPurified MBP‐HP1α‐6xHis was subjected to pulldown by GST or GST‐Haspin‐N350, followed by anti‐HP1α immunoblotting and CBB staining. Note the presence of a significant amount of partial GST‐Haspin protein.
-
EPurified MBP‐HP1α‐6xHis was subjected to pulldown by GST or Haspin (80‐130)‐GST for 1 h, followed by anti‐MBP immunoblotting and CBB staining.
-
FLysates of nocodazole‐arrested mitotic HeLa cells transiently expressing HP1α‐Flag and Haspin‐GFP (WT or ΔPxVxL) were subjected to immunoprecipitation followed by immunoblotting. L. exp., long exposure.
-
GLysates of asynchronous HeLa, Haspin KO clone D2 and HP1 DKO clone 2A4 stably expressing the indicated fusion proteins were immunoblotted.
-
H, IThe indicated stable cell lines were exposed to MG132, then fixed at the indicated time points for DNA staining, and quantified in around 200 cells. Means and ranges are shown (H; n = 2). After treatment with MG132 for 6 h, using mitotic chromosome spreads, the percentage of cells with cohesion loss was determined in around 100 cells (I).
-
JLysates of asynchronous HeLa, Haspin KO clone D2 cells with or without stable expression of Haspin‐GFP (WT or ΔPxVxL) were immunoblotted.
-
KThe indicated stable cell lines were analyzed for metaphase chromosome alignment in around 200 cells as in (H). Means and ranges are shown (n = 2).
-
LThe indicated stable cell lines were analyzed for sister‐chromatid cohesion in around 100 cells as in (I).
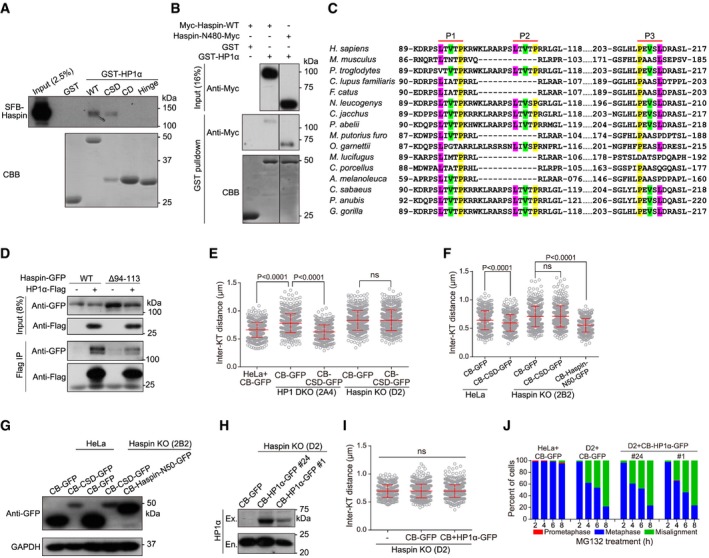
Figure EV4. Haspin binds HP1 CSD and is required for HP1‐dependent cohesion protection (related to Fig 4).
-
ALysates of nocodazole‐arrested mitotic HeLa cells stably expressing SFB‐Haspin were subjected to pulldown by GST or GST‐fused HP1α (WT and the indicated domains/fragments), followed by anti‐Flag immunoblotting and CBB staining for GST proteins.
-
BLysates of nocodazole‐arrested mitotic HeLa cells transiently expressing Myc‐Haspin or Haspin‐N480‐Myc were subjected to pulldown by GST or GST‐fused HP1α.
-
CAlignment of Haspin N‐terminal sequences surrounding three PxVxL motifs.
-
DLysates of nocodazole‐arrested mitotic HeLa cells transiently expressing HP1α‐Flag and Haspin‐GFP (WT or Δ94–113) were subjected to immunoprecipitation followed by immunoblotting. Parts of the images are shown in Fig 4A.
-
EThe indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
-
F, GHeLa and Haspin KO clone 2B2 cells transiently expressing the indicated fusion proteins were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (F) (unpaired t‐test). Asynchronous cell lysates were immunoblotted (G).
-
HLysates of asynchronous Haspin KO clone D2 cells stably expressing the indicated fusion proteins were immunoblotted. Ex., exogenous, En., endogenous.
-
IHaspin KO clone D2 cells stably expressing the indicated fusion proteins were treated with nocodazole for 3 h. Mitotic chromosome spreads were stained with CENP‐C antibodies and DAPI. The inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test).
-
JHeLa and Haspin KO clone D2 cells stably expressing the indicated fusion proteins were exposed to MG132, then fixed at the indicated time points for DNA staining, and quantified in around 100 cells.
The defect of Haspin in the co‐IP with the HP1α‐W174A mutant suggests that Haspin might have the PxVxL motif (consensus [P/L]‐X‐V‐X‐[M/L/V]) which interacts with the hydrophobic pocket of CSD dimer 54, 58. We noticed in the N‐terminus of human Haspin a short fragment, 94‐LTVTPKRWKLRARPSLTVTP‐113, which contains two potential PxVxL motifs (P1 and P2 in short, Fig EV4C). Indeed, MBP‐HP1α was efficiently pulled down by Haspin residues 80–130 fused to GST (Fig 4E). Since deleting residues 94–113 of Haspin did not eliminate the co‐IP of Haspin with HP1α (Fig EV4D), we additionally deleted another conserved PxVxL motif, 108‐PEVSL‐112 (P3, Fig EV4C). We confirmed that the Haspin‐ΔPxVxL‐GFP mutant lacking these three potential PxVxL motifs was defective in the co‐IP with HP1α‐Flag (Fig 4F). Thus, HP1α CSD directly interacts with the N‐terminus of Haspin in a PxVxL motif‐dependent manner.
Haspin is required for HP1 to protect centromeric cohesion
Given that HP1α interacts with Haspin, and that the cohesion defects in HP1 DKO cells are reminiscent of those in Haspin KO cells 43, we next investigated whether there is a functional link between HP1 and Haspin in centromeric cohesion protection. Interestingly, stable expression of CB‐CSD in Haspin KO (clone D2) cells was incapable of maintaining metaphase chromosome bi‐orientation, sister‐chromatid cohesion, and proper inter‐KT distance (Figs 4G–I and EV4E). Similarly, transient expression of CB‐CSD did not restore proper inter‐KT distance in another Haspin KO clone 2B2 (Fig EV4F and G). In contrast, as a positive control 43, stable expression of CENP‐B‐fused Haspin residues 1–50 (CB‐Haspin‐N50‐GFP) was able to do so. Similarly, stable expression of CB‐HP1α also failed to support proper inter‐KT distance and metaphase chromosome alignment in Haspin KO cells (Fig EV4H–J). Thus, even when forced targeted to centromeres, HP1α requires Haspin to protect centromeric cohesion.
Furthermore, we stably expressed Haspin‐GFP (WT or the ΔPxVxL mutant) into Haspin KO cells (Fig 4J). As we previously reported 43, 44, due to the weakened centromeric cohesion, Haspin KO cells showed strong defects in maintaining chromosome bi‐orientation and sister‐chromatid cohesion during MG132‐induced metaphase arrest (Fig 4K and L). Importantly, these defects were efficiently rescued by Haspin‐GFP but not by Haspin‐ΔPxVxL‐GFP. Thus, the interaction with Haspin is important for HP1 to protect centromeric cohesion in mitosis.
HP1 promotes centromeric localization of Haspin
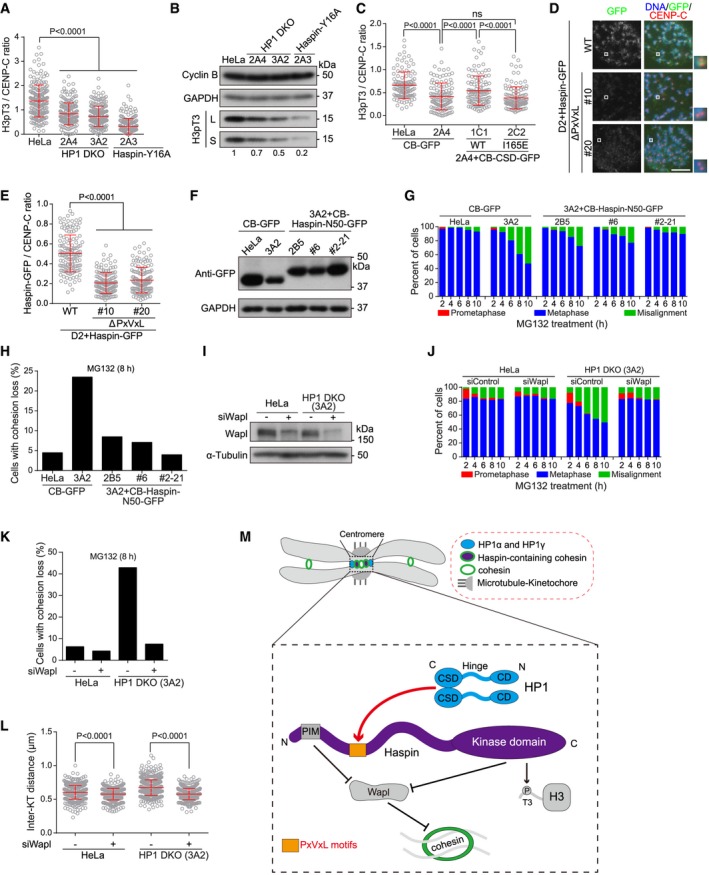
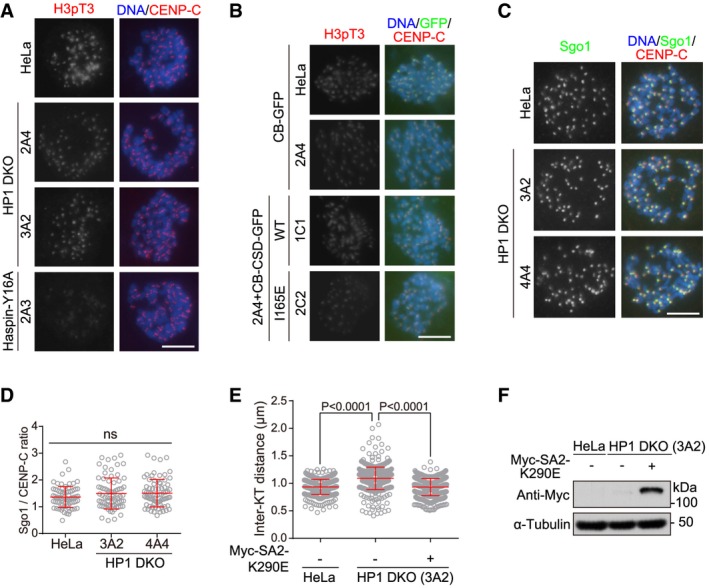
The cohesin regulatory subunit Pds5B promotes Haspin localization at centromeres through binding Haspin at its N‐terminal Pds5‐interacting motif (PIM) 42, 43, which is similar to the Pds5‐binding YSR motifs identified in Wapl and Sororin 59. We further found that the PIM mutant of Haspin deficient in binding Pds5B is largely impaired in supporting proper centromeric cohesion 43, indicating that centromeric localization is a prerequisite for Haspin to protect cohesion. We then examined the centromeric localization of Haspin in HP1 DKO cells. During mitosis, Haspin localizes predominantly to centromeres and phosphorylates histone H3 at Thr3 (H3pT3), which is particularly concentrated at centromeres 40, 57, 60. We thus used centromeric H3pT3 as the indicator for the extent of centromeric localization of endogenous Haspin. Immunofluorescence microscopy showed that centromeric H3pT3 on mitotic chromosomes was significantly lower in HP1 DKO cells than in control HeLa cells (Figs 5A and EV5A). Consistently, immunoblotting of lysates prepared from nocodazole‐arrested mitotic cells showed that total H3pT3 was reduced by 30–50% in HP1 DKO cells (Fig 5B). Moreover, stable expression of CB‐CSD (see Fig 3G), but not the I165E mutant, largely increased centromeric H3pT3 in mitotic HP1 DKO cells (Figs 5C and EV5B). These results suggest that HP1α and HP1γ are required for the full localization of Haspin at mitotic centromeres. Consistently, compared to Haspin‐GFP, the Haspin‐ΔPxVxL‐GFP mutant was around 2.2‐fold less concentrated at mitotic centromeres (Fig 5D and E). Our observations in HeLa cells are in line with the report that H3pT3 and Hrk1 localization were abolished specifically at the heterochromatic regions in Swi6‐deleted fission yeast 57.
Figure 5. HP1 promotes centromeric localization of Haspin to antagonize Wapl.
-
AHeLa, HP1 DKO clones, endogenous Haspin‐Y16A mutant expressing cells were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. The centromeric H3pT3/CENP‐C immunofluorescence intensity ratio was determined on around 200 chromosomes in 13 cells (unpaired t‐test) (see example images in Fig EV5A).
-
BLysates of nocodazole‐arrested mitotic cells described in (A) were immunoblotted. S, short exposure. L, long exposure.
-
CHeLa and HP1 DKO clone 2A4 cells stably expressing the indicated fusion proteins were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. The centromeric H3pT3/CENP‐C immunofluorescence intensity ratio was determined on around 200 chromosomes in 10 cells (unpaired t‐test) (see example images in Fig EV5B).
-
D, EThe indicated stable cell lines were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. Example images are shown (D). The centromeric GFP/CENP‐C immunofluorescence intensity ratio was determined on around 140 chromosomes in 10 cells (E) (unpaired t‐test). Scale bar, 10 μm.
-
FLysates of asynchronous HeLa and HP1 DKO clone 3A2 cells stably expressing the indicated fusion proteins were immunoblotted.
-
G, HThe indicated stable cell lines were exposed to MG132, then fixed at the indicated time points for DNA staining, and quantified in around 100 cells (G). After treatment with MG132 for 8 h, using mitotic chromosome spreads, the percentage of cells with cohesion loss was determined in around 100 cells (H).
-
I–LHeLa and HP1 DKO clone 3A2 cells transfected the indicated siRNAs were immunoblotted (I); or were exposed to MG132, fixed at the indicated time points for DNA staining, and quantified in around 100 cells (J); or were used to prepare chromosome spreads after 8‐h treatment with MG132, and then, the percentage of cells with cohesion loss was determined in around 100 cells (K); or were treated with nocodazole for 3 h to make chromosome spreads, stained with CENP‐C antibodies and DAPI, and then, the inter‐KT distance was measured on over 400 chromosomes in over 20 cells (L) (unpaired t‐test).
-
MSchematic depiction of the role for HP1 in localizing Haspin at mitotic centromeres to antagonize the cohesin‐release activity of Wapl.
Figure EV5. HP1 promotes centromeric localization of Haspin to antagonize Wapl (related to Fig 5).
-
A, BExample images of cells described in Fig 5A and C.
-
C, DHeLa and the indicated HP1 DKO clones were treated with nocodazole for 3 h. Mitotic chromosome spreads were immunostained. Example images are shown (C). The centromeric Sgo1/CENP‐C immunofluorescence intensity ratio was determined on around 100 chromosomes in 10 cells (D) (unpaired t‐test).
-
E, FChromosome spreads prepared from nocodazole‐arrested mitotic HeLa and HP1 DKO clone 3A2 cells transiently expressing Myc‐SA2‐K290E were immunostained. Then, the inter‐KT distance was measured on over 400 chromosomes in over 20 cells (unpaired t‐test) (E), and lysates of asynchronous cells were immunoblotted (F).
We previously showed that the PIM of Haspin competes with the YSR motif of Wapl for binding Pds5B, and that centromeric targeting of the PIM‐containing fragment of Haspin as the form of CB‐Haspin‐N50‐GFP rescues the centromeric cohesion defect in Haspin KO cells (see Fig 4G–I) 43. We hypothesized that if HP1 protects centromeric cohesion through localizing Haspin, artificially targeting Haspin to centromeres might bypass the need for HP1 in cohesion protection. Indeed, stable expression of CB‐Haspin‐N50‐GFP was sufficient to maintain proper metaphase chromosome alignment and sister‐chromatid cohesion in HP1 DKO clone 3A2 cells (Fig 5F–H).
Previous studies showed that Swi6/HP1 has a pivotal function in localizing Sgo1 at mitotic centromeres through their direct association in fission yeast 30, and probably also in mammalian cells 19, 29, 30, 35. However, it seemed that Sgo1 localization at inner centromeres was not conspicuously altered in nocodazole‐arrested mitotic HP1 DKO cells (Fig EV5C and D), suggesting that HP1 may not play a critical role in localizing Sgo1 at mitotic centromeres of human cells 61. Indeed, a HP1‐binding‐deficient mutant of Sgo1 was functional in centromeric cohesion protection, and localized normally to mitotic centromeres in HeLa cells 32.
Inhibiting Wapl activity bypasses the need for HP1 in centromeric cohesion protection
Wapl releases the majority of cohesin from chromosome arms in early mitosis to allow sister‐chromatid resolution 2, 3. Wapl activity in cohesin release is inhibited at mitotic centromeres by Haspin 43, 44, Sororin and Sgo1 59, 62, 63, 64, 65, 66, 67. Given that HP1 promotes centromeric localization of Haspin, we wondered whether aberrantly increased local Wapl activity, as a result of delocalization of centromeric Haspin, might account for the centromeric cohesion defects in HP1 DKO cells. Interestingly, Wapl depletion by siRNA prevented metaphase chromosome misalignment and PCS in HP1 DKO clone 3A2 cells (Fig 5I–K). Moreover, proper inter‐KT distance was restored in HP1 DKO cells by Wapl depletion (Fig 5L), or by transient expression of Myc‐SA2‐K290E (Fig EV5E and F), a Wapl‐binding‐deficient SA2 mutant which is resistant to being released by Wapl 63. Thus, Wapl inhibition rescues centromeric cohesion defects in HP1 DKO cells.
Conclusion
Taken together, our results indicate that HP1α and HP1γ play a redundant role in the protection of centromeric cohesion in human cells. We propose that, together with Pds5B, HP1 ensures the full occupancy of Haspin at mitotic centromeres, thereby enabling Haspin to prevent Wapl‐Pds5B interaction and Wapl‐mediated cohesin release (Fig 5M). This study reveals a molecular mechanism by which HP1 links centromeric heterochromatin to the protection of sister‐chromatid cohesion at mitotic centromeres of mammalian cells.
Materials and Methods
Cell culture, plasmids, siRNA, transfection, and drug treatments
All cells were cultured in DMEM supplemented with 1% penicillin/streptomycin and 10% FBS (Gibco), and maintained at 37°C with 5% CO2. The HeLa cell line, originally from Dr. Jonathan Higgins laboratory at Brigham and Women's Hospital and Harvard Medical School, has been used in a number of studies for the regulation of mitotic chromosome segregation. HeLa cells stably expressing SFB‐Haspin and HP1‐Flag proteins were isolated and maintained in puromycin (Calbiochem) at 1.0 and 0.5 μg/ml, respectively. Cells stably expressing H2B‐GFP, CENP‐B fusion proteins, and Haspin‐GFP (WT or mutants) were isolated and maintained in blasticidin (Sigma) at 3 and 1.5 μg/ml, respectively. Haspin KO cells (clones D2 and 2B2) and endogenous Haspin‐Y16A cells (clone 2A3) were previously described 43.
The plasmids of Haspin‐GFP, SFB‐Haspin, pBos‐CENP‐B‐Haspin‐N50‐GFP, and Myc‐SA2‐K290E were previously described 43, 44. pBos‐HP1α‐GFP was constructed by replacing the H2B fragment in pBos‐H2B‐GFP (Clontech) with the KpnI‐/BamHI‐digested PCR fragments of HP1α gene. HP1α‐Flag‐6xHis and HP1γ‐Flag‐6xHis were constructed by replacing the GFP fragment in pEF‐IRES‐P‐EGFP‐Flag‐6xHis with the NotI/SfaAI‐digested PCR fragments of HP1α and HP1γ genes. To make pBos‐CENP‐B‐HP1 (WT or mutants)‐GFP constructs, the PCR fragments were subcloned into the BamHI site of pBos‐CENP‐B‐GFP. Haspin‐N480‐Myc‐6xHis was constructed by the PCR fragments encoding Haspin residues 1‐480 into the KpnI and EcoRV sites of pEF6‐Myc‐6xHis (Invitrogen). All point mutations were introduced with the QuikChange II XL site‐directed mutagenesis kit (Agilent Technologies). All plasmids were sequenced to verify desired mutations and absence of unintended mutations.
Cells were transfected twice with siRNA in a 24‐h interval. The following siRNA duplexes were ordered from Integrated DNA Technologies (IDT): siWapl (5′‐CGGACUACCCUUAGCACAAdTdT‐3′), siHP1α (5′‐CCUGAGAAAAACUUGGAUUdTdT‐3′). Plasmid and siRNA transfections were done with Fugene 6 (Promega) and Lipofectamine RNAi MAX (Invitrogen), respectively. Cells were arrested in prometaphase with 1.0 μM nocodazole (Selleckchem). Drugs used in this study were STLC (5 μM, Tocris Bioscience) and MG132 (10 μM, Sigma). Mitotic cells were collected by selective detachment with “shake‐off”.
Antibodies
Rabbit polyclonal antibodies used were to H3pT3 (B8633, Dr. Jonathan Higgins), Wapl (A300‐268A, Bethyl; for immunoblotting), cyclin B1 (clone D5C10, Cell Signaling Technology, CST), GFP (A11122, Invitrogen), MBP (E8032, New England BioLabs), and GAPDH (14C10, CST). Mouse monoclonal antibodies used were to HP1α (MAB3446 for immunoblotting; MAB3584 for immunostaining), HP1β (MAB3448), HP1γ (MAB3450), all from Millipore, and to α‐tubulin (T‐6047, Sigma), 6xHis‐tag (GNI4110‐HS, GNI), Flag‐tag (M2, Sigma), Myc‐tag (4A6, Millipore), and Sgo1 (3C11, Abnova). Guinea pig polyclonal antibodies against CENP‐C were from MBL (PD030). Human centromere autoantibodies were from Immunovision. Secondary antibodies for immunoblotting were goat anti‐rabbit or horse anti‐mouse IgG‐HRP (CST). Secondary antibodies for immunostaining were donkey anti‐rabbit IgG‐Alexa Fluor 488 or Cy3 (Jackson ImmunoResearch), anti‐mouse IgG‐Alexa Fluor 488 or 546 (Invitrogen), anti‐human IgG‐Alexa Fluor 647 (Jackson ImmunoResearch), goat anti‐guinea pig IgG‐Alexa Fluor 647 (Invitrogen).
CRISPR/Cas9‐mediated editing of HP1 genes in HeLa cells
To express single‐guide RNAs (sgRNAs) for human HP1 genes, oligonucleotides were ordered from IDT, annealed, and cloned into the BbsI site of dual Cas9 and sgRNA expression vector pX330 or pX462 (Dr. Feng Zhang laboratory, Addgene, #42230 and #62987). The plasmids were transfected into HeLa cells using Fugene 6 (Promega). After 48‐h incubation, cells were split individually to make a clonal cell line with selection using 1 μg/ml puromycin for 2–3 days. The sgRNA target DNA sequences preceding a 5′‐NGG PAM (protospacer adjacent motif) are shown in Appendix Fig S3. HP1α KO clone 1D4 was obtained by means of double‐nicking with the Cas9‐D10A mutant and a pair of sgRNAs targeting two sequences (5′‐AGAACTGTCAGCTGTCCGCTtgg‐3′, 17 bases downstream of the start codon, and 5′‐GGATGAGGAGGAGTATGTTGtgg‐3′) which would largely reduce potential off‐target effects. HP1γ was further knocked out in HP1α KO clone 1D4 cells using sgRNA targeting sequence of 5′‐TTTTCCACGACAAATTCTTCagg‐3′ (82 bases downstream of the start codon of HP1γ gene; for clone 2A4, the PAM sequence in lower case), or using a pair of sgRNAs targeting two sequences (5′‐TTTTCCACGACAAATTCTTCagg‐3′ and 5′‐CTAGATCGACGTGTAGTGAAtgg‐3′; for clones 3A2 and 4A4). The HP1γ KO clone 3C3 was obtained from HeLa cells with a sgRNA targeting a sequence (5′‐ACGTGTAGTGAATGGGAAAGtgg‐3′) which is 114 bases downstream of the start codon. The HP1β KO clone 2A5 was obtained from HeLa cells using a pair of sgRNAs targeting two sequences (5′‐CTTTGCCCTTTACCACTCGAcgg‐3′, 87 bases downstream of the start codon, and 5′‐GGAGTACCTCCTAAAGTGGAagg‐3′). Clones with loss of HP1 proteins were isolated by immunostaining and confirmed by immunoblotting. The genomic DNA PCR fragments were subcloned into pBluescript II KS (−) and transformed into E. coli, and then, certain numbers of bacterial colonies were sequenced to confirm the gene disruption. The PCR primers were as follows: HP1α forward 5′‐CCCTTTTCATCCCATCAAGA‐3′ and reverse 5′‐TCCACAAGCGGAGAGATTTT‐3′; HP1β forward 5′‐AGAGTGCTCCCTAATGCCCT‐3′ and reverse 5′‐ATAAGCCATGAAGGTGGGGAC‐3′; HP1γ forward 5′‐AAGGTGTCTGCCCTGGGATA‐3′ and reverse 5′‐GACACCTACAGGAAAGCAGGT‐3′.
Fluorescence microscopy, time‐lapse live cell imaging, and statistical analysis
Cells grown on coverslips were fixed with 2% paraformaldehyde (PFA) in PBS for 10 min followed by extraction with 0.1 or 0.5% Triton X‐100 in PBS for 5 min. Mitotic HeLa cells obtained by shake‐off were re‐attached to glass coverslips by Cytospin at 1,500 rpm at 5 min, pre‐extracted for 5 min with 1% Triton X‐100 in PHEM (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, and 2 mM MgCl2, pH 6.9), and then fixed with 4% PFA in PHEM for 20 min. For immunostaining HP1, cells were fixed with 1% PFA in PBS containing 0.2% Triton X‐100 for 10 min, then in 50 mM NH4Cl for 30 min, and in 0.2% Triton X‐100 in PBS for 10 min 31. In general, to produce chromosome spreads, mitotic cells obtained by selective detachment were incubated in 75 mM KCl for 10 min, re‐attached to glass coverslips by Cytospin, and then fixed with 2% PFA in PBS for 20 min followed by extraction with 0.1% Triton X‐100 in PBS for 5 min. Fixed cells were stained with primary antibodies for 1–2 h and secondary antibodies for 1 h, all with 3% BSA in PBS with 0.1–0.5% Triton X‐100 and at room temperature. DNA was stained for 10 min with DAPI.
Fluorescence microscopy was carried out at room temperature using a Nikon ECLIPSE Ni microscope with a Plan Apo Fluor 60× Oil (NA 1.4) objective lens and a Clara CCD (Andor Technology). The inter‐KT distance was measured using the centromere marker CENP‐C or ACA on over 20 kinetochores per cell in at least 20 cells. Distance was determined by drawing a line from the outer kinetochore extending to the outer edge of its sister kinetochore. The length of the line was calculated using the imaging software of NIS‐Elements BR (Nikon). Quantification of fluorescent intensity was carried out with ImageJ (NIH) using images obtained at identical illumination settings. Briefly, on chromosome spreads, the average pixel intensity of H3pT3, HP1α, Haspin‐GFP, or Sgo1 staining at inner centromeres, defined as regions falling within a 3‐pixel‐diameter circle at paired centromeres, was determined using ImageJ. ACA or CENP‐C intensity was determined within a 9‐pixel‐diameter circle encompassing single centromere dots. After background correction, the ratio of H3pT3/CENP‐C, HP1α/ACA, Haspin‐GFP/CENP‐C, or Sgo1/CENP‐C intensity was calculated for each centromere. The average number of chromosomes in our HeLa cell line is 62.6. Cohesion loss was defined as over 20% sister‐chromatid pairs (> 25 separated chromatids) in a cell were separated (Appendix Fig S5).
Time‐lapse live cell imaging was carried out with the GE DV Elite Applied Precision DeltaVision system (GE Healthcare) equipped with Olympus oil objectives of 40× (NA 1.35) UApo/340 and an API Custom Scientific CMOS camera, and Resolve3D softWoRx imaging software. Cells expressing H2B‐GFP were plated in four‐chamber glass‐bottomed 35‐mm dishes (Cellvis) coated with poly‐D‐lysine, and filmed in a climate‐controlled and humidified environment (37°C and 5% CO2). Images were captured every 3 min (Movies EV1 and EV2), every 5 min (Movies EV3,EV4 and EV5), and every 6 min (Movies EV6 and EV7). The acquired images were processed using Adobe Photoshop and Adobe Illustrator. Statistical analyses were carried out by unpaired t‐test using GraphPad Prism 6.
Immunoblotting and immunoprecipitation
SDS–PAGE, immunoblotting, and immunoprecipitation were carried out using standard procedures. Cell lysates were prepared in standard SDS sample buffer or Lamond buffer for immunoblotting. For the co‐immunoprecipitation, cells were lysed in P150 buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% NP‐40, or 0.5% Triton X‐100) with 2 mM MgCl2, 10% glycerol, 1 mM dithiothreitol (DTT), protease inhibitor cocktail (P8340, Sigma), 1 mM PMSF, 0.1 μM okadaic acid (Calbiochem), 10 mM NaF, 20 mM β‐glycerophosphate, and benzonase (Merck). After removal of insoluble materials by high‐speed centrifugation, lysates were precleared with GammaBind G Sepharose (17‐0885‐02, GE Healthcare). Lysates were incubated with antibodies for 3 h at 4°C before addition of GammaBind G Sepharose for a further 1 h. Beads were washed several times with the lysis buffer, boiled in standard SDS sample buffer, and subject to immunoblotting.
Protein purification and GST pulldown
Plasmids expressing MBP‐ or GST‐fused HP1α proteins were constructed by subcloning PCR fragments into pGEX‐MBP‐6xHis and pGEX‐4T1, respectively. The plasmid for GST‐Haspin‐N350 was constructed by subcloning Haspin residues 1–350 into pGEX‐4T1. The plasmids were transformed into BL21 (DE3)‐competent cells (Stratagene). Cells were grown in LB broth under antibiotic selection at 37°C until OD600 at 0.6–0.7, and protein expression was induced with 0.1–0.4 mM IPTG at 16°C for 16 h. Cells were lysed by sonication in buffer A (50 mM Tris–HCl, pH 8.0, 300 mM NaCl, for MBP fusion proteins) or buffer B (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1% Triton X‐100 for GST fusion proteins). The lysate was clarified by centrifugation and incubated with Amylose Resin (BioLab) or Glutathione Sepharose 4B (GE Healthcare) in lysis buffer. The resin was washed with lysis buffer or eluted with 10 mM maltose or 10 mM glutathione.
For GST‐HP1 pulldown of Haspin from lysates, nocodazole‐arrested mitotic HeLa cells expressing exogenous Haspin were lysed in P150 buffer. The lysates were precleared with Glutathione Sepharose 4B beads and then incubated with GST fusion proteins immobilized to Glutathione Sepharose 4B beads for 2 h. For GST‐Haspin‐N350 or Haspin (80‐130)‐GST pulldown of purified recombinant MBP‐HP1α‐6xHis, the recombinant proteins were incubated for 1 h with bead‐immobilized GST fusion proteins in 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X‐100. Following all these pulldown assays which were conducted at 4°C, the beads were washed 3–5 times with the same buffer and subjected to analysis by immunoblotting and CBB staining.
Author contributions
QY and QC designed and performed the majority of the experiments and analysis with the contributions from CL and HY. ZZ, XX, MZ, FQ, and LZ contributed reagents. FW conceived and supervised the project, designed the experiments, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Dr. Xiangwei He for critically reading the manuscript and the LSI core facility for technical assistance. This work was supported by grants to FW from National Key Research and Development Program of China (2017YFA0503600) and the National Natural Science Foundation of China (31571393, 31771499, 31561130155, 31322032, and 31371359), the Natural Science Foundation of Zhejiang Province (LR13C070001 and Y17C070004 to HY), and the Fundamental Research Funds for the Central Universities. FW is a recipient of the National Thousand Young Talents Award.
EMBO Reports (2018) 19: e45484 29491004
See also: https://doi.org/10.15252/embr.201845980 (April 2018)
References
- 1. Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K (2008) The cohesin ring concatenates sister DNA molecules. Nature 454: 297–301 [DOI] [PubMed] [Google Scholar]
- 2. Gandhi R, Gillespie PJ, Hirano T (2006) Human Wapl is a cohesin‐binding protein that promotes sister‐chromatid resolution in mitotic prophase. Curr Biol 16: 2406–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM (2006) Wapl controls the dynamic association of cohesin with chromatin. Cell 127: 955–967 [DOI] [PubMed] [Google Scholar]
- 4. Waizenegger IC, Hauf S, Meinke A, Peters JM (2000) Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103: 399–410 [DOI] [PubMed] [Google Scholar]
- 5. Haarhuis JH, Elbatsh AM, Rowland BD (2014) Cohesin and its regulation: on the logic of X‐shaped chromosomes. Dev Cell 31: 7–18 [DOI] [PubMed] [Google Scholar]
- 6. Peters JM, Nishiyama T (2012) Sister chromatid cohesion. Cold Spring Harb Perspect Biol 4: a011130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bloom KS (2014) Centromeric heterochromatin: the primordial segregation machine. Annu Rev Genet 48: 457–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allshire RC, Madhani HD (2017) Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol http://doi.org/10.1038/nrm.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC (2001) Requirement of heterochromatin for cohesion at centromeres. Science 294: 2539–2542 [DOI] [PubMed] [Google Scholar]
- 10. Nonaka N, Kitajima T, Yokobayashi S, Xiao G, Yamamoto M, Grewal SI, Watanabe Y (2002) Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol 4: 89–93 [DOI] [PubMed] [Google Scholar]
- 11. Eissenberg JC, Elgin SC (2014) HP1a: a structural chromosomal protein regulating transcription. Trends Genet 30: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koch B, Kueng S, Ruckenbauer C, Wendt KS, Peters JM (2008) The Suv39h‐HP1 histone methylation pathway is dispensable for enrichment and protection of cohesin at centromeres in mammalian cells. Chromosoma 117: 199–210 [DOI] [PubMed] [Google Scholar]
- 13. Canzio D, Larson A, Narlikar GJ (2014) Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol 24: 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- 15. Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- 16. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113 [DOI] [PubMed] [Google Scholar]
- 17. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD et al (2000) Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- 18. Brasher SV, Smith BO, Fogh RH, Nietlispach D, Thiru A, Nielsen PR, Broadhurst RW, Ball LJ, Murzina NV, Laue ED (2000) The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J 19: 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ (2000) Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol 10: 517–525 [DOI] [PubMed] [Google Scholar]
- 20. Maison C, Almouzni G (2004) HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 5: 296–304 [DOI] [PubMed] [Google Scholar]
- 21. Hirota T, Lipp JJ, Toh BH, Peters JM (2005) Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438: 1176–1180 [DOI] [PubMed] [Google Scholar]
- 22. Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD (2005) Regulation of HP1‐chromatin binding by histone H3 methylation and phosphorylation. Nature 438: 1116–1122 [DOI] [PubMed] [Google Scholar]
- 23. Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y (2003) Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci 116: 3327–3338 [DOI] [PubMed] [Google Scholar]
- 24. Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B (1999) Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 108: 220–234 [DOI] [PubMed] [Google Scholar]
- 25. Higgins JM, Prendergast L (2016) Mitotic mysteries: the case of HP1. Dev Cell 36: 477–478 [DOI] [PubMed] [Google Scholar]
- 26. Tanno Y, Susumu H, Kawamura M, Sugimura H, Honda T, Watanabe Y (2015) The inner centromere‐shugoshin network prevents chromosomal instability. Science 349: 1237–1240 [DOI] [PubMed] [Google Scholar]
- 27. Guenatri M, Bailly D, Maison C, Almouzni G (2004) Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol 166: 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shimura M, Toyoda Y, Iijima K, Kinomoto M, Tokunaga K, Yoda K, Yanagida M, Sata T, Ishizaka Y (2011) Epigenetic displacement of HP1 from heterochromatin by HIV‐1 Vpr causes premature sister chromatid separation. J Cell Biol 194: 721–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Serrano A, Rodriguez‐Corsino M, Losada A (2009) Heterochromatin protein 1 (HP1) proteins do not drive pericentromeric cohesin enrichment in human cells. PLoS ONE 4: e5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamagishi Y, Sakuno T, Shimura M, Watanabe Y (2008) Heterochromatin links to centromeric protection by recruiting shugoshin. Nature 455: 251–255 [DOI] [PubMed] [Google Scholar]
- 31. Abe Y, Sako K, Takagaki K, Hirayama Y, Uchida KS, Herman JA, DeLuca JG, Hirota T (2016) HP1‐assisted Aurora B kinase activity prevents chromosome segregation errors. Dev Cell 36: 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang J, Chaudhary J, Dong H, Kim S, Brautigam CA, Yu H (2011) Mitotic centromeric targeting of HP1 and its binding to Sgo1 are dispensable for sister‐chromatid cohesion in human cells. Mol Biol Cell 22: 1181–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hahn M, Dambacher S, Dulev S, Kuznetsova AY, Eck S, Worz S, Sadic D, Schulte M, Mallm JP, Maiser A et al (2013) Suv4‐20h2 mediates chromatin compaction and is important for cohesin recruitment to heterochromatin. Gene Dev 27: 859–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiyomitsu T, Iwasaki O, Obuse C, Yanagida M (2010) Inner centromere formation requires hMis14, a trident kinetochore protein that specifically recruits HP1 to human chromosomes. J Cell Biol 188: 791–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nozawa RS, Nagao K, Masuda HT, Iwasaki O, Hirota T, Nozaki N, Kimura H, Obuse C (2010) Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol 12: 719–727 [DOI] [PubMed] [Google Scholar]
- 36. Chakraborty A, Prasanth KV, Prasanth SG (2014) Dynamic phosphorylation of HP1alpha regulates mitotic progression in human cells. Nat Commun 5: 3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X, Song Z, Huo Y, Zhang J, Zhu T, Wang J, Zhao X, Aikhionbare F, Duan H, Wu J et al (2014) Chromatin protein HP1 interacts with the mitotic regulator borealin protein and specifies the centromere localization of the chromosomal passenger complex. J Biol Chem 289: 20638–20649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hengeveld RCC, Vromans MJM, Vleugel M, Hadders MA, Lens SMA (2017) Inner centromere localization of the CPC maintains centromere cohesion and allows mitotic checkpoint silencing. Nat Commun 8: 15542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ainsztein AM, Kandels‐Lewis SE, Mackay AM, Earnshaw WC (1998) INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol 143: 1763–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dai J, Sultan S, Taylor SS, Higgins JM (2005) The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Gene Dev 19: 472–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dai J, Sullivan BA, Higgins JM (2006) Regulation of mitotic chromosome cohesion by Haspin and Aurora B. Dev Cell 11: 741–750 [DOI] [PubMed] [Google Scholar]
- 42. Goto Y, Yamagishi Y, Shintomi‐Kawamura M, Abe M, Tanno Y, Watanabe Y (2017) Pds5 regulates sister‐chromatid cohesion and chromosome bi‐orientation through a conserved protein interaction module. Curr Biol 27: 1005–1012 [DOI] [PubMed] [Google Scholar]
- 43. Zhou L, Liang C, Chen Q, Zhang Z, Zhang B, Yan H, Qi F, Zhang M, Yi Q, Guan Y et al (2017) The N‐terminal non‐kinase‐domain‐mediated binding of haspin to Pds5B protects centromeric cohesion in mitosis. Curr Biol 27: 992–1004 [DOI] [PubMed] [Google Scholar]
- 44. Liang C, Chen Q, Yi Q, Zhang M, Yan H, Zhang B, Zhou L, Zhang Z, Qi F, Ye S et al (2018) A kinase‐dependent role for Haspin in antagonizing Wapl and protecting mitotic centromere cohesion. EMBO Rep 19: 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y et al (2013) Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang T, Wei JJ, Sabatini DM, Lander ES (2014) Genetic screens in human cells using the CRISPR‐Cas9 system. Science 343: 80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ (2011) Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr Biol 21: 1018–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gorbsky GJ (2013) Cohesion fatigue. Curr Biol 23: R986–R988 [DOI] [PubMed] [Google Scholar]
- 51. Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S et al (2008) Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci USA 105: 3443–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elbatsh AMO, Medema RH, Rowland BD (2014) Genomic stability: boosting cohesion corrects CIN. Curr Biol 24: R571–R573 [DOI] [PubMed] [Google Scholar]
- 53. McKinley KL, Cheeseman IM (2017) Large‐scale analysis of CRISPR/Cas9 cell‐cycle knockouts reveals the diversity of p53‐dependent responses to cell‐cycle defects. Dev Cell 40: 405–420.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED (2004) Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J 23: 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nielsen AL, Oulad‐Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R (2001) Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell 7: 729–739 [DOI] [PubMed] [Google Scholar]
- 56. Pluta AF, Saitoh N, Goldberg I, Earnshaw WC (1992) Identification of a subdomain of CENP‐B that is necessary and sufficient for localization to the human centromere. J Cell Biol 116: 1081–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamagishi Y, Honda T, Tanno Y, Watanabe Y (2010) Two histone marks establish the inner centromere and chromosome bi‐orientation. Science 330: 239–243 [DOI] [PubMed] [Google Scholar]
- 58. Smothers JF, Henikoff S (2000) The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr Biol 10: 27–30 [DOI] [PubMed] [Google Scholar]
- 59. Ouyang Z, Zheng G, Tomchick DR, Luo X, Yu H (2016) Structural basis and IP6 requirement for Pds5‐dependent cohesin dynamics. Mol Cell 62: 248–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT, Gorbsky GJ, Higgins JM (2010) Histone H3 Thr‐3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 330: 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marston AL (2015) Shugoshins: tension‐sensitive pericentromeric adaptors safeguarding chromosome segregation. Mol Cell Biol 35: 634–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu H, Rankin S, Yu H (2013) Phosphorylation‐enabled binding of SGO1‐PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat Cell Biol 15: 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hara K, Zheng G, Qu Q, Liu H, Ouyang Z, Chen Z, Tomchick DR, Yu H (2014) Structure of cohesin subcomplex pinpoints direct shugoshin‐Wapl antagonism in centromeric cohesion. Nat Struct Mol Biol 21: 864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nishiyama T, Sykora MM, Huis in ‘t Veld PJ, Mechtler K, Peters JM (2013) Aurora B and Cdk1 mediate Wapl activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc Natl Acad Sci USA 110: 13404–13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K et al (2010) Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 143: 737–749 [DOI] [PubMed] [Google Scholar]
- 66. Rankin S, Ayad NG, Kirschner MW (2005) Sororin, a substrate of the anaphase‐promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell 18: 185–200 [DOI] [PubMed] [Google Scholar]
- 67. Schmitz J, Watrin E, Lenart P, Mechtler K, Peters JM (2007) Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol 17: 630–636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5